

Российский физиологический журнал им. И.М. Сеченова, 2021, T. 107, № 4-5, стр. 568-583
Применение метода экспансионной микроскопии в нейробиологии
К. З. Деревцова 1, *, Е. И. Пчицкая 1, А. В. Раковская 1, И. Б. Безпрозванный 1, 2, **
1 Лаборатория молекулярной нейродегенерации Санкт-Петербургского политехнического университета Петра Великого
Санкт-Петербург, Россия
2 Отделение физиологии Юго-Западного Медицинского Центра Техасского университета
Даллас, США
* E-mail: derevtcova19@yandex.ru
** E-mail: mnlabspb@gmail.com
Поступила в редакцию 20.01.2021
После доработки 15.02.2021
Принята к публикации 16.02.2021
Аннотация
Многие биологические исследования требуют анализа ультраструктурных изменений на уровне клеточных органелл и молекул. Разрешающая способность современных конфокальных микроскопов ограничена дифракционный пределом (200–300 нм), в связи с чем изучать с помощью стандартной флуоресцентной микроскопии столь малые объекты невозможно. Методы микроскопии сверхвысокого разрешения требуют дорогостоящего оборудования и являются технически трудными в использовании, что в свою очередь ограничивает их повсеместное применение. Однако в последнее время появились методы, позволяющие увеличить разрешение микроскопии не за счет усовершенствования системы регистрации изображения, а посредством физического изотропного расширения биологического образца с помощью управляемого химического процесса. Благодаря этому методу, получившему название экспансионная или расширительная микроскопия (ExM), стало возможным получать трехмерные изображения образцов с разрешением достаточным для изучения отдельных органелл клетки с использованием обычного конфокального микроскопа. В статье рассмотрены методические особенности применения ExM при изучении образцов тканей головного мозга с приведением алгоритма, по которому можно осуществлять адаптацию стандартного протокола под цели и задачи конкретного исследования. Кроме того, рассматривается история возникновения данного метода, его основные принципы и примеры использования в различных областях биологии и медицины, а также отражены будущие направления для совершенствования данной технологии.
ИСТОРИЯ ВОЗНИКНОВЕНИЯ И РАЗВИТИЕ МЕТОДА ЭКСПАНСИОННОЙ МИКРОСКОПИИ
В 1970-х годах было открыто и исследовано явление расширения полиэлектролитных гелей с помощью воды [1]. Гель, представляющий собой сеть из полиэлектролита, подвергается гидролизу, ионизируется и при помещении в воду образовавшиеся противоионы создают осмотическое давление, приводящее к увеличению геля в размере до десятков раз [2]. Профессор Boyden и его коллеги предприняли попытку заключить биологический образец в гель, после чего расширить его в воде, используя данную особенность [3]. Предварительно, для достижения анизотропности расширения, образец подвергался гидролизу белков с помощью протеиназы К. Для визуализации клеточных структур были использованы антитела, конъюгированные с содержащим флюрофор ДНК-олигонуклеотидом. ДНК-олигонуклеотид сшивался с гелем через метакрилоиловую группу (methacryloyl group), благодаря чему флуоресцентная метка сохраняла свою локализацию относительно геля при расширении. Такое физическое увеличение расстояния между наноразмерными структурами и молекулами позволяет разнести их в пространстве на большие расстояния и, таким образом, преодолеть дифракционный предел обычного конфокального микроскопа. Разработанный метод получил название экспансионная микроскопия (Expansion Microscopy, ExM) [3]. Авторы методики исследовали ее на предмет погрешности при расширении и пришли к выводу, что она значительно меньше потенциального увеличения разрешения микроскопии, достигаемого при данном подходе. Авторам удалось расширить гистологический препарат в 5 раз и достичь латерального разрешения в 70 нм с помощью обычного оптического микроскопа. Для сравнения – в оптической микроскопии латеральное разрешение определяется формулой d = 0.61λ/NA, где NA = n sin α – числовая апертура объектива, n – коэффициент преломления среды, α – угол между оптической осью объектива и наиболее отклоняющимся лучом, попадающим в объектив (апертурный угол), λ – длина световой волны. Таким образом, с использованием иммерсионного масла и объективов с большой апертурой (NA = 1.4) в латеральной плоскости можно достичь максимального разрешения около 200 нм при использовании длины волны возбуждения в диапазоне между 400 и 650 нм, характерной для конфокальной микроскопии [4]. При этом размеры надмолекулярных структур нейрона, таких как микротрубочки, рибосомы, рецепторные белки, ядерные поры не превышают 150 нм, а клеточные мембраны имеют толщину менее 10 нм [5]. Таким образом, использование экспансионной микроскопии открывает возможность визуализации и изучения структуры таких малых объектов без необходимости использовать микроскопию сверхвысокого разрешения.
Далее исследовательская группа под руководством профессора Boyden модернизировала метод экспансионной микроскопии для детектирования РНК, посредством сшивки молекул данного типа с гелем через низкомолекулярный линкер [6]. После расширения геля РНК визуализируется с помощью флуоресцентной in situ гибридизации (FISH) с точностью до одной молекулы как в культивируемых клетках, так и в срезах головного мозга. Метод получил название ExFISH (Expansion FISH, экспансионная флуоресцентная in situ гибридизация), и он позволил изучать структуру и местоположение РНК с высоким разрешением, и что также важно, в толстых образцах, которые ранее были недоступны для проведения FISH и визуализации на большой глубине. Позднее в комбинации с экспансионной микроскопией была успешно применена ДНК in situ гибридизация [7].
Синтез конъюгированных с олигонуклеотидом антител является дорогостоящей процедурой, которую необходимо выполнять для каждого нового исследуемого белка. Поэтому следующим шагом в развитии данного метода стала его модификация, позволяющая напрямую сшивать белковые молекулы с гелем. Это открыло возможность для использования экспрессируемых в клетках флуоресцентных белков и стандартного иммуноокрашивания с помощью вторичных флуоресцентно-меченных антител для визуализации структур в ExM. Для этого было предложено использовать малые молекулы MA-NHS (N-гидроксисукцинимидиловый эфир метакриловой кислоты) [7] и Акрилоил-X (6-(акрилоил)амино)гексановая кислота, сложный эфир сукцинимидила, AcX) [8]. При этом подходе количество сшивок антитела с гелем оказалось достаточным для сохранения флуоресценции путем удержания частей белка в геле после протеолиза с использованием протеиназы К. Метод получил название удерживающая белки экспансионная микроскопия (Protein retention expansion microscopy, ProExpM). Подобный качественный скачок привел к широкому распространению данного метода, который позволяет получать трехмерные изображения образцов с наноразмерной точностью.
Следующим этапом в развитии технологии стало увеличение возможного расширения образца с целью достижения более высокого разрешения объектов. Итеративная экспансионная микроскопия (iExM) позволила расширить образец до 20 раз и достичь разрешения до 25 мкм на стандартных флуоресцентных микроскопах [10]. Для этого гель после расширения снова помещался в гелевую матрицу и подвергался повторному расширению. В данном методическом подходе использовались конъюгированные с ДНК антитела как в оригинальном протоколе [3]. С помощью этого метода авторы визуализировали синаптические белки и дендритные шипики в головном мозге мыши. В оптимизации протокола, проведенной другой исследовательской группой, благодаря улучшению процесса гелеобразования и сшивки белков с матриксом геля удалось достичь десятикратного увеличения образца [9]. Данный подход совместим для использования с флуоресцентными белками.
МЕТОДЫ ИССЛЕДОВАНИЯ
Все эксперименты проводились на 18 самцах и самках мышей трансгенной линии M Thy1-GFP, экспрессирующих флуоресцентный белок GFP в отдельных нейронах. Кроме того, в работе были использованы 3 самца линии мышей 5xFAD-М, также экспрессирующих флуоресцентный белок GFP в отдельных нейронах и содержащих тройную мутацию в гене, кодирующем APP белок, и двойную мутацию в гене, кодирующем пресенилин (обнаруживаемые у человека при наследственных формах болезни Альцгеймера). План экспериментальных исследований на животных был рассмотрен и утвержден комиссией по биоэтике Института биомедицинских систем и биотехнологий ФГАОУ ВО “Санкт-Петербургский политехнический университет Петра Великого” и соответствует положениям “Европейской конвенции о защите позвоночных животных, используемых для экспериментальных и других научных целей” (Страсбург, 1986) и принципам Хельсинкской декларации Всемирной медицинской ассоциации о гуманном обращении с лабораторными животными (1996).
Для проведения перфузии животных анестезировали путем внутрибрюшинного введения смеси золетила и ксилазина (40 мкл золетила, Zoletil Virbac, Франция + + 160 мкл 0.9%-ного раствора NaCl и 30 мкл ксилазина + 170 мкл раствора NaCl). Итоговую смесь вводили в дозе 150 мкл животным внутрибрюшинно, далее, если это было необходимо, вводили дополнительно эту смесь под холку животному в объеме 20–30 мкл.
Изготовление фронтальных срезов мозга
Фиксацию ткани проводили с помощью интракардиальной перфузии с использованием перистальтического насоса. Для перфузии в качестве промывочного буфера использовали фосфатно-солевой буфер, в качестве фиксирующего раствора – 4%-ный раствор параформальдегида. Далее аккуратно извлекали мозг и помещали его в 4%-ный раствор параформальдегида для дальнейшей фиксации. Мозг фиксировали неделю, далее изготавливали фронтальные срезы мозга различной толщины (50, 80 и 100 мкм) с помощью вибрационного микротома Campden 5100MZ-plus и помещали их в 1.5%-ный раствор параформальдегида.
Флуоресцентное мечение объектов
Детектирование исследуемых белков может быть выполнено путем стандартной процедуры иммуноокрашивания с помощью иммуноглобулинов, коньюгированных с флюрофорами. Для ProExM могут быть использованы наработанные в клетке флуоресцентные белки, сигнал которых может быть усилен с помощью иммуноокрашивания с первичными антителами против данного флуоресцентного белка, где флюорофор вторичных антител обладает схожим с ним спектром возбуждения и испускания света. Для детекции ДНК или РНК используют флуоресцентно-меченные зонды (флуоресцентная in situ гибридизация) [6, 7]. Процедура окрашивания проводится в соответствии с протоколами, предлагаемыми производителями.
Для удаления остатков параформальдегида срезы после фиксации помещали в раствор 100 мM ${\text{N}}{{{\text{H}}}_{{\text{4}}}}{\text{Cl}}$ в фосфатно-солевом буфере, и затем осуществляли пермеабилизацию фиксированной ткани, выдерживая ее в течение 15 мин в 0.1%-ном (вес/объем) растворе Triton X-100 в фосфатно-солевом буфере. Далее срезы отмывали в фосфатно-солевом буфере 3 раза по 5 мин, а затем помещали в блокирующий буфер (5%-ный раствор бычьего сывороточного альбумина в фосфатно-солевом буфере) на 6 ч при комнатной температуре. Далее срезы повторно отмывали в фосфатно-солевом буфере 3 раза по 5 мин и далее инкубировали в растворе, содержащем первичные кроличьи анти-GFP (Sigma, #G-1544) в разведении 1/1000 на ночь на шейкер при 4°С. Затем удаляли раствор с антителами и промывали в фосфатно-солевом буфере 4 раза по 30 мин для удаления несвязанных первичных антител. Далее инкубировали ткани с вторичными антителами (иммунофлуоресцентные антитела Life Technologies, Alexa Fluor 488, #A11008) в разведении 1/2000 в блокирующем буфере на шейкере на низкой скорости в течение ночи при комнатной температуре или при 4°C. После окрашивания удаляли раствор с антителами и промывали в фосфатно-солевом буфере 4 раза по 30 мин. Далее применяли протокол для экспансионной микроскопии.
Протокол экспансионной микроскопии
Все варианты протокола ExM имеют схожую логическую схему, результатом применения которой является получение изотропно расширенных образцов тканей или культуры клеток. Протокол экспансионной микроскопии состоит из семи этапов: флуоресцентное мечение объектов, закрепление, полимеризация, гомогенизация, расширение, визуализация и проверка расширенного образца на наличие искажений при расширении [3]. Далее рассмотрим подробно каждый этап.
Закрепление (анкеровка). Это процесс химической обработки образца для обеспечения корректного сшивания использованных для детекции объектов исследования, флуоресцентных зондов, флюорофоров и белков с гелем. Этот этап очень важен, так как способствует физическому удержанию флюорофоров в толще геля, сохраняя при этом их исходное топографическое расположение. Первоначально этот этап реализовывали с помощью технологии связывания антител, конъюгированных с ДНК-олигонуклеотидами. Антитела, связанные с комплементарными ДНК-олигонуклеотидами, также содержали в своем составе акрилатную группу, которая “сшивала” ДНК олигонуклеотиды с гелевым матриксом [3]. В настоящее время эта процедура заменена на более удобный метод прямой сшивки белков при помощи малых молекул MA-NHS [8] или Acryloyl-X [10], которые напрямую связываются с белками через N-сукцинимидилметакрилат и, таким образом, через дополнительную акрилатную группу закрепляются в матрице геля. В наших протоколах мы осуществляли этап закрепления перед этапом гелеобразования, для чего окрашенные срезы помещали в раствор 1 мМ MA-NHS (Sigma, #730300-1G) в фосфатно-солевом буфере на 1 ч при комнатной температуре.
Полимеризация. Этап, при котором в камеру, где находится исследуемый образец, заливают в раствор для полимеризации. Как правило, для лучшего проникновения геля в образец ткани он предварительно выдерживается в растворе для полимеризации небольшой промежуток времени в условиях, предотвращающих начало полимеризации. Затем образцы переносятся из раствора и закрепляются в фиксированных формах для заливки (рис. 1–3). Далее происходит полимеризации геля, в результате которой гель с образцом твердеет.
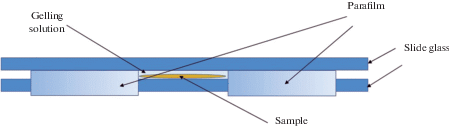
Рис. 2.
Фотография предметного стекла с расположенным по центру срезом, подготовленным для дальнейшей полимеризации в термостате. (a) – вид сбоку, (b) – вид сверху.


Необходимо, чтобы все стоковые растворы для полимеризации и гомогенизации были приготовлены заранее. Составы стоковых растворов для полимеризации приведены ниже в таблицах: “Stock X” (табл. 1), “4HT”, “TEMED”, “APS” (табл. 2). Состав лизирующего буфера для гомогенизации приведен в табл. 3.
Таблица 1.
Концентрации компонентов для приготовления стокового мономерного раствора
| Раствор мономеров (“Stock X”) |
Концентрация в стоковом
растворе (г/100 мл раствора) |
Количество (мл) |
|---|---|---|
| Полиакрилат натрия (Sigma, #408220-100G) |
38 (33 wt % из-за более высокой плотности) | 2.25 |
| Акриламид (Sigma, #A9099-500G) |
×50 | 0.5 |
| N, N'-метиленбисакриламид (VWR Life Science AMRESCO, #Am-O172-0.05) |
2 | 0.75 |
| Хлорид натрия | 29.2 (5 M) | 4 |
| Фосфатно-солевой буфер, 10×кратный | 10 | 1 |
| Вода | до 100 мл | 0.9 |
| Общий объем | 100 мл | 9.4 |
Таблица 2.
Концентрации компонентов для приготовления стоковых растворов TEMED, APS, 4HT
| Название раствора | Химическое вещество | Концентрация для стокового раствора (г/100 мл) |
|---|---|---|
| TEMED | Тетраметилэтилендиамин (Helicon, #L-0847) |
10 |
| APS | Пероксодисульфат аммония (Sigma, #A3678-25G) |
10 |
| 4HT | 4-Hydroxy-TEMPO (Sigma, #176141-1G) |
0.5 |
Таблица 3.
Концентрации реагентов для приготовления лизирующего буфера для гомогенизации
| Реагент | Количество | Конечная концентрация (100 мл раствора) |
|---|---|---|
| Triton X-100 | 2.5 мл | 0.50 г |
| EDTA, disodium (Дина́триевая соль этилендиаминтетрау́ксусной кислоты́, 0.5 M, pH 8) | 1 мл | 0.2 мл |
| Tris Cl (1 M) водный раствор, pH 8 | 25 мл | 5 мл |
| Хлорид натрия | 23.38 г | 4.67 г |
| Вода | Довести до 500 мл | |
| Протеиназа K (800 Е/мл) | 1 : 100 разведение | 800 Е (= 8 Е/мл) |
| Общий объем | 500 мл |
Для приготовления полимеризующего раствора смешиваем стоковые растворы в следующих пропорциях 47 : 1 : 1 : 1. Растворение стоковых растворов происходит в следующей последовательности: сначала к раствору Stock X добавляем 4HT, затем TEMED, и в последнюю очередь APS. Таким образом, для 1 мл гелеобразующего раствора брали:
940 мкл Stock X (мономерный раствор),
20 мкл 4НТ (раствор, ингибирующий полимеризацию),
20 мкл TEMED (раствор, ускоряющий полимеризацию),
20 мкл APS (раствор, инициирующий полимеризацию).
Далее в полученный раствор помещали срезы мозга и выдерживали на холодном блоке в течение 30 мин. Для заливки среза в гелеобразующий раствор конструировали камеру из предметного стекла и двух ограничителей с боков. В качестве ограничителей использовали пленку парафилм (Parafilm), закрепленную с двух сторон предметного стекла в три слоя (рис. 1). Сверху накрывали предметным стеклом, также обернутым в пленку, чтобы улучшить адгезию между двумя прижатыми стеклами (рис. 1).
Срезы помещали по центру этой камеры (рис. 2). Затем заливали гелеобразующий раствор и аккуратно закрывали сверху предметным стеклом, исключая смещение среза в стороны (рис. 2). Залитые в камерах срезы аккуратно переносили в контейнер и помещали термостат при температуре 37°C на 1.5 ч, и после полимеризации срезы освобождали из камер и помещали в лизирующий буфер на 4–8 ч в зависимости от толщины среза.
Гомогенизация. Далее для обеспечения корректного изотропного расширения препарата проводится его ферментативная или тепловая денатурация. Этот шаг предотвращает деформационные изменения образца во время последующего расширения. Осуществляется путем инкубации с участием фермента протеиназы K, а также с помощью автоклавирования в лизирующем буфере, содержащем 5% Triton X-100 и 1% SDS, в течение 1 ч при температуре 121°C [8]. Требуется подбор условий гомогенизации для обеспечения равномерного расширения образца с сохранением максимально возможной флуоресценции объектов. Для гомогенизации мы использовали протеиназу К в концентрации 800 Е (8 Е/мл).
Расширение. После гомогенизации гель с образцом аккуратно переносится в емкости большего размера и подвергается гидролитическому расширению (рис. 4). Расширение достигается за счет вымывания ионов, удерживающих расширение геля, из гелевой матрицы с помощью дважды дистиллированной воды (ddH2O). Классический протокол ExM включает в себя три повтора погружения геля с препаратом в ddH2O, что в конечном итоге, приводит к его 4-кратному увеличению [3]. Современные протоколы позволяют расширять исследуемый образец более чем в 10 раз [11, 12].
Рис. 4.
Соотношение размеров исходного образца и увеличенного образца в геле, полученного с помощью анизотропного расширения полиэлектролитного геля. Слева – срезы мозга до увеличения, справа – срез мозга после увеличения. Линейкой обозначен реальный размер образцов.

Мы проводили расширение геля, добавляя дистиллированную воду по 20 мин 3 раза. Следует отметить, что с расширенными срезами ввиду их хрупкости следует обращаться очень аккуратно. Переносить срез на стекло можно с помощью кисти и покровного стекла, как показано на рис. 3.
Следуя описанному выше протоколу, удалось увеличить исследуемый образец более чем в 4.5 раза (рис. 4).
Визуализация. Далее осуществляется этап визуализации расширенного образца с помощью любого доступного конфокального или эпифлуоресцентного микроскопа [3, 11]. На этом этапе образцы геля с заключенным в его матрицу препаратом исследуемой ткани аккуратно переносили на стекла и анализировали на конфокальном микроскопе фирмы Thorlabs. Как уже отмечалось выше, применение протокола экспансионной микроскопии позволило увеличить образец более чем в 4 раза (рис. 4).
В настоящее время описаны успешные примеры использования микроскопии сверхвысокого разрешения для анализа гелей после экспансионной микроскопии (SIM, STED, STORM), что позволяет достичь еще большего разрешения [13–16]. Так как препарат, залитый в гель, после расширения имеет большие размеры, для визуализации необходимо разделить его на более мелкие составляющие, также возможно выбрать область для визуализации, представляющую интерес в данном конкретном эксперименте. Во время получения изображений гель должен находиться в водном растворе или влажной среде для предотвращения его высыхания и последующего сжимания. Наиболее удобным способом является визуализация геля в водной среде с помощью водного объектива. Для закрепления геля возможно использовать камеру для прижизненной микроскопии, или сконструировать новую, например, из чашки Петри. Для лучшего сцепления геля с поверхностью ее можно обработать раствором поли-D-лизина. Возможно визуализировать гель с использованием масляных объективов с высокой числовой апертурой, при этом проложить между образцом и объективом тонкое покровное стекло. Тем не менее, из-за значительной толщины геля не всегда возможно использовать данный способ.
Проверка расширенного образца на наличие искажений при расширении. После визуализации полученных образцов необходимо провести экзаменацию на наличие искажений нормальной формы и расположения исследуемых биологических объектов. Для выполнения данного анализа помимо ручного или полуавтоматического совмещения изображений образца до и после расширения доступны программные продукты для автоматического определения возникающих в ходе ExM искажений [11]. Гели со значительными искажениями объектов не используются для анализа. В этом случае требуется дополнительная оптимизация процессов анкеровки и гомогенизации для получения стабильно воспроизводимого результата. Также необходимо оценить степень достигнутого увеличения образца путем анализа изменений в размере/весе геля или, используя информацию о размере исследуемых объектов до и после расширения [11].
Поскольку в задачи исследования не входило измерение размерных характеристик изучаемых объектов, на которые могло оказать влияние искажение в результате расширения, мы оценивали препарат на возможное искажение формы нейрона и его частей (шипики, ядро) только визуально.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Методику экспансионной микроскопии проводили, используя 5 вариантов протокола. Важным критерием отбора подходящего протокола являлась способность максимально сохранять сигнал, испускаемый несколькими флюорофорами, наряду с расширением, достаточным для удовлетворительной визуализации клеточных органелл. За основу был взят протокол для proExM [17], при этом варьировали продолжительность инкубирования в растворах для сшивки с полиэлектролитным гелем и в лизирующем буфере, а также изменяли концентрацию протеиназы К в лизирующем буфере в зависимости от толщины среза.
Таким образом, удалось показать, что продолжительность инкубирования в растворе для закрепления, содержащем MA-NHS, не влияла на эффективность свечения флюорофоров. Испытание различных вариантов инкубирования с протеиназой К для ферментированной гомогенизации показало, что сокращение времени инкубирования приводит к недостаточному расширению препарата (рис. 6).
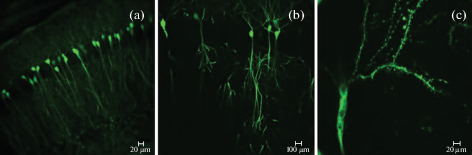
При варианте протокола № 1 продолжительность инкубирования в растворе для закрепления, содержащем MA-NHS, составила 12 ч, а ферментативная обработка протеиназой К была сокращена до 2-х ч в термостате при 37°C (рис. 5). Использование этого протокола позволило расширить препарат примерно в 2 раза, что является недостаточным для оценки морфологии шипиков и детектирования ультраструктурных изменений в клетке.
Рис. 5.
Конфокальные изображения нейронов и дендритов, локализованных в CA1 области гиппокампа мышей линии M Thy1-GFP, до применения экспансионной микроскопии (a) и после (b) и (c). (a, b) – объектив с 20-кратным увеличением, (c) – с 60-кратным увеличением. Длина масштабного отрезка 20 мкм. Иммунодетекцию проводили первичными кроличьими антителами к GFP белку (Sigma, #G-1544) и иммунофлуоресцентными антителами Alexa Fluor 488 (Life Technologies, #A11008).

Применение второго варианта протокола, при котором продолжительность инкубирования в растворе для закрепления, содержащем MA-NHS, составила 1 ч, а ферментативная обработка протеиназой К была сокращена до 2-х ч в термостате при 37°C, привела к аналогичному результату: препарат также удалось расширить примерно в 2 раза.
Рис. 6.
Конфокальные изображения нейронов и дендритов, локализованных в CA1 области гиппокампа мышей линии M Thy1-GFP, до применения экспансионной микроскопии (a) и после (b) и (c). (a, c) – объектив с 20-кратным увеличением, (b) – с 60-кратным увеличением. Длина масштабного отрезка 100 и 20 мкм. Иммунодетекцию проводили первичными кроличьими антителами к GFP белку (Sigma, #G-1544) и иммунофлуоресцентными антителами Alexa Fluor 488 (Life Technologies, #A11008).
Вариант протокола № 3: продолжительность инкубирования в растворе для закрепления, содержащем MA-NHS, составила 12 ч, а ферментативная обработка протеиназой К проводилась в течение 4-х часов при комнатной температуре (рис. 6). Сокращение длительности ферментативной обработки с целью сохранения сигнала, испускаемого флюорофором, привело к недостаточному расширению препарата.
Аналогичный результат недостаточного расширения был получен при применении варианта протокола № 4, при котором продолжительность инкубирования в растворе для закрепления, содержащем MA-NHS, составила 1 ч, а ферментативная обработка протеиназой К проводилась в течение 4-х ч при комнатной температуре.
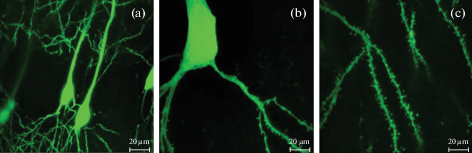
Таким образом, в результате подбора условий для оптимального расширения образца с сохранением удовлетворительного уровня свечения флюорофора оптимальная продолжительность инкубирования в растворе для закрепления, содержащем MA-NHS, составила 1 ч, а ферментативная обработка протеиназой К – в течение 5-ти ч при комнатной температуре. Это позволило осуществить расширение образца и сохранить достаточный уровень экстинкции флюорофора в толще геля (рис. 7). С помощью этого варианта удалось добиться увеличения структуры нейронов в 3.5–4 раза, что позволило визуализировать мельчайшие детали их морфологии, включая дендритные шипики, и при этом сохранить интенсивность свечения флюорофоров, которые мы использовали в исследовании (рис. 7). Это, в свою очередь, позволило визуализировать объекты, экспрессирующие флюорофор, без дополнительного инкубирования с иммуноглобулинами к GFP уже после этапа расширения. Расширенные препараты, залитые в гелевый матрикс, как правило, помещены в большие емкости (обычно чашка Петри), что многократно увеличивает расход антител, используемых для дополнительного усиления сигнала флюорофора. Конечная стоимость такого исследования сильно возрастает. Кроме того, обработка антителами расширенных образцов достаточно трудоемка и занимает больше времени.
Рис. 7.
Конфокальные изображения нейронов CA1 области гиппокампа мышей линии M Thy1-GFP до (a) и после (b, c) применения метода экспансионной микроскопии получены на микроскопе Thorlabs c увеличением 60×. Длина масштабного отрезка 20 мкм. Иммунодетекцию проводили первичными кроличьими антителами к GFP белку (Sigma, #G-1544) и иммунофлуоресцентными антителами Alexa Fluor 488 (Life Technologies, #A11008).
Подобранный протокол обработки тканей позволил также провести окрашивание срезов, содержащих эндогенный флюорофор, с антителами, конъюгированными с флюорофором, имеющим другой спектр возбуждения и испускания света. Для этой цели было проведено расширение срезов мозга, которые дополнительно окрашивали первичными антителами к IP3R. Были подобраны кроличьи поликлональные иммуноглобулины фирмы Abcam (кат. номер: ab5804) и вторичные Alexa Fluor® 594, которые подходили для использования с ExM. Для окрашивания использовали срезы мозга мышей линии 5xFAD-M, нейроны которого экспрессируют флуоресцентный белок GFP. Благодаря снижению продолжительности инкубирования с протеиназой К, удалось сохранить удовлетворительный сигнал, испускаемый белком GFP после расширения срезов, что позволило получить двухцветную микрофотографию, полученную слиянием двух каналов (Alexa Fluor® 594 и GFP) (рис. 8).
Рис. 8.
Конфокальные изображения нейронов, локализованных в области задней теменной коры, экспрессирующих белки IP3R и GFP, после экспансионной микроскопии (в качестве первичных антител – кроличьи поликлональные иммуноглобулины (Abcam, ab5804) и GFP на срезах мозга мышей линии 5xFAD-M). (a) – микрофотография с нейронами, экспрессирующими зеленый флуоресцентный белок GFP, (b) – микрофотография с нейронами, окрашенными антителами на IP3R, вторичными – Alexa Fluor 594, (c) – двухцветная микрофотография, полученная слиянием двух каналов (Alexa Fluor 594 и GFP). На срезах мозга представлены нейроны, локализованные в ассоциативной зоне задней теменной коры, слой 6b (PTLp), координаты относительно точки Bregma: AP = –0.8 мм, ML = ±2.00 мм и DV = –2.0 мм. Увеличение объектива ×10. Масштабный отрезок равен 100 мкм.

Таким образом, примененный адаптированный протокол экспансионной микроскопии позволил осуществить расширение препаратов без значительной потери уровней экстинкции как эндогенных флюорофоров, так и введенных на этапе иммуногистохимического окрашивания, что дает возможность применять этот метод при изучении колокализации различных белковых комплексов и молекул с использованием нескольких флуоресцентных меток, в том числе и эндогенно синтезируемых, без дополнительной иммунодетекции флуоресцентными антителами.
Отличием от стандартных протоколов является более бережная обработка ферментами на стадии расширения срезов, которая позволяет снизить разрушительное действие протеиназ на иммуноглобулины, используемые для иммуногистохимической детекции объектов. Кроме того, такая обработка позволяет сохранять сигнал, испускаемый эндогенно синтезируемым флюорофором, что освобождает исследователя от необходимости дополнительного подкрашивания срезов с целью визуализации морфологии. Подобным же образом применяемый протокол подойдет и для “тройной” флуоресценции, где в качестве одного из флюорофоров будет использоваться тот, что уже экспрессируется в клетках, а два остальных флюорофора будут введены на этапе окрашивания препаратов. Эти приемы позволят прежде всего минимизировать расходы и сократить трудоемкость.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Появление метода ExM сделало возможным изучение наноразмерных структур и комплексов, таких как отдельные органеллы клетки, используя обычный микроскоп, ограниченный пределом дифракции. Таким образом, исследование колокализации регуляторных молекул, а также характера межсинаптических контактов между нервными клетками стало доступным для изучения на расширенных образцах. Кроме того, оптическая прозрачность получаемых образцов позволила беспрепятственно отслеживать в толще препарата трехмерную структуру нейрона, а также реконструировать путь от тела нейрона к самым разветвленным и дистальным частям клетки (аксоны, дендриты), что, конечно, дало возможность существенно повысить качество анализа. Экспансионная микроскопия позволила также, расширив биологический образец, сделать возможным выявление эпитопов, которые могли быть скрыты в белковых комплексах, путем отделения белков друг от друга. Эта особенность создала широкие перспективы применения этого метода для одновременного иммуногистохимического окрашивания близкорасположенных белков и молекул РНК.
Проводимые исследования с использованием метода ExFISH с целью картирования мРНК и других нуклеиновых кислот в области нейронов и проводящих путей также продемонстрировали свою актуальность при анализе точной локализации экспрессирующихся регуляторных молекул. Этот метод позволил с точностью до нанометра визуализировать совместное местоположение и идентичность молекул мРНК и белков [6].
С помощью протокола proExM авторам исследования удалось провести картирование и проанализировать архитектонику синапсов в области полосатого тела мыши [18], а также в мозге дрозофилы [19] и рыбок данио [20]. Этим же методом были установлены изменения характеристик астроцитов, локализованных вблизи кровеносных сосудов в образцах головного мозга пациентов с эпилепсией [21]. ProExM также оказался полезным при выполнении исследования планарий Schmidtea mediterranea, которое выявило новый тип глиальных клеток у этого организма [22].
Таким образом, экспансионная микроскопия оказалась особенно полезна для исследований в области нейробиологии. Однако, как и в случае с любой новой технологией, попытки исследователей использовать этот метод в своих работах могут требовать адаптирования существующего протокола для конкретной научной области. Различными исследовательскими группами постоянно вносятся разнообразные технические усовершенствования, расширяющие возможности этого метода.
Основным недостатком метода является деградация флюорофоров, что ограничивает допустимый предел расширения ввиду необходимости соблюсти баланс между потенциальным улучшением разрешения и качеством получаемого изображения. Ограничением метода также является пространственная ошибка, возникающая из-за неравномерности расширения геля и увеличивающаяся пропорционально кратности расширения образца. Так как образец расширяется и по оси z, одним из ограничений методики является невозможность получить изображение при высоких степенях расширения из-за относительно короткой рабочей длины объективов с большой кратностью увеличения и высокой числовой апертурой [23].
В лаборатории создателя метода Boyden было предложено сменить гелевую матрицу для заключения образца в ExM с полиакрилатных гелей, обладающих сетчатой структурой, на гели, состоящие из мономеров тетраэдрической формы. Данные гели обладают более однородной структурой, большей способностью к расширению со значительно меньшей пространственной ошибкой. Преимуществом такого гелевого матрикса также является сохранение флуоресценции широко используемого флюорофора Alexa 647, который не совместим с полиакрилатными гелями [24].
Другой исследовательской группой с целью улучшить сохранение флюорофоров были разработаны трифункциональные анкеры [25]. Они не подвергаются деградации во время полимеризации, гомогенизации или денатурации и легко окрашиваются органическими красителями перед расширением. Предлагаемый анкер состоит из трех частей: коннектора для связывания анкера с антителами или ферментными метками SNAP и CLIP, линкера для связывания с матрицей геля и репортерного участка, содержащего биотин или дигоксигенин для визуализации. После гомогенизации гель окрашивается флуоресцентно-меченным стрептавидином или антителами к дигоксигенину, благодаря чему визуализируется объект интереса. Результаты авторов методики указывают, что такой подход позволяет добиться существенно большей яркости флуоресценции по сравнению с обычной ExM.
С использованием системы биотин-стрептавидин было предложено помимо белков и нуклеиновых кислот изучать и другие молекулы в экспансионной микроскопии, например, липиды, гликаны и малые молекулы, благодаря устойчивости стрептавидина к обработке ферментами и полимеризации [26]. В данной модификации ExM для визуализации биомолекул используется клик-мечение, которое основано на парах функциональных групп, быстро и специфически взаимодействующих друг с другом, например, азид-алкин. Биомолекула, метаболически меченная алкином, взаимодействует с биотин-азидом, а затем с флуоресцентно-меченным стрептавидином. Далее подготовка геля и визуализация осуществляется согласно стандартной методике. Подобная модификации ExM открывает новые возможности для ее использования в имаджинге биологических объектов.
В целом, можно заключить, что методика экспансионной микроскопии удобна в применении для различного рода научно-исследовательских задач. Применяемый в нашей работе протокол показал свою гибкость и простоту в использовании. Реактивы, используемые для выполнения этого метода, общедоступны для лабораторного использования и не требуют специального оборудования и оснащения. В зависимости от целей исследования условия обработки тканей можно варьировать, применяя практически любые флуоресцентные красители.
Подобранный в нашей лаборатории протокол позволил снизить разрушительное действие протеиназ на иммуноглобулины, применяемые для иммуногистохимической детекции объектов, что в свою очередь отменило необходимость дополнительного окрашивания препарата после расширения с целью усилить недостаточный уровень экстинции, наблюдаемый вследствие деградации флюорофоров.
Таким образом, мы можем заключить, что этот метод является доступным и легко реализуемым в рамках стандартного набора инструментов, которыми обычно укомплектована научная лаборатория. Главным достоинством метода является то, что ExM улучшает разрешение изображений, получаемых с использованием стандартного микроскопа, путем физического увеличения образца. При применении апробированного в работе протокола было достигнуто 4-кратное увеличение нейронов мозга мышей, позволившее отразить тонкую структуру клетки с возможностью визуализации дендритных шипиков, клеточных органелл. В будущем мы планируем с помощью этого метода исследовать морфологические изменения в нейронах и отростках на фоне нейродегенеративного процесса, а также при других формах патологии.
Наиболее перспективным, на наш взгляд, направлением в настоящий момент является комбинация ExM с известными технологиями микроскопии сверхвысокого расширения [13–15, 27–29], что позволит достичь еще большего разрешения и визуализировать мельчайшие детали клеточных органелл. Однако пока методы флуоресцентной микроскопии сверхвысокого разрешения весьма сложно и дорогостояще реализуются. Возможно, что дальнейшее развитие методики ExM обещает в скором времени нивелировать существующие недостатки, что сделает ее еще более удобной для использования в нейробиологическом эксперименте.
Список литературы
What Is a Polymer? Live Science. Accessed 17 Feb 2021. https://www.livescience.com/60682-polymers.html
Tanaka T. (1978) Collapse of Gels and the Critical Endpoint. Phys. Rev. Lett. 40: 820–823. https://doi.org/10.1103/PhysRevLett.40.820
Chen F., Tillberg P.W., Boyden E.S. (2015) Expansion microscopy. Science. (80) 347: 543–548. https://doi.org/10.1126/science.1260088
Лежнев Э.И., Попова И.И., Кузьмин С.В., Слащев С.М. (2001). Kонфокальная лазерная сканирующая микроскопия: принципы, устройство, применение (часть 2). Научное приборостроение. 11(3): 26–42. [Lezhnev E.I., Popova I.I., Kuzmin S.V., Slashchev S.M. (2001). Confocal laser scanning microscopy: principles, device, application (part 2). Scientific Instrumentation. 11(3): 26–42 (In Russ)].
Геннис Р. (ред) (1997) Биомембраны. Молекулярная структура и функции. М. Мир [Biomembranes. Molecular structure and function (ed R. Gennis) (In Russ)]. ISBN 5-03-002419-0
Chen F., Wassie A.T., Cote A.J., Sinha A., Alon S., Asano S., Daugharthy E.R., Chang J.B., Marblestone A., Church G.M., Raj A., Boyden E.S. (2016) Nanoscale imaging of RNA with expansion microscopy. Nat. Methods. 13: 679–684. https://doi.org/10.1038/nmeth.3899
Zhao Y., Bucur O., Irshad H., Chen F., Weins A., Stancu A.L., Oh E.Y., Distasio M., Torous V., Glass B., Stillman I.E., Schnitt S.J., Beck A.H., Boyden E.S. (2017) Nanoscale imaging of clinical specimens using pathology-optimized expansion microscopy. Nat. Biotechnol. 35:757–764. https://doi.org/10.1038/nbt.3892
Chozinski T.J., Halpern A.R., Okawa H., Kim H.J., Tremel G.J., Wong R.O.L., Vaughan J.C. (2016) Expansion microscopy with conventional antibodies and fluorescent proteins. Nat. Methods. 13: 485–488. https://doi.org/10.1038/nmeth.3833
Chang J.B., Chen F., Yoon Y.G., Jung E.E. Babcock H., Kang J.S., Asano S., Suk H.J., Pak N., Tillberg P.W., Wassie A.T., Cai D., Boyden E.S. (2017) Iterative expansion microscopy. Nat. Methods. 14: 593–599. https://doi.org/10.1038/nmeth.4261
Tillberg P.W., Chen F., Piatkevich K.D., Zhao Y., Yu C.C., English B.P., Gao L., Martorell A., Suk H.J., Yoshida F., Degennaro E.M., Roossien D.H., Gong G., Seneviratne U., Tannenbaum S.R., Desimone R., Cai D., Boyden E.S. (2016) Protein-retention expansion microscopy of cells and tissues labeled using standard fluorescent proteins and antibodies. Nat. Biotechnol. 34: 987–992.https://doi.org/10.1038/nbt.3625
Truckenbrodt S., Maidorn M., Crzan D., Wildhagen H., Kabatas S., Rizzoli S.O. (2018) X10 expansion microscopy enables 25-nm resolution on conventional microscopes. EMBO Rep. 19: (9): e45836. https://doi.org/10.15252/embr.201845836
Cipriano B.H., Banik S.J., Sharma R., Rumore D., Hwang W., Briber R.M., Raghavan S.R. (2014) Superabsorbent hydrogels that are robust and highly stretchable. Macromolecules. 47: 4445–4452. https://doi.org/10.1021/ma500882n
Cahoon C.K., Yu Z., Wang Y., Guo F., Unruh J.R., Slaughter B.D. Hawley R.S. (2017) Superresolution expansion microscopy reveals the three-dimensional organization of the Drosophila synaptonemal complex. Proc. Natl. Acad. Sci. USA. 114: E6857–E6866. https://doi.org/10.1073/pnas.1705623114
Halpern A.R., Alas G.C.M., Chozinski T.J., Paredez A.R., Vaughan J.C. (2017) Hybrid Structured Illumination Expansion Microscopy Reveals Microbial Cytoskeleton Organization. ACS Nano. 11: 12677–12686. https://doi.org/10.1021/acsnano.7b07200
Gao M., Maraspini R., Beutel O., Zehtabian A., Eickholt B., Honigmann A., Ewers H. (2018) Expansion Stimulated Emission Depletion Microscopy (ExSTED). ACS Nano. 12: 4178–4185. https://doi.org/10.1021/acsnano.8b00776
Gambarotto D., Zwettler F.U., Le Guennec M., Schmidt-Cernohorska M., Fortun D., Borgers S., Heine J., Schloetel J.G., Reuss M., Unser M., Boyden E.S., Sauer M., Hamel V., Guichard P. (2019) Imaging cellular ultrastructures using expansion microscopy (U-ExM). Nat. Methods. 16: 71–74. https://doi.org/10.1038/s41592-018-0238-1
Asano S.M., Gao R., Wassie A.T., Tillberg P.W., Chen F., Boyden E.S. (2018) Expansion Microscopy: Protocols for Imaging Proteins and RNA in Cells and Tissues. Curr. Protoc. Cell. Biol. 80(1): e56. https://doi.org/10.1002/cpcb.56
Crittenden J.R., Tillberg P.W., Riad M.H., Shima Y., Gerfen C.R., Curry J., Housman D.E., Nelson S.B., Boyden E.S., Graybiel A.M. (2016) Striosome-dendron bouquets highlight a unique striatonigral circuit targeting dopamine-containing neurons. Proc. Natl. Acad. Sci. USA. 113: 11318–11323. https://doi.org/10.1073/pnas.1613337113
Mosca T.J., Luginbuhl D.J., Wang I.E., Luo L. (2017) Presynaptic LRP4 promotes synapse number and function of excitatory CNS neurons. Elife. 6: 27347. https://doi.org/10.7554/eLife.27347
Freifeld L., Odstrcil I., Förster D., Ramirez A., Gagnon J.A., Randlett O., Costa E.K., Asano S., Celiker O.T., Gao R., Martin-Alarcon D.A., Reginato P., Dick C., Chen L., Schoppik D., Engert F., Baier H., Boyden E.S. (2017) Expansion microscopy of zebrafish for neuroscience and developmental biology studies. Proc. Natl. Acad. Sci. USA. 114: E10799–E10808. https://doi.org/10.1073/pnas.1706281114
Deshpande T., Li T., Herde M.K., Becker A., Vatter H., Schwarz M.K., Henneberger C., Steinhäuser C., Bedner P. (2017) Subcellular reorganization and altered phosphorylation of the astrocytic gap junction protein connexin43 in human and experimental temporal lobe epilepsy. Glia. 65: 1809–1820. https://doi.org/10.1002/glia.23196
Wang I.E., Lapan S.W., Scimone M.L. Clandinin T.R., Reddien P.W. (2016) Hedgehog signaling regulates gene expression in planarian glia. Elife. 5: e16996. https://doi.org/10.7554/eLife.16996
Truckenbrodt S., Sommer C., Rizzoli S.O., Danzl J.G. (2019) A practical guide to optimization in X10 expansion microscopy. Nat. Protoc. 14 :832–863. https://doi.org/10.1038/s41596-018-0117-3
Gao R., Yu C.C. (Jay), Gao L., Piatkevich K.D., Neve R.L., Upadhyayula S., Boyden E.S. (2019) A highly homogeneous expansion microscopy polymer composed of tetrahedron-like monomers. bioRxiv. 814111
Shi X., Li Q., Dai Z., Tran A.A., Feng S., Ramirez A.D., Lin Z., Wang X., Chow T.T., Seiple I.B., Huang B. (2019) Label-retention expansion microscopy. bioRxiv. 687954
Sun D., Fan X., Shi Y., Zhang H., Huang Z., Cheng B., Tang Q., Li W., Zhu Y., Bai J., Liu W., Li Y., Wang X., Lei X., Chen X. (2021) Click-ExM enables expansion microscopy for all biomolecules. Nat. Methods. 18: 107–113. https://doi.org/10.1038/s41592-020-01005-2
Wang Y., Yu Z., Cahoon C.K., Parmely T., Thomas N., Unruh J.R., Slaughter B.D., Hawley R.S. (2018) Combined expansion microscopy with structured illumination microscopy for analyzing protein complexes. Nat. Protoc. 13: 1869–1895. https://doi.org/10.1038/s41596-018-0023-8
Gambarotto D., Zwettler F.U., Cernohorska M., Fortun D., Borgers S., Heine J., Schloetel J.G., Reuss M., Unser M., Boyden E.S., Sauer M., Hamel V., Guichard P. (2018) Imaging beyond the super-resolution limits using ultrastructure expansion microscopy (UltraExM). bioRxiv. 308270
Tong Z., Beuzer P., Ye Q., Axelrod J., Hong Z., Cang H. (2016) Ex-STORM: Expansion Single Molecule Super-resolution Microscopy. https://doi.org/10.1101/049403
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова