

Российский физиологический журнал им. И.М. Сеченова, 2021, T. 107, № 4-5, стр. 533-567
Влияние диеты как фактора экспозома на работу головного мозга
А. А. Федотова 1, 2, А. Б. Тяглик 2, А. В. Семьянов 1, 2, 3, *
1 Московский государственный университет им. М.В. Ломоносова
Москва, Россия
2 Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова РАН
Москва, Россия
3 Первый Московский государственный медицинский университет им. И.М. Сеченова
Москва, Россия
* E-mail: alexeysemyanov@gmail.com
Поступила в редакцию 29.01.2021
После доработки 17.02.2021
Принята к публикации 20.02.2021
Аннотация
В обзоре рассмотрена концепция экспозома, который представляет собой совокупность взаимодействующих друг с другом факторов среды, оказывавших влияние на организм в течение всей жизни. Приведена классификация факторов экспозома, объединенных в три основные группы: внутренняя среда, образ жизни, внешняя среда. Особое внимание уделено анализу влияния диеты как фактора экспозома на работу головного мозга. Рассмотрены три основных режима питания, различающихся в зависимости от количества калорий и соотношения макронутриентов (жиров, белков и углеводов), входящих в их состав. Проанализированы основные молекулярные и клеточные механизмы кетогенной диеты, ограничения калорий и западной диеты в отношении функционирования головного мозга. Обсуждается ограниченность накопленных данных о влиянии диеты на нейрон-астроцитарные взаимодействия в мозге. Отдельная глава посвящена рассмотрению взаимосвязей между различными факторами экспозома в контексте влияния диеты, что часто упускают в исследованиях. Указывается на необходимость комплексного анализа работы головного мозга, позволяющего проследить функциональные взаимосвязи на разных уровнях организации (молекулярном, клеточном, органном). Это поможет систематизировать накопленные знания и положит начало разработке терапевтических подходов на основе индивидуального экспозома.
ГЛОССАРИЙ
Внешняя среда – совокупность физических, химических и биологических факторов окружающей среды, влияющих на организм (например, радиация, климатические условия, химическое загрязнение, инфекционные агенты, микробиота).
Внутренняя среда – совокупность эндогенных факторов, которые определяются генетической программой организма и эпигеномом (например, гормональный фон, пол, возраст, уровень метаболизма).
Диета с сокращением числа потребляемых калорий (ограничение калорий) – диета, характеризующаяся ограничением среднего дневного потребления калорий ниже обычного (на 10–50%), без нарушения баланса основных питательных веществ.
Западная диета – диета, характеризующаяся превышением необходимого для жизнедеятельности организма количества потребляемых калорий, при этом 30–35% энергии обеспечивается за счет потребляемых жиров, 50–55% – за счет углеводов, 15% – за счет белков.
Кетогенная диета – диета, состав которой характеризуется кетогенным соотношением макронутриентов (жиров, белков, углеводов), при котором 70–80% энергии обеспечивается за счет потребляемых жиров, 15–25% – за счет белков, 5% – за счет углеводов.
Кетогенное соотношение диеты – это соотношение количества (выраженного в граммах) жиров к углеводам и белкам; чем выше значение данного показателя, тем сильнее степень кетоза.
Кетоз – метаболическое состояние, характеризующееся увеличением в крови уровня кетоновых тел, при котором снабжение организма энергией зависит в большей степени от жировых запасов, чем от глюкозы.
Метаболизм – совокупность всех химических превращений, происходящих в живой системе посредством серии последовательных катализируемых ферментами реакций (метаболических путей).
Образ жизни – совокупность индивидуальных привычек организма, включающих такие факторы, как употребление наркотических веществ, диета, уровень стресса и физической активности.
Ось взаимодействия – функциональная взаимосвязь между различными элементами организма, существующая на разных уровнях организации (органном, клеточном, молекулярном).
Экспозом – совокупность взаимодействующих между собой факторов (внешней среды, внутренней среды и образа жизни), оказывавших влияние на организм в течение всей жизни.
Ca2+ события – ограниченные во времени и пространстве области повышения уровня Ca2+ в клетке или клеточных сетях [1].
ВВЕДЕНИЕ
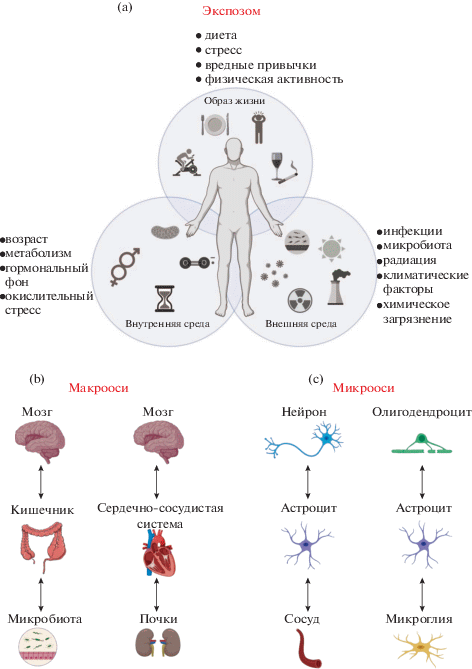
Концепция экспозома возникла как стратегия изучения взаимодействия генов и окружающей среды [2, 3]. Дополняя геном, экспозом обеспечивает исчерпывающее описание истории воздействия среды на онтогенез организма. Экспозом – это совокупность взаимодействующих между собой факторов, оказывавших влияние на организм в течение всей жизни. В структуре экспозома можно выделить три группы воздействий на организм: внутренняя среда, образ жизни, внешняя среда (рис. 1a). К первой группе относятся эндогенные факторы, характеризующие внутреннее состояние организма (метаболизм, гормональный профиль, воспаление, окислительный стресс, перекисное окисление липидов, процесс старения). Вторая группа объединяет широкий спектр воздействий на организм, связанных с образом жизни (наркотические вещества, физическая активность, диета, стресс, медицинские вмешательства, прием лекарственных препаратов, прививки и т.д.). К третьей группе относятся факторы окружающей среды (микробиота, инфекции, химические загрязнения, радиация, климатические факторы, социальное окружение). Одни и те же факторы могут быть отнесены к разным группам, например, микробиота может рассматриваться как фактор образа жизни или как фактор внешней среды. Следовательно, важно принимать во внимание перекрытия между тремя основными группами факторов экспозома и рассматривать их как взаимосвязанные.
Рис. 1.
Экспозом и примеры функциональных макро- и микроосей взаимодействия, на которые он воздействует. (a) – схема экспозома как совокупности факторов, оказывавших влияние на организм человека в течение жизни (в центре). Факторы разделены на три группы: образ жизни, внутренняя среда и внешняя среда, которые представлены в виде перекрывающихся кругов. Область перекрытия кругов показывает, что факторы из разных групп сложным образом взаимодействуют друг с другом. Эффект каждого фактора на организм зависит от наличия и истории взаимодействия с другими факторами экспозома, например, факторы образа жизни дают разный эффект в зависимости от пола и возраста. К первой группе факторов относятся эндогенные факторы, характеризующие внутреннее состояние организма (метаболизм, гормональный профиль, окислительный стресс, процесс старения и т.д.). Вторая группа объединяет широкий спектр воздействий на организм, связанных с образом жизни организма (употребление наркотических веществ, физическая активность, диета, стресс и т.д.). К третьей группе относятся факторы окружающей среды (микробиота, инфекции, химические загрязнения, радиация, климатические факторы и т.д.). (b) – оси взаимодействия между органами (макрооси): микробиота-кишечник–мозг, отвечает за контроль и интеграцию функций кишечника и головного мозга, включая иммунный ответ, кишечную проницаемость и нейроэндокринную регуляцию; почки–сосудистая система–мозг, вовлекается в снижение почечной функции, обусловленное нарушениями питания и артериальной гипертензией. (c) – оси взаимодействия между клетками (микрооси): нейрон–астроцит–кровеносный сосуд, вовлекается в регулирование локального кровотока в зависимости от активности конкретной области головного мозга; олигодендроцит–астроцит–микроглия, регулирует формирование, повреждение и восстановление миелина.
Концепция экспозома получила широкое распространение в области психиатрии, где экспозом определяется как совокупность факторов внешней среды, которые вносят вклад в развитие психических заболеваний [4]. Описан аддитивный эффект взаимодействия между риском развития шизофрении при полигенной предрасположенности и влиянием факторов экспозома. Данный результат указывает на необходимость учитывать экспозом в патогенезе заболевания и корректировать его в терапевтических целях [5]. Недавно в сфере поведенческой нейронауки был предложен термин “психоэкспозом”, описывающий воздействие факторов окружающей среды на степень психологической устойчивости. Предлагается использование данных детального анализа влияния психоэкспозома на поведение человека, которое ляжет в основу разработки образовательных и терапевтических программ, способствующих развитию адаптивного поведения [6]. Концепция экспозома также была применена для описания факторов, влияющих на развитие болезни Альцгеймера [7].
Функционирование отдельных органов описывают в рамках специфических разделов физиологии и зачастую не принимают во внимание их взаимодействия (нервные, гуморальные, гормональные) с другими органами. Такие взаимодействия между органами в ответ на воздействие факторов экспозома также называют осями взаимодействия (рис. 1b). Однако факторы экспозома могут оказывать воздействия и на оси, формируемые на клеточном уровне. Таким образом, можно определить макрооси как взаимодействия на уровне органов и микрооси как взаимодействия на уровне клеток (рис. 1b, c). Например, макроось легкие–мозг вовлечена в проникновение нейротоксинов в организм человека при наличии загрязнения воздуха и табачного дыма; макроось почки–сосудистая система–мозг вовлекается в снижение почечной функции, обусловленное нарушениями питания и артериальной гипертензией [7]. Аналогичные связи существуют между ЦНС и кишечником, в основе двустороннего взаимодействия которых лежат нейроэндокринные и иммунологические процессы. Кишечный микробиом, рассматриваемый специалистами гастроэнтерологии как часть кишечного экспозома, является одним из основных компонентов макрооси микробиота–кишечник–мозг [8]. Данная макроось отвечает за контроль и интеграцию функций кишечника и головного мозга и связывает эмоциональные и когнитивные центры мозга с функциями кишечника, включая иммунный ответ, кишечную проницаемость и нейроэндокринную регуляцию [9]. Таким образом, представляется актуальным исследование работы мозга в рамках концепции экспозома.
Диета является одним из ключевых факторов экспозома, оказывающих влияние на функционирование ЦНС. Метаболиты, поступающие в организм с пищей в виде липидов, белков и углеводов, используются клетками мозга в качестве основных источников энергии. Растущее число случаев ожирения по всему миру и связанные с ним когнитивные дисфункции [10] указывают на острую необходимость анализа эффектов, вызываемых различными диетами в отношении работы ЦНС.
Долгое время предпринимались попытки описать функции головного мозга только с точки зрения функционирования нейронов. Однако мозг является сложной системой, состоящей из разных типов клеток, взаимодействующих друг с другом и отвечающих на изменения окружающей среды [11]. К этим клеткам относятся глиальные клетки и клетки кровеносной системы. Глиальные клетки – это гетерогенная морфологически и функционально группа клеток, включающая микроглию и макроглию. Макроглия включает астроциты и олигодендроциты. На клеточном уровне могут быть определены микрооси взаимодействия. Например, микроось нейрон–астроцит–кровеносный сосуд (рис. 1c) регулирует локальный кровоток в зависимости от активности конкретной области головного мозга. Микроось олигодендроцит–астроцит–микроглия регулирует формирование, повреждение и восстановление миелина (рис. 1c) [12]. Факторы экспозома также оказывают свое действие посредством таких клеточных микроосей. Целью настоящего обзора является рассмотрение и обобщение имеющихся научных данных об изменениях, происходящих в ЦНС под влиянием различных режимов питания как одного из факторов экспозома, в частности, в контексте микрооси нейрон–астроцит.
ОСОБЕННОСТИ МЕТАБОЛИЗМА В МОЗГЕ
Метаболизм – совокупность всех химических превращений, происходящих в живой системе посредством серии последовательных катализируемых ферментами реакций (метаболических путей). Превращение предшественника в конечный продукт идет через серию промежуточных продуктов, или метаболитов. Ацетилкофермент А (ацетил-КоА) является ключевым промежуточным продуктом метаболизма: он возникает при распаде белков, липидов и углеводов, служит в качестве предшественника многих соединений (жирных кислот, из которых из которых впоследствии синтезируются жиры, гликолипиды, фосфолипиды и другие производные, кетоновых тел, изопреноидов) и поглощается в катаболическом пути, известном как цикл Кребса (цикл трикарбоновых кислот) (рис. 2) [13].
Рис. 2.
Взаимосвязь обмена белков, жиров и углеводов. Под действием ферментов желудочно-кишечного тракта белки расщепляются до аминокислот (протеолиз). Аминокислоты участвуют в дальнейших превращениях. Выделяют кетогенные и глюкогенные аминокислоты. Глюкогенные аминокислоты при деградации образуют пируват и другие вещества, являющиеся промежуточными метаболитами цикла Кребса (2-оксоглутарат, сукцинил-КоА, фумарат, оксалоацетат, не показаны). Эти вещества при недостатке углеводов в организме превращаются в глюкозу (глюконеогенез). Глюкоза может запасена в виде гликогена, который в дальнейшем может расщепляться с образованием глюкозы. Продукты распада кетогенных аминокислот (ацетоацетат и ацетил-КоА) используются для синтеза кетоновых тел (ацетоацетата, β-гидроксибутирата и ацетона). Жиры метаболизируются с образованием глицерина и жирных кислот. Фосфорилирование глицерина приводит к образованию промежуточных продуктов (не показаны), включаемых в реакции глюконеогенеза. Катаболизм жирных кислот приводит к образованию ацетоацетил-КоА и ацетил-КоА, способных превращаться друг в друга. Они метаболизируется с образованием кетоновых тел. Углеводы расщепляются в пищеварительной системе до моносахаридов. Основным моносахаридом является глюкоза. Глюкоза метаболизируется с образованием ацетил-КоА, вступающего в цикл Кребса.
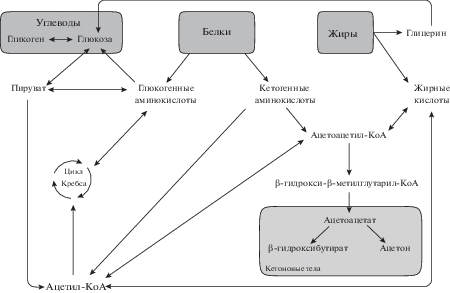
В условиях, когда расщепление липидов и углеводов сбалансировано, ацетил-КоА включается в цикл Кребса. Процесс зависит от доступности оксалоацетата. В отсутствие углеводов или при нарушении их использования концентрация оксалоацетата снижается, поскольку оксалоацетат расходуется на образование глюкозы и поэтому не может конденсироваться с ацетил-КоА в цикле Кребса. В таких условиях расщепление жиров преобладает, и путь метаболизма ацетил-КоА смещается в сторону образования кетоновых тел – ацетоацетата и β-гидроксибутирата. Ацетон, образующийся из ацетоацетата, также является кетоновым телом. Кетоновые тела служат в качестве метаболического источника энергии, а также как субстраты процессов синтеза холестерина, жирных кислот и миелиногенеза. В ранний постнатальный период ацетоацетат и β-гидроксибутират, образующиеся в результате окисления содержащихся в материнском молоке липидов, являются предпочтительными субстратами [14]. Это обусловлено высокими энергетическими затратами, которые требуются для развития и процессов миелинизации в тканях мозга. Основным органом для образования кетоновых тел является печень. Из митохондрий печени кетоновые тела диффундируют в кровь и переносятся к периферическим тканям. Сердечная мышца и корковый слой почек предпочтительно используют в качестве источника энергии ацетоацетат, а не глюкозу. В противоположность этому глюкоза является главным энергетическим субстратом для мозга в условиях сбалансированного питания. При голодании и диабете мозг адаптируется к использованию ацетоацетата [14]. Среди диет, приводящих к смещению метаболизма в сторону образования кетоновых тел (состояние кетоза), наиболее распространенными являются две – кетогенная диета и диета с сокращением числа потребляемых калорий (далее, ограничение калорий) (табл. 1). При этом развивается кетоз – метаболическое состояние, при котором снабжение организма энергией зависит в большей степени от жировых запасов, чем от глюкозы [15]. Кетогенная диета является низкоуглеводной диетой, состав которой характеризуется кетогенным соотношением макронутриентов, при котором 70–80% энергии обеспечивается за счет жиров, 15–25% – за счет белков, 5% – за счет углеводов [16]. Кетогенное соотношение показывает отношение количества жиров (выраженного в граммах) к углеводам и белкам; чем выше значение данного показателя, тем сильнее степень кетоза [17, 18]. Кетогенное соотношение позволяет определить, по какому пути будет протекать метаболизм: глюкоцентрическому или липоцентрическому. Критическим является процент белка в пище, поскольку высокое содержание белка может приводить к глюконеогенезу в условиях стресса и недостатка питательных веществ, увеличивая глюкоцентричность метаболизма [19]. Ограничение калорий заключается в ограничении среднего дневного потребления калорий ниже обычного (на 10–50%), без нарушения баланса основных питательных веществ [20–22].
Таблица 1.
Особенности кетогенной диеты, ограничения калорий и западной диеты
| Кетогенная диета | Ограничение калорий | Западная диета | |
|---|---|---|---|
| Особенности состава диеты | Диета с кетогенным соотношением макронутриентов, при котором 70–80% энергии обеспечивается за счет жиров, 15–25% – за счет белков, 5% – за счет углеводов [16] | Сбалансированный состав, ограничение потребляемых калорий (на 10–50%) [20] | Превышение необходимого для жизнедеятельности организма количества потребляемых калорий, при этом 30–35% энергии обеспечивается за счет жиров, 50–55% – за счет углеводов, 15% – за счет белков [23, 24] |
| Гликолиз | Снижение | Снижение | Увеличение |
| Липидный обмен | Активация липолиза и β-окисления жирных кислот | Активация липолиза и β-окисления жирных кислот | Подавление липолиза, запасание жиров |
Жиры являются основной формой депонирования энергии. Важной особенностью жиров является то, что при их гидролизе образуются два функционально различных продукта – жирные кислоты и глицерин. Глицерин в условия голодания или ограничения калорий используется в процессе глюконеогенеза и тем самым участвует в обеспечении глюкозой клеток мозга и других глюкозозависимых клеток. При окислении жирных кислот образуется аденозинтрифосфат (АТФ), используемый большинством тканей. В норме у взрослых животных количество жира в организме сохраняется в течение длительного времени на относительно постоянном уровне. Постоянство поддерживается, поскольку биосинтез и окисление триацилглицеролов (нейтральных жиров) протекают одновременно (для этих процессов устанавливается стационарное состояние). При ограничении калорий и в условиях кетогенной диеты активируется липолиз в жировой ткани, что приводит к увеличению концентрации жирных кислот в крови. Около 50% жирных кислот перерабатывается в печени в кетоновые тела. Однако мозг использует их в качестве источника энергии в меньшей степени, что связано с двумя факторами. Во-первых, скорость продукции АТФ в результате окисления жирных кислот меньше, чем при использовании в качестве энергетического субстрата глюкозы. Во-вторых, процесс переноса жирных кислот через гематоэнцефалический барьер (ГЭБ) относительно медленный [25, 26]. Тем не менее, после прохождения через ГЭБ жирные кислоты могут быть использованы как астроцитами, так и нейронами. Изменения метаболизма жирных кислот в астроцитах могут быть связаны с развитием патологий (болезнь Хантингтона, болезнь Альцгеймера) [27]. Модуляция процесса окисления жирных кислот предлагается в качестве терапевтического подхода при лечении ишемии и глиобластомы [27]. Кетоновые тела поступают в мозг, используя облегченную диффузию при помощи переносчиков монокарбоксилата (monocarboxylate transporter, МСТ). Транспорт кетоновых тел через ГЭБ зависит от изоформы переносчика и концентрации кетоновых тел в крови. Транспорт кетоновых тел, в отличие от транспорта глюкозы, не увеличивается при повышенной нейрональной активности [28]. Исследования на грызунах показали, что MCT представлены во всем мозге, хотя экспрессия МСТ сильно варьирует в зависимости от типа клеток. Изоформа MCT1 экспрессируется в эндотелиальных клетках и астроцитах [29]. Астроциты также экспрессируют MCT4. МСТ1 и МСТ4 обладают низким сродством к β-гидроксибутирату [30]. Нейроны почти исключительно экспрессируют изоформу MCT2, которая обладает высоким сродством к β-гидроксибутирату. Наличие MCT2 в постсинапсе вместе с повышенной плотностью митохондрий [31] позволяет предположить, что эта изоформа играет важную роль в синаптической передаче. Таким образом, нейроны и (в меньшей степени) астроциты обладают способностью поглощать кетоновые тела. Профиль экспрессии MCT1 и MCT2 в различных структурах мозга консервативен у людей и грызунов [32].
Для полного понимания энергетических процессов в клетках мозга необходимо рассмотреть сотни различных реакций, однако мы остановимся только на ключевых процессах. Человеческий мозг составляет около 2% от общей массы тела, но энергетические затраты, необходимые для его работы, составляют примерно 25% от общих энергетических потребностей организма. Основным энергетическим субстратом для мозга является глюкоза. Метаболизм глюкозы в головном мозге аналогичен метаболизму в других тканях и включает три основных направления: гликолиз, протекающий в цитозоле клеток мозга, запасание глюкозы в форме гликогена и пентозофосфатный путь окисления глюкозы [33].
Гликолиз – это универсальный центральный путь катаболизма глюкозы. Ферментативное расщепление молекулы глюкозы в процессе гликолиза приводит к образованию пирувата. В зависимости от условий пируват может превращаться в лактат (анаэробные условия) или в ацетил-КоА (аэробные условия). Глиальные клетки, в особенности астроциты и олигодендроциты, метаболизируют глюкозу преимущественно гликолитическими путями, образуя из глюкозы лактат и пируват [33]. Лактат и пируват проникают через специализированный транспортер МСТ2 в нейроны, где метаболизируются в митохондриях в ходе цикла Кребса и окислительного фосфорилирования с образованием АТФ. В цитозоле нейронов присутствуют разные изоформы фермента лактатдегидрогеназы, катализирующего реакцию превращения лактата в пируват. Поддержание работы нейронов и нейропередачи в отсутствие глюкозы за счет лактата и пирувата играет нейропротекторную роль в условиях гипогликемии и ишемии [14]. Увеличение лактата встречается и в норме в ходе интенсивной физической нагрузки, концентрация лактата в крови может составлять от 3 до 10 мМ, а уровень окисления лактата в мозге может составлять до 20–25% от общей энергетической потребности мозга [14]. Высказываются различные гипотезы относительно использования лактата в головном мозге [34]. Ранее считалось, что даже при достаточном количестве глюкозы лактат является основным субстратом для поддержания активности нейронов [35]. Согласно современным данным, глюкоза также используется нейронами в качестве источника энергии, причем окисление глюкозы происходит в равной степени в астроцитах и нейронах in vivo [36, 37]. Усиление работы Na+/K+-аденозинтрифосфатазы (Na+/K+-АТФ-азы) в результате активации нейронов приводит к увеличению потребления АТФ, что активирует как гликолитический путь, так и окислительное фосфорилирование в нейронах и астроцитах. Производство лактата в головном мозге во время активности нейронов происходит в результате гликолитического метаболизма, временно превышающего скорость окислительного фосфорилирования. Накопление лактата рассматривается как потенциально вредное, поэтому лактат должен либо удаляться через кровоток, либо потребляться клетками мозга, находящимися в неактивном состоянии [34]. Напротив, гипотеза лактатного шаттла предполагает, что глутамат, помимо модуляции возбудимости нейронов, стимулирует гликолиз, то есть утилизацию глюкозы [38]. Увеличение потребления глюкозы, вызванное активностью мозга, происходит в основном в астроцитах. Они метаболизируют глюкозу в процессе гликолиза для производства лактата. Специфический профиль экспрессии генов в астроцитах определяет тот факт, что в процессе аэробного гликолиза пируват преимущественно превращается в лактат и почти не используется в цикле Кребса [33]. Образуемый астроцитами лактат расходуется впоследствии для поддержания окислительного метаболизма активных нейронов [34, 39]. Основная критика гипотезы лактатного шаттла заключается в отсутствие прямых данных о том, что мозг использует лактат как основной источник энергии in vivo [40, 41]. Отмечается, что клеточные источники лактата в мозге остаются неизвестными и рискованно полагать, что лактат всегда образуется в астроцитах [40]. Накопленные экспериментальные данные скорее подтверждают, что в ответ на стимуляцию нейроны обладают способностью увеличивать собственный гликолиз, а также экспортировать, а не импортировать лактат [37].
Астроциты, в отличие от нейронов, могут сохранять запасы энергии в виде гликогена при избытке энергетического субстрата (глюкозы, пирувата или лактата). Использование гликогена в головном мозге может быть напрямую связано с низким уровнем внеклеточной глюкозы. Гликоген впоследствии может быть мобилизован с помощью гликогенфосфорилазы в виде глюкозы и метаболизирован до пирувата [42]. Хотя в нейронах активно работает ген, кодирующий гликогенсинтазу (ключевой фермент в метаболизме гликогена), готовая гликогенсинтаза постоянно расщепляется убиквитиновой системой клетки. Тем не менее гликоген способен накапливаться в нейронах в небольших количествах и полностью метаболизироваться с помощью гликогенфосфорилазы [43]. Накопление гликогена в нейронах сопровождает некоторые неврологические заболевания, например, болезнь Лафора [44]. Причиной агрегации гликогена при этом заболевании являются мутации в генах, кодирующих белки, участвующие в метаболизме гликогена (лафорин и малин) [45, 46]. Заболевание характеризуется нейродегенерацией, наличием генерализованных и фокальных эпилептических приступов, экстрапирамидными нарушениями, расстройством высших психических функций, деменцией [47].
Метаболизм мозга считается практически полностью окислительным [42]. Тем не менее, гетерогенность клеток мозга предполагает наличие в них разных метаболических систем. Для разных типов клеток характерен индивидуальный метаболических профиль, который меняется с возрастом. Например, нейроны могут использовать кетоновые тела, лактат и пируват в качестве альтернативных источников энергии, тогда как астроциты во взрослом мозге в большей степени зависят от глюкозы. В ранний постнатальный период (10 дней) альтернативные субстраты энергии усиливают активность астроцитов [48]. При развитии мозга источники энергии (глюкоза, кетоновые тела) используются для биосинтеза макромолекул, необходимых для пролиферации нейронов, образования синапсов и миелинизации [49]. Самым энергозатратным процессом во взрослом головном мозге является поддержание ионных градиентов через плазматическую мембрану, необходимые для генерации потенциала действия и нейротрансмиссии. Поддержание градиентов преимущественно происходит за счет работы Na+/K+-АТФ-азы, локализованной в мембране нейронов и астроцитов. На работу данного ионного насоса приходится примерно 50% энергии, образующейся при окислении глюкозы в нервной системе. 80–85% от общей потребляемой энергии отражает нейротрансмиссию глутамата, а 10–15% – энергетические затраты на поддержание потенциала покоя. Нейроны представляют собой более энергозатратный тип клеток (80–85% потребностей мозга в энергии), чем клетки глии (5–15% потребностей) [14]. В астроцитах энергия используется для поддержания ионного обмена, работы транспортеров (например, для выведения лактата через MCT4) и поддержания гомеостаза во внеклеточном пространстве [50].
При высоком кетогенном соотношении макронутриентов метаболизм сдвигается в сторону расщепления жиров, и мозг использует альтернативные источники энергии – кетоновые тела (β-гидроксибутират, ацетоацетат, ацетон). Образующийся β-гидроксибутират метаболизируется в ацетил-КоА и поступает в цикл Кребса [30]. Важно отметить, что на данный момент нет однозначных данных о снижении уровня глюкозы при кетогенной диете в мозге здоровых животных [51]. Потребление глюкозы клетками мозга здоровых людей при кетогенной диете снижается, поскольку часть потребности митохондрий в глюкозе реализуется за счет использования в качестве источника энергии кетоновых тел. У людей с болезнью Альцгеймера в условиях кетогенной диеты, напротив, несмотря на наличие кетоза и метаболизацию кетоновых тел, потребление глюкозы клетками мозга не снижается [52–54].
Когда углеводы, липиды или белки поступают в количествах, превосходящих энергетические потребности организма, избыток калорий запасается в виде триацилглицеролов (жиров). Накопленный таким образом жир может быть в будущем использован для получения энергии, что позволяет организму адаптироваться к условиям голодания. Одной из диет, при которой потребление калорий превышает необходимое для жизнедеятельности организма, является западная диета. Она характеризуется превышением необходимого для жизнедеятельности организма количества потребляемых калорий, при этом энергия обеспечивается в основном за счет углеводов (50–55%) и жиров (30–35%) и в меньшей степени за счет белков (15%) [23, 24]. Таким образом, в отличие от кетогенной диеты, для западной диеты наряду с высоким содержанием жиров характерно высокое содержание углеводов. В организме имеется механизм, препятствующий перерасходу питательных веществ. Избыточное окисление жирных кислот ингибирует окисление глюкозы, а ее избыток подавляет распад жиров и окисление жирных кислот (при этом сама глюкоза переходит в жиры) [13]. В результате западная диета приводит к ожирению.
Хотя нейроны и астроциты различаются по метаболическому профилю, для них характерно метаболическое взаимодействие. Метаболизм глюкозы находится в пространственно-временной зависимости от нейрональной активности в областях мозга, задействованных в выполнении конкретных задач. У крыс во время плавания наблюдается увеличение метаболизма в мозжечке (область мозга, участвующая в координации движений) в области нейропиля [55]. В этой области колокализуются пресинаптические и постсинаптические окончания нейронов и астроциты, окружающие синаптические контакты. Такая морфологическая связь между астроцитами и нейронами, получившая название трехчастного синапса [56], усиливается функциональной взаимосвязью. В ответ на высвобождаемый нейронами глутамат, астроциты выделяют полученный из глюкозы лактат. Лактат используется для поддержания активности нейронов в качестве энергетического субстрата. Глюкоза обеспечивает основу для обновления нейронального пула глутамата. С помощью астроцитарной глутаминсинтазы глутамат превращается в глутамин. Глутамат, захватываемый астроцитами во время синаптической передачи, проходит такой же путь (глутамат-глутаминовый цикл). Таким образом, глутаматергическая передача осуществляется благодаря тесному морфофункциональному взаимодействию астроцитов и нейронов [14].
Астроциты образуют сеть посредством соединения через щелевые контакты, что обеспечивает поддержание внеклеточного гомеостаза за счет буферизации ионов Ca2+, K+, молекул глутамата и глюкозы [57]. Считается, что астроциты критически вовлечены в транспорт глюкозы во внеклеточное пространство и поддержание концентрации глюкозы на постоянном уровне (0.5–1.5 мМ). Капилляры головного мозга окружены тонкими астроцитарными отростками, посредством которых происходит поглощение энергетических субстратов. Чтобы достичь областей мозга, удаленных от кровоснабжения, энергетические субстраты могут проходить через астроцитарные щелевые контакты, проницаемые для глюкозы, лактата, глутамата, глутамина. Образующиеся при активации нейронов промежуточные продукты метаболизма выступают в роли сигнальных молекул, вовлеченных в метаболическую передачу сигналов о необходимости пополнения запасов энергетических субстратов и усилении местного кровотока. Основными сигнальными агентами являются ионы K+ (высвобождаемые после потенциалов действия и во время синаптической передачи), глутамат (захватываемый астроцитами через глутаматные транспортеры, а также оказывающий влияние посредством активации расположенных на мембране астроцитов метаботропных рецепторов к глутамату), аммиак, уровень лактата, концентрация кислорода и агенты, способные к диффузии (NO, H2O2, супероксид-анион) [50].
Таким образом, нейроны и астроциты различаются по метаболическому профилю, однако образуемая ими микроось нейрон–астроцит–сосуд представляет собой единую метаболическую единицу. Диеты с различным содержанием жиров, белков и углеводов вызывают смещение метаболизма в сторону образования кетоновых тел (кетогенная диета, ограничение калорий) или в сторону усиления запасания жиров (западная диета) (табл. 1). В связи с этим необходимо провести анализ молекулярных механизмов кетогенной диеты, ограничения калорий и западной диеты и происходящих под их влиянием изменений в работе нейронов и астроцитов.
ВЛИЯНИЕ КЕТОГЕННОЙ ДИЕТЫ НА РАБОТУ МОЗГА
Увеличение окислительного метаболизма кетоновых тел в митохондриях при кетогенной диете подавляет гликолиз (табл. 1, рис. 3) [58]. Происходит метаболический сдвиг, выражающийся в увеличении продукции АТФ митохондриями и уменьшении выработки АТФ в процессе гликолиза в цитоплазме.
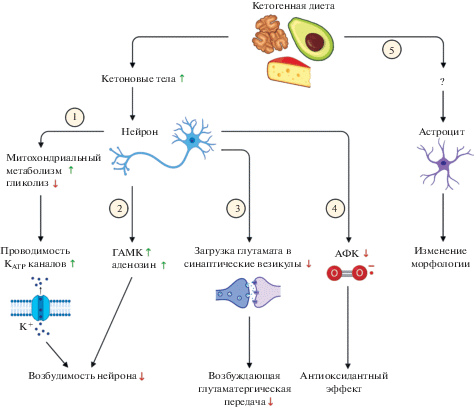
Рис. 3.
Механизмы действия кетогенной диеты на работу мозга. Кетогенная диета смещает метаболизм в сторону образования кетоновых тел. (1) – в нейронах кетоновые тела метаболизируются в митохондриях. Происходит увеличение продукции АТФ митохондриями и уменьшение выработки АТФ в процессе гликолиза. В результате активируются АТФ-зависимые калиевые каналы, которые в нормальном состоянии заблокированы гликолитической АТФ. Повышение калиевой проводимости приводит к снижению возбудимости нейронов. (2) – кетогенная диета приводит к увеличению уровня ГАМК. Активация хлорных каналов ГАМКА-рецепторов вызывает гиперполяризацию нейронов и снижение их возбудимости. Активация аденозинкиназы на фоне кетогенной диеты приводит к повышению уровня аденозина. Аденозин подавляет нейрональную активность через A1-рецептор. (3) – кетогенная диета приводит к снижению возбуждающей передачи. Кетоновое тело ацетоацетат ингибирует везикулярный переносчик глутамата VGLUT2, что снижает эффективность загрузки глутамата в синаптические везикулы. (4) – кетоновые тела β-гидроксибутират и ацетоацетат выступают в роли антиоксидантов. β-гидроксибутират ингибирует продукцию АФК митохондриями и запускает антиоксидантную программу, приводящую к активации экспрессии антиоксидантных ферментов. (5) – кетогенная диета влияет на морфологию астроцитов. Наблюдается уменьшение разветвленности и размера астроцитов. При этом отсутствие маркеров глиоза не указывает на воспалительную реакцию, свидетельствуя о метаболической активации астроцитов в ответ на кетоз. АТФ – аденозинтрифосфат, АФК – активные формы кислорода, ГАМК – гамма-аминомасляная кислота, KATP – АТФ-зависимые калиевые каналы, VGLUT2 – vesicular glutamate transporter 2 (везикулярный транспортер глутамата 2).
Молекулярные эффекты кетогенной диеты
Воздействие кетогенной диеты на KATP-каналы. Производство АТФ в результате гликолиза играет важную роль в регуляции процессов на плазматической мембране, включая работу Na+/K+-АТФ-азы и АТФ-зависимых калиевых каналов (KATP-каналов) [59]. KATP-каналы широко представлены в головном мозге, в частности, в гипоталамусе [60] и гиппокампе [61], и являются связующим звеном между метаболизмом и возбудимостью нейронов. АТФ, образующийся в ходе гликолиза, ингибирует KATP-каналы [62]. При возбуждении в нейрон поступает избыток ионов Na+, которые затем удаляются Na+/K+-АТФ-азой для восстановления ионного баланса. Расходование молекул АТФ этим насосом в одном субмембранном компартменте с KATP-каналами приводит к их активации [61]. Например, повышенная активность респираторных нейронов ствола мозга мышей активирует KATP-каналы за счет работы Na+/K+-АТФ-азы [63]. Кетогенная диета также приводит к уменьшению уровня АТФ вблизи мембраны, вызывая активацию KATP-каналов. Увеличение уровня β-гидроксибутирата усиливает активность KATP-каналов в гранулярных нейронах зубчатой фасции гиппокампа крыс [61], вероятно, за счет снижения уровня гликолитического АТФ. При этом блокада Na+/K+-АТФ-азы строфантидином предотвращает активацию KATP-каналов [61]. Таким образом, кетогенная диета приводит к изменению метаболического состояния нейронов и снижению их возбудимости за счет увеличения калиевой проводимости.
Влияние кетогенной диеты на путь mTOR. Путь mTOR (mechanistic target of rapamycin, механистическая мишень рапамицина) отвечает за контроль синтеза белков, транскрипции, аутофагии, метаболизма, биогенеза и сохранения органелл в клетке [64]. Белок mTOR выполняет роль сенсора питательных веществ, его активность может подавляться при снижении количества доступной глюкозы или увеличении уровня кетоновых тел [65]. Кетогенная диета в течение 16 нед. у мышей в возрасте 12–14 нед. снижает экспрессию белка mTOR и увеличивает уровень эндотелиальной синтазы оксида азота, которая регулирует работу макрооси сердечно-сосудистая система–мозг. На фоне диеты у мышей наблюдается усиление мозгового кровотока в области вентромедиального гипоталамуса, что может способствовать снижению риска развития болезни Альцгеймера за счет выведения β-амилоида [66]. Путь mTOR играет центральную роль в развитии рака [67]. Ингибирование пути mTOR является мощным противоопухолевым воздействием. В модели глиобластомы in vivo у мышей использование кетогенной диеты в течение одного месяца значительно снижает рост опухолевых клеток и увеличивает выживаемость животных. Опосредованные диетой противоопухолевые эффекты коррелируют со снижением экспрессии белка mTOR [68]. Таким образом, применение кетогенной диеты может значительно улучшить прогноз данного онкологического заболевания и увеличить процент выживаемости за счет ингибирования пути mTOR.
Антиоксидантное действие кетогенной диеты. Кетоновые тела играют важную роль в антиоксидантной защите, регулируя баланс активных форм кислорода (АФК) [69]. Как L-, так и D-изоформа β-гидроксибутирата и ацетоацетат выступают в роли антиоксидантов, нейтрализуя АФК [70]. Антиоксидантный эффект β-гидроксибутирата [71] обусловлен наличием гидроксильной группы [70]. Кроме того, β-гидроксибутират ингибирует продукцию АФК митохондриями [69]. На изолированных митохондриях нейронов неокортекса крыс показано, что сочетание β-гидроксибутирата и ацетоацетата снижает выработку АФК [72]. β-гидроксибутират также запускает антиоксидантную программу через регуляцию факторов транскрипции FOXO1, FOXO3 и NRF2, которые активируют экспрессию антиоксидантных ферментов [69].
Нейромедиаторы, вовлеченные в действие кетогенной диеты. Состояние кетоза активирует работу глутаматдекарбоксилазы – фермента, катализирующего преобразование глутамата в ГАМК. Это приводит к снижению уровня глутамата и повышению уровня ГАМК [73]. Последнее вызывает гиперполяризацию нейронов за счет увеличения хлорной проводимости через каналы ГАМКА-рецепторов [73]. Гиперполяризация снижает возбудимость нейронов посредством влияния на потенциал-зависимые каналы, участвующие в формировании потенциала действия. Кроме того, на фоне кетогенной диеты увеличивается захват глутамата астроцитами из синаптической щели и внеклеточного пространства [74]. Астроциты возвращают глутамат в пресинаптический нейрон в виде глутамина (глутамат-глутаминовый цикл). Ацетоацетат снижает высвобождение глутамата нейронами за счет блокады везикулярного переносчика глутамата VGLUT2 (который обеспечивает загрузку глутамата в везикулы), конкурируя с хлоридом за участок аллостерической регуляции. Это приводит к снижению возбуждающей передачи в нейронах поля CA1 гиппокампа [74]. Таким образом, влияние кетогенной диеты на уровни ГАМК и глутамата снижает возбудимость нейронов и способствует уменьшению возбуждающей глутаматергической передачи.
Влияние кетогенной диеты на аденозин. Кетогенная диета вызывает повышение уровня аденозина в головном мозге [75]. Известно, что увеличение количества аденозина наблюдается при возрастании энергетических потребностей клеток мозга, например, при повышенной активности нейронов [76]. Кетогенная диета снижает уровень аденозинкиназы – фермента, катализирующего превращение аденозина в аденозинмонофосфат, что приводит к увеличению содержания внеклеточного аденозина в головном мозге [75]. В гиппокампе и коре головного мозга мышей внеклеточный аденозин подавляет активность нейронов через подтип 1 аденозинового рецептора (A1-рецептор), который расположен на пре- и постсинаптической мембранах [77]. Таким образом, опосредованные аденозином эффекты кетогенной диеты приводят к уменьшению возбудимости нейронов.
Клеточные эффекты кетогенной диеты
В мозге крыс в возрасте 6 мес. кетогенная диета приводит к морфологическим изменениям глиальных клеток. Происходит увеличение ветвления отростков микроглии и уменьшение размеров и разветвленности астроцитов [78]. Такие морфологические изменения астроцитов характерны для воспаления, однако маркеры глиоза в исследовании не обнаружены. Следовательно, изменения морфологии астроцитов следует интерпретировать как реакцию на кетоз, а не как воспалительный процесс. Это указывает на важную роль глиальных клеток в метаболизме мозга. Тем не менее, количество исследований, посвященных влиянию кетогенной диеты на морфофункциональные характеристики глии, ограничено. Основная часть работ по изучению клеточных механизмов кетогенной диеты посвящена нейронам. Нейропротекторный эффект кетогенной диеты показан в различных моделях заболеваний. В мышиной модели бокового амиотрофического склероза (линия SOD1-G93AВ) кетогенная диета снижает степень атрофии двигательных нейронов в спинном мозге и замедляет развитие нарушений двигательной координации [79]. Митохондриальная дисфункция играет центральную роль в нейродегенерации при данном заболевании, которая компенсируется за счет способности β-гидроксибутирата стимулировать выработку АТФ [79]. В экспериментальных моделях инсульта и ишемии головного мозга кетогенная диета приводит к снижению степени атрофии нейронов у крыс [80, 81]. У мышей нейропротекторный эффект кетогенной диеты заключается в уменьшении объема ишемического инсульта и усилении местного кровотока [82] за счет увеличения уровня аденозина в головном мозге [76]. Кетоновые тела способствуют уменьшению глутаматергической передачи и предупреждают повреждение нервных клеток за счет снижения эксайтотоксичности глутамата [83].
Системные эффекты и терапевтическое значение кетогенной диеты
Кетогенная диета широко известна своим противоэпилептическим действием. Применение кетогенной диеты для лечения эпилепсии было предложено в начале 20-х годов прошлого века, и с 1990-х эта диета используется как способ лечения лекарственно-резистентных форм эпилепсии [84]. Уменьшение глутаматергической передачи на фоне кетогенной диеты приводит к снижению возбудимости нейронов [74]. В различных моделях эпилепсии противоконвульсивный эффект кетогенной диеты связывают со снижением порога возникновения, уменьшением интенсивности эпилептического приступа и его отсрочкой [85]. Тем не менее хроническое использование β-гидроксибутирата на органотипической культуре гиппокампа не приводит к блокаде фармакологически индуцированной эпилептиформной активности [86]. Это может быть связано с физиологическими особенностями органотипической ткани. Один из механизмов снижения возбудимости нейронов опосредован аденозиновой сигнализацией [87]. Так, в пилокарпиновой модели эпилепсии у крыс на фоне кетогенной диеты наблюдается длительное снижение частоты возникновения приступов. Инъекция глюкозы или DPCPX, блокатора A1-рецептора, нивелирует этот эффект [88]. В той же модели эпилепсии у крыс наблюдается уменьшение количества дистальных отростков астроцитов и изменение кальциевой активности в этих клетках [89]. Следовательно, особую важность представляют исследования влияния кетогенной диеты на астроцитарную морфологию при эпилепсии. Данные клинических исследований указывают на снижение количества приступов у детей в краткосрочном (3 мес.) и долгосрочном (более 6 мес.) периодах на фоне кетогенной диеты [90, 91]. Остается нерешенным вопрос, какие молекулярные механизмы кетогенной диеты обеспечивают снижение эпилептической активности у людей.
Кетогенная диета может применяться при лечении нейродегенеративных заболеваний, включая болезнь Альцгеймера [83] и болезнь Паркинсона [21]. Наблюдаемый у пациентов с болезнью Альцгеймера гипометаболизм глюкозы может быть частично компенсирован за счет кетогенной диеты, восполняющей уровень АТФ за счет продукции кетоновых тел [83]. Поскольку нейродегенеративные заболевания сопровождаются окислительным стрессом, развивающемся за счет продукции АФК, антиоксидантный эффект кетоновых тел может играть важную роль для улучшения прогноза заболевания [92]. Кетогенная диета приводит к достоверному улучшению баллов унифицированной шкалы оценки заболевания Международного общества изучения двигательных расстройств (UPDRS) при болезни Паркинсона, способствует снижению тремора в состоянии покоя, улучшению равновесия, походки, эмоционального фона [93]. Защитный эффект β-гидроксибутирата против нейротоксичности, вызванной нейротоксином 1-метил-4-фенил-1,2,3,6-тетрагидропиридином, предполагает, что продолжительное увеличение уровня кетоновых тел помогает отсрочить прогрессирование идиопатической формы болезни Паркинсона [94, 95]. У детей в возрасте от 4 до 10 лет с расстройством аутистического спектра кетогенная диета приводит к достоверному улучшению показателей рейтинговой шкалы детского аутизма (CARS), однако диета менее эффективна при тяжелой форме заболевания [96].
Несмотря на отмеченное положительное влияние кетогенной диеты, ее долгосрочные эффекты могут быть негативными. В литий-пилокарпиновой модели эпилепсии у молодых крыс (в возрасте 53–57 дней на момент тестирования) обнаружено значительное уменьшение размеров головного мозга на фоне диеты [97], а также нарушение зрительно-пространственной памяти. Это проявляется в увеличении латентного периода при поиске скрытой платформы в водном лабиринте Морриса. Общая частота эпилептических приступов при этом снижается [97]. Необходимы дополнительные исследования для оценки долгосрочных последствий применения кетогенной диеты в отношении работы мозга.
Таким образом, влияние кетогенной диеты обусловлено производством кетоновых тел. Кетоновые тела усиливают митохондриальный метаболизм, снижают возбудимость нейронов (за счет активации KATP-каналов, увеличения уровней ГАМК и аденозина) и возбуждающую глутаматергическую передачу, являются антиоксидантами. Описанные молекулярные и клеточные механизмы опосредуют противоэпилептический и нейропротекторный эффект кетогенной диеты (рис. 3).
ВЛИЯНИЕ ДИЕТЫ С СОКРАЩЕНИЕМ ЧИСЛА ПОТРЕБЛЯЕМЫХ КАЛОРИЙ НА РАБОТУ МОЗГА
Ограничение калорий, как и кетогенная диета, приводит к переходу от углеводного метаболизма к липидному и сопровождается усиленным образованием кетоновых тел.
Молекулярные эффекты ограничения калорий
Выделяют четыре основных молекулярных механизма, лежащих в основе влияния ограничения калорий на работу мозга: (1) путь mTOR, (2) путь протеинкиназы, активируемой аденозинмонофосфатом (AMPK), (3) сигнальный путь IGF-1/инсулина (insulin-like growth factor 1, инсулиноподобный фактор 1) и (4) путь сиртуина [20].
Влияние ограничения калорий на путь mTOR. Мутации, встречающиеся в пути mTOR, связаны с дисплазией, эпилепсией, расстройствами развития ЦНС, включая туберозный склероз, синдром Каудена, нейрофиброматоз типа 1 [98]. Предполагается, что противоэпилептический эффект ограничения калорий может быть частично обусловлен ингибированием пути mTOR. Ограничение калорий на 15% уменьшает фосфорилирование протеинкиназы B и рибосомного белка S6 в неокортексе и гиппокампе у крыс, что свидетельствует об ингибировании каскада mTOR. Отмечается повышение порога потенциала действия, оказывающее противосудорожный эффект [99]. Таким образом, ингибирование пути mTOR в результате ограничения калорий опосредует противоэпилептический эффект данной диеты.
Влияние ограничения калорий на путь AMPK. AMPK – это фермент, принимающий участие в регуляции энергетического гомеостаза клетки. При уменьшении энергетических запасов в клетке происходит переключение клеточной программы пролиферации и роста в сторону катаболизма питательных веществ. У мышей в возрасте 8 мес. при ограничении калорий на 20% в течение 2 мес. отмечается увеличение экспрессии фермента AMPK. На поведенческом уровне это проявляется в улучшении гиппокамп-зависимого пространственного обучения в водном лабиринте Морриса [100]. У крыс при индукции эпилепсии по типу киндлинга ограничение калорий на 15% вызывает увеличение фосфорилирования AMPK в гиппокампе и неокортексе. Повышенная активность AMPK приводит к ингибированию пути mTOR [64]. Такие молекулярные изменения опосредуют противосудорожный эффект ограничения калорий в этой модели эпилепсии [99]. Таким образом, за счет увеличения активности пути AMPK ограничение калорий оказывает нейропротекторное действие и противосудорожный эффект, опосредованный ингибированием пути mTOR.
Влияние ограничения калорий на сигнальный путь IGF-1/инсулина. Сигнальный путь IGF-1/инсулина играет важную роль в функционировании ЦНС. При развитии головного мозга активность пути IGF-1/инсулина обеспечивает выживаемость нейронов за счет активации нейротрофических факторов. При старении активность данного пути снижается, что связано с увеличением уровня окислительного стресса и необходимостью активации антиоксидантной защиты [101]. На фоне ограничения калорий на 30% у мышей при старении наблюдается снижение экспрессии белка IGF-1 и его рецептора в гиппокампе. Снижение активности пути IGF-1/инсулина приводит к увеличению экспрессии фактора транскрипции FOXO3 [101]. FOXO3 способствует снижению окислительного стресса, оказывая нейропротекторное действие. В модели инсульта у крыс линии Sprague–Dawley ограничение калорий на 30% приводит к значительному снижению уровня инсулина и IGF-1 в сыворотке крови [102]. Ограничение калорий в течение 8 нед. (начиная с возраста 20 мес.) перед индукцией инсульта улучшает восстановление, связанное с кожной чувствительностью, сенсомоторной интеграцией и пространственной памятью (тест на оценку функциональной асимметрии, тест “вращающийся стержень” и водный лабиринт Морриса соответственно) [102]. Таким образом, сигнальный путь IGF-1/инсулина опосредует нейропротекторный эффект ограничения калорий при старении и травмах головного мозга.
Влияние ограничения калорий на путь сиртуина. Сиртуиновые белки являются никотинамидадениндинуклеотид (НАД+)-зависимыми деацетилазами, которые регулируют активность белков, вовлеченных в метаболизм и обеспечение выживаемости клеток [20]. Сниженная экспрессия сиртуина 1 ассоциирована с процессами нейродегенерации. Например, уменьшение уровня сиртуина 1 наблюдается у пациентов с болезнью Альцгеймера [103]. Ограничение калорий, напротив, приводит к увеличению экспрессии данного белка в головном мозге мышей [104], что связывают с нейропротекторным эффектом данной диеты [20].
Клеточные эффекты ограничения калорий
Влияние ограничения калорий на процесс аутофагии. Аутофагия – катаболический процесс, который обеспечивает удаление неправильно свернутых или агрегированных белков и поддерживает гомеостаз органелл в цитоплазме клетки. Нарушение аутофагии сопровождает болезни Альцгеймера, Паркинсона, Крейтцфельдта–Якоба, мультисистемную атрофию, связанные с нарушением структуры белков и их аккумуляцией [105]. Между процессом аутофагии (катаболизм) и активностью пути mTOR (анаболизм) существует отрицательная обратная связь: увеличение активности пути mTOR ингибирует процесс аутофагии [106]. Ограничение калорий на 30% в течение 10 мес. у мышей уменьшает экспрессию mTOR в гиппокампе, что указывает на увеличение уровня аутофагии. В поведении это проявляется в виде улучшения пространственной памяти в водном лабиринте Морриса [107]. В модели черепно-мозговой травмы у мышей краткосрочное (1 мес.) ограничение калорий на 30% приводит к уменьшению экспрессии mTOR и глиального кислого фибриллярного белка (glial fibrillary acidic protein, GFAP) в гиппокампе. Это свидетельствует об увеличении процесса аутофагии и уменьшении реактивности астроцитов, что сопровождается улучшением гиппокамп-зависимой памяти в водном лабиринте Морриса [108]. Таким образом, ограничение калорий играет защитную роль при старении и травмах ЦНС за счет активации процесса аутофагии.
Влияние ограничения калорий на морфофункциональные свойства астроцитов. Недавно обнаружено, что ограничение калорий на 30% в течение одного месяца до начала эксперимента изменяет морфофункциональные характеристики астроцитов мышей в возрасте 2–3 мес. [109]. На фоне диеты отмечается увеличение объемной доли дистальных перисинаптических астроцитарных листочков. Изменение морфологии астроцитов способствует повышению эффективности захвата ими К+ и глутамата из синаптической щели. Эти изменения усиливают синаптическую пластичность, увеличивая амплитуду долговременной потенциации в гиппокампе. Наблюдается увеличение длительности Ca2+-событий в астроцитах одновременно с уменьшением их размера, снижается амплитуда интенсивности кальциевого сигнала. Изменение описанных пространственно-временных характеристик кальциевой динамики в астроцитах свидетельствует о функциональных перестройках астроцитов [109].
Системные эффекты и терапевтическое значение ограничения калорий
В многочисленных исследованиях, от дрозофилы до человека, ограничение калорий связывают с увеличением средней продолжительности жизни и замедлением ухудшения когнитивных функций в процессе старения [110]. Отмечается, что как небольшое ограничение калорий (на 10%), так и существенное ограничение (на 40%) увеличивают продолжительность жизни [111]. Поскольку известно, что ограничение потребления белка также может влиять на продолжительность жизни, важное значение имеет анализ экспериментальных работ с различной степенью ограничения калорий с учетом состава диеты. На основе такого комплексного анализа сделан вывод о том, что положительный эффект диеты с сокращением числа потребляемых калорий достигается именно за счет ограничения калорий, а не вследствие снижения потребления белка (или сахарозы, и, возможно, жиров, в пище) [112].
Противоэпилептический эффект ограничения калорий. Ограничение калорий широко применяется в различных моделях эпилепсии в качестве терапии. Ограничение калорий на 40% в течение 6 мес. у крыс предотвращает дегенерацию ГАМКергических нейронов в гиппокампе и энторинальной коре, которая наблюдается после введения каиновой кислоты, вызывающей судорожные приступы [98]. У крыс при индукции эпилепсии по типу киндлинга в амигдале ограничение калорий на 15% увеличивает порог возникновения эпилептической активности и снижает ее продолжительность [99]. У мышей линии EL, используемой в качестве модели эпилепсии, ограничение калорий на 15% приводит к снижению частоты возникновения эпилептических приступов, что сопровождается понижением уровня глюкозы в крови [113]. Таким образом, ограничение калорий обладает противоэпилептическим действием, которое может быть опосредовано уменьшением гликолитического обмена в мозге.
Эффекты ограничения калорий при старении. С середины 30-х годов ХХ века известно, что длительное ограничение калорий (в течение 700–900 дней) увеличивает продолжительность жизни у крыс, причем у самцов эффект выражен сильнее, чем у самок [114]. Положительный эффект ограничения калорий при старении показан в 20-летнем лонгитюдном исследовании на макаках-резусах. При ограничении калорий наблюдается уменьшение числа случаев диабета и замедление атрофии серого вещества головного мозга [115], при этом возраст животных на момент начала диеты является определяющим фактором ее эффективности [116]. Ограничение калорий на 40% у самок мышей препятствует возрастным нарушениям двигательно-моторной координации в тесте “вращающийся стержень” и оказывает положительный эффект на процессы формирования и хранения пространственной памяти в водном лабиринте Морриса. Позднее начало диеты (в возрасте 66 нед.) не приводит к улучшению пространственного обучения [117]. Ограничение калорий на 35% у крыс препятствует возрастному снижению пластичности в гиппокампе, сопровождающемуся нарушением долговременной потенциации. Замедление старения головного мозга при ограничении калорий связывают с поддержанием нормального кровотока и целостности белого вещества. Эффект обеспечивается за счет увеличения нейрогенеза в гиппокампе и снижения окислительного стресса при помощи нейропротекторных факторов, включая белок теплового шока HSP-70 и глюкозорегулируемый белок теплового шока GRP-78 [118].
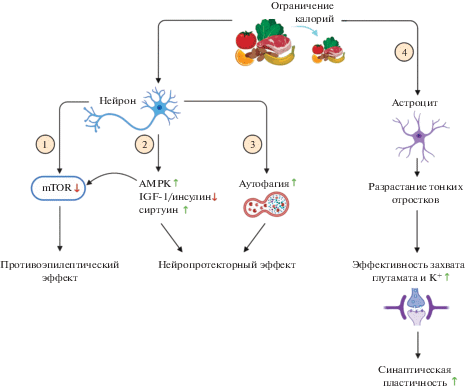
Таким образом, ограничение калорий, как и кетогенная диета, приводит к образованию кетоновых тел. Это способствует снижению окислительного стресса и оказывает нейропротекторный эффект (рис. 4). В отличие от кетогенной диеты ограничение калорий не оказывает влияние на KATP-каналы [119]. Противосудорожное действие обеспечивается молекулярным путем mTOR, ингибирование которого может дополнительно происходить за счет активации AMPK. На клеточном уровне ограничение калорий усиливает процесс аутофагии и изменяет морфофункциональные характеристики астроцитов.
Рис. 4.
Механизмы действия диеты с сокращением числа потребляемых калорий. (1) – путь mTOR отвечает за контроль синтеза белков, транскрипции, аутофагии, метаболизма, биогенеза и сохранения органелл в клетке. Ограничение калорий приводит к ингибированию каскада mTOR. Отмечается повышение порога потенциала действия, что оказывает противосудорожный эффект. (2) – повышение активности AMPK при ограничении калорий также приводит к ингибированию пути mTOR, что оказывает противосудорожное действие. Кроме того, увеличение экспрессии AMPK на фоне ограничения калорий связывают с нейропротекторным эффектом. Снижение активности сигнального пути IGF-1/инсулина при ограничении калорий приводит к увеличению экспрессии фактора транскрипции FOXO3 (не показан). FOXO3 способствует снижению окислительного стресса, оказывая нейропротекторный эффект. Сиртуин регулирует активность белков, вовлеченных в метаболизм и обеспечение выживаемости клеток. Уровень сиртуина увеличивается при ограничении калорий, что связывают с нейропротекторным эффектом этой диеты. (3) – при ограничении калорий активируется процесс аутофагии. Аутофагия направлена на удаление неправильно свернутых или агрегированных белков и поддержание гомеостаза органелл в клетке. Повышение процесса аутофагии на фоне диеты, особенно при старении, опосредует нейропротекторный эффект. (4) – ограничение калорий влияет на морфофункциональные характеристики астроцитов. Происходит увеличение количества тонких астроцитарных отростков, что приводит к повышению эффективности захвата К+ и глутамата из синаптической щели и увеличению амплитуды синаптической потенциации. AMPK – протеинкиназа, активируемая аденозинмонофосфатом, IGF-1 – инсулиноподобный фактор 1, mTOR –механистическая мишень рапамицина.
ВЛИЯНИЕ ЗАПАДНОЙ ДИЕТЫ НА РАБОТУ МОЗГА
В отличие от кетогенной диеты, западная диета характеризуется превышением необходимого для жизнедеятельности организма количества потребляемых калорий, при этом большая часть энергии обеспечивается за счет жиров и углеводов [23]. Это приводит к смещению метаболизма в сторону усиления запасания жиров.
Молекулярные эффекты западной диеты
Молекулярные механизмы западной диеты изучены мало. Западная диета может вызывать проявления воспалительной реакции, связанные с увеличением уровня окислительного стресса, что отрицательно влияет на когнитивные функции. Западная диета приводит к увеличению наработки мРНК, кодирующих провоспалительные цитокины в гиппокампе, включая фактор некроза опухоли-α и интерлейкин-1β. Изменения в воспалительном профиле гиппокампа проявляются в виде нарушения пространственного распознавания у крыс [120]. Увеличение уровня провоспалительных факторов при западной диете связывают со сниженной экспрессией BDNF (brain-derived neurotrophic factor, нейротрофический фактор мозга) в гиппокампе и коре. BDNF контролирует процесс синаптической пластичности, лежащей в основе процессов обучения и памяти [121]. Механизм влияния западной диеты на уровень экспрессии BDNF остается неизвестным. Molteni с соавт. [121] показали, что физическая активность может модулировать эффекты западной диеты. Западная диета нарушает экспрессию белков, обеспечивающих нейропластичность (BDNF, синапсина 1, GAP-43 и CREB). Нарушение их экспрессии и пространственного восприятия не наблюдается, если крысам предоставлять доступ к беговому колесу в течение 2 мес. в ходе диеты. Наличие физической активности в условиях западной диеты снижает уровень окислительного стресса. Было выдвинуто предположение, что окислительный стресс является ранним эффектом такой диеты. В результате происходит формирование АФК, что приводит к уменьшению экспрессии BDNF и когнитивному дефициту (рис. 5) [122].
Рис. 5.
Механизм действия западной диеты на работу мозга. На фоне западной диеты повышаются уровни провоспалительных факторов IL1β и TNF-α. Провоспалительные факторы приводят к увеличению образования АФК, что способствует развитию окислительного стресса. Окислительный стресс запускает реактивную перестройку астроцитов. Морфологические и функциональные изменения астроцитов, связанные с реактивностью, приводят к нарушению синаптической пластичности. Это проявляется в уменьшении экспрессии BDNF в нейронах. АФК – активные формы кислорода. BDNF – brain-derived neurotrophic factor (нейротрофический фактор мозга), IL1β – Interleukin 1β (интерлейкин 1β), TNF-α – tumor necrosis factor alpha (фактор некроза опухоли α).
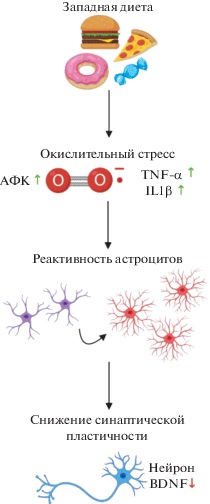
Клеточные эффекты западной диеты
Одним из ключевых последствий применения западной диеты является нарушение проницаемости ГЭБ, приводящее к метаболическим и когнитивным расстройствам [123–125]. Наблюдается снижение активного транспорта через ГЭБ гормонов гипоталамуса, участвующих в регуляции пищевого поведения – грелина [126], отвечающего за чувство голода, и лептина, контролирующего чувство насыщения [127]. Помимо гормональных изменений, нарушение проницаемости ГЭБ на фоне западной диеты связывают с когнитивным дефицитом в модели гиппокамп-зависимого ассоциативного обучения у крыс [123–125]. Дисфункция ГЭБ усугубляет течение болезни Альцгеймера, поскольку способствует накоплению β-амилоида в лимбической системе и неокортексе [128, 129]. β-амилоид является маркером данного заболевания и отражает степень патологии. Сопутствующее нейровоспаление, в свою очередь, может модулировать проницаемость ГЭБ [130–132].
Кроме того, западная диета приводит к повышению активации микроглии [133], что проявляется в снижении эффективности выполнения гиппокамп-зависимого теста “радиальный лабиринт” у крыс [134].
Системные эффекты западной диеты
Метаболический синдром и инсулинорезистентность. Инсулин контролирует необходимый уровень экспрессии транспортеров глюкозы и их доставку в клеточную мембрану [135], что важно для нормального функционирования мозга. Избыток веса, вызванный применением западной диеты, связан с нарушением инсулиновой сигнализации. У мышей это проявляется в снижении уровня экспрессии нейрональных транспортеров глюкозы GLUT1 и GLUT3 на 40% по сравнению с контролем, что вызывает дефицит поступления глюкозы в головной мозг [136]. У мышей в возрасте 2 мес., подвергнутых западной диете в течение 3 мес., наблюдается ожирение в сочетании с системной инсулинорезистентностью и нарушением регуляции липидного обмена. Животные демонстрируют депрессивно-подобное поведение, при этом признаки тревожности и ухудшения памяти отсутствуют [137]. Высокий уровень экспрессии рецептора инсулина в гиппокампе и коре головного мозга [138] указывает на важную роль инсулина в регуляции синаптической пластичности, лежащей в основе процессов обучения и памяти. Периферическая инсулинорезистентность, вызванная западной диетой, приводит к ухудшению этих процессов и связана с повышенным риском развития деменции у людей [139]. У мышей западная диета приводит к ухудшению гиппокамп-зависимой памяти в тесте Y-образный лабиринт, при этом степень функциональных изменений в мозге зависит от процентного содержания жиров в пище [140]. Западная диета широко распространена в современном обществе и способствует росту случаев ожирения по всему миру. Под влиянием западной диеты развивается метаболический синдром, то есть состояние организма, при котором наблюдаются три или более из перечисленных патологических состояний: абдоминальное ожирение, высокое содержание глюкозы и/или холестерина в крови, повышенное кровяное давление, инсулинорезистентность [141]. Развитие инсулинорезистентности, одного из ключевых проявлений метаболического синдрома, традиционно рассматривается с глюкоцентрической точки зрения, согласно которой глюкотоксичность играет ведущую роль при данной патологии. Однако в настоящее время известно, что липотоксичность за счет увеличения уровня жирных кислот в крови также вносит значительный вклад в развитие инсулинорезистенции при метаболическом синдроме (липоцентрическая гипотеза) [142]. Вредное воздействие западной диеты обусловлено ее аддиктогенностью [143]. Часто на фоне ожирения двигательная активность организма снижается. Особенностью метаболизма углеводов является уменьшение расходования энергии в состоянии покоя. Таким образом, на фоне западной диеты организм переходит в режим быстрого насыщения как на метаболическом, так и на поведенческом уровне. Сокращение углеводов при кетогенной диете, напротив, приводит к снижению свободного потребления энергии и увеличению расхода энергии в покое и при активности [144].
Западная диета приводит к увеличению риска развития деменции [128]. Западная диета значительно усугубляет болезнь Альцгеймера, способствуя усилению процессов нейродегенерации [145] и увеличивая число реактивных астроцитов, то есть астроцитов, претерпевающих морфофункциональные перестройки в ответ на повреждение, заболевание или инфекцию ЦНС [146]. У мышей линии APPswe/PS1, являющихся моделью болезни Альцгеймера, на фоне западной диеты ухудшается формирование пространственной памяти (в тесте “T-образный лабиринт”) и способности к обучению (в тесте на распознавание объектов) [147]. В модели нейродегенерации, вызванной повреждением спинного мозга, западную диету используют для оценки влияния метаболического синдрома на процессы восстановления после травмы. У мышей до индукции травмы западная диета (60% Ккал жира в течение 7 нед.) способствует астроглиозу и потере миелина в неповрежденном спинном мозге, а после травмы значительно ухудшает сенсомоторное восстановление, усугубляет потерю олигодендроцитов, уменьшает рост аксонов и увеличивает уровень микроглиоза [148].
Таким образом, западная диета увеличивает окислительный стресс и способствует нарушению проницаемости ГЭБ. При данной диете наблюдается снижение пластичности нейронов, астроглиоз, воспалительные процессы, что в совокупности приводит к ухудшению когнитивных функций и прогноза нейродегенеративных заболеваний.
ВЗАИМОСВЯЗЬ ДИЕТЫ И ДРУГИХ ФАКТОРОВ ЭКСПОЗОМА
Помимо рассмотренных факторов экспозома (возраст, пол, состав и продолжительность режима питания, уровень физической активности), модулирующих эффекты диеты на работу ЦНС, существует ряд других факторов, изученных в меньшей степени (влияние материнской диеты на потомство, обогащение среды, стресс, микробиота).
Влияние режима питания на потомство
У мышей режим питания самок во время беременности и в период вскармливания оказывает влияние на работу мозга, поведение и метаболизм у потомства. Содержание самок в условиях западной диеты в эти периоды приводит к метаболическим изменениям у потомства. В краткосрочном и долгосрочном периодах эти изменения проявляются в переедании, развитии резистентности к лептину, увеличении массы тела и развитии предпочтения к еде, содержащей высокое количество жиров и углеводов [149]. На молекулярном уровне у потомства усиливаются воспалительные процессы и снижается уровень экспрессии BDNF в гиппокампе [149]. Применение западной диеты за несколько недель до наступления беременности и во время нее вызывает структурные изменения в мозге потомства, происходящие за счет нарушений в программе нейрональной дифференциации. Это приводит к потере пространственной идентичности и аксональной селективности кортикальных нейронов [150]. На поведенческом уровне отмеченные эффекты западной диеты проявляются в виде ухудшения пространственной памяти в гиппокамп-зависимых тестах (лабиринт Морриса, лабиринт Барнс) [149]. Отмечается изменение нормального социального поведения, связанное со снижением количества окситоцин-экспрессирующих нейронов у потомства мышей [151].
Влияние уровня обогащения среды
Условия содержания животных также оказывают влияние на процессы обучения и формирования памяти. Высокая степень обогащения среды оказывает положительное влияние (например, в ходе нормального старения), а низкая – отрицательное (например, при индуцированных травмах мозга). Следовательно, есть основания предполагать, что отрицательное влияние западной диеты может быть скорректировано при помощи обогащения стандартных лабораторных условий содержания животных. Нивелирование отрицательного эффекта западной диеты на инструментальное поведение крыс при содержании в условиях многофакторной стимулирующей среды подтверждает наличие отмеченного ранее взаимодействия между факторами экспозома [152]. Это имеет важное значение для применения данных подходов в клинической практике.
Влияние факторов стресса
Другой фактор экспозома – стресс – непосредственно связан с эффектами применения диеты в отношении работы головного мозга. Стресс вызывает изменения в макрооси гипоталамус–гипофиз–надпочечники, стимулируя выработку гормонов (инсулин, лептин, грелин), сигнализация которых нарушена при западной диете [153]. Эти гормоны вовлечены в пищевое поведение и активируют области мозга, отвечающие за мотивацию и ответ на стресс. Увеличение количества гормонов (глюкокортикоиды, кортикорелин) и нейротрансмиттеров (норадреналин) в ответ на воздействие стрессогенных факторов может приводить к активации системы вознаграждения в мозге (прилежащее ядро, полосатое тело). Это способствует увеличению потребления жирной и сладкой пищи, характерной для западной диеты [153]. Долговременное ограничение калорий приводит к развитию психологического стресса, что на молекулярном уровне соответствует увеличению уровня кортизола. В поведенческих тестах при долговременном ограничении калорий наблюдается повышение тревожности у мышей. Это проявляется в снижении количества посещений открытых рукавов в тесте “приподнятый крестообразный лабиринт” и уменьшении спонтанной двигательной активности [117]. Таким образом, в зависимости от типа диеты стресс может быть либо причиной, либо следствием изменений в функционировании нервной системы.
Влияние микробиоты
Эффекты диеты на работу ЦНС невозможно рассматривать отдельно от влияния кишечной микробиоты – фактора экспозома, входящего в состав макрооси микробиота–кишечник–мозг (рис. 1b). В головном мозге и пищеварительном тракте западная диета вызывает снижение передачи сигналов, регулируемых ретиноевой кислотой и желчными кислотами, рецепторы которых участвуют в контроле метаболизма и процессов воспаления. Дисбактериоз и нарушение регуляции синтеза желчных кислот в печени на фоне западной диеты сопровождаются системным воспалением, активацией микроглии и снижением нейропластичности [133]. Дисбактериоз, связанный с сокращением числа бактерий, продуцирующих короткоцепочечные жирные кислоты [154] на фоне западной диеты в сочетании с социальной изоляцией может приводить к депрессивно-подобному поведению у мышей [155]. Пропионовая кислота, относящаяся к классу короткоцепочечных жирных кислот, в модели рассеянного склероза уменьшает воспаление в ЦНС и приводит к снижению уровня демиелинизации, способствуя сокращению степени атрофии аксонов [154].
Таким образом, при изучении влияния диеты на работу мозга необходимо принимать во внимание комбинацию факторов экспозома и степень их взаимодействия.
ЗАКЛЮЧЕНИЕ
Рассмотрение физиологии организма с позиции экспозома позволяет определить причинно-следственные связи между комбинацией факторов, оказывавших влияние на организм в течение его жизни. Рассмотренные в обзоре механизмы диет как одного из факторов экспозома, а также анализ их применения позволяют выделить молекулярные механизмы их воздействия на работу головного мозга. Кетогенная диета и ограничение калорий имеют сходные молекулярные механизмы, поскольку обе приводят к метаболическому сдвигу в сторону образования кетоновых тел из жирных кислот. Обе диеты оказывают нейропротекторный эффект в условиях недостатка углеводов (рис. 3, 4). При западной диете, напротив, избыток жиров на фоне большого количества углеводов увеличивает окислительный стресс. Это способствует нарушению проницаемости ГЭБ и развитию астроглиоза, который приводит к снижению пластичности у нейронов. Несмотря на наличие морфофункциональной связи между нейронами и астроцитами (рис. 1c), исследования влияния диеты на морфологию и физиологию астроцитов крайне немногочисленны. Данные об изменениях нейрон-глиальных взаимодействий под влиянием различных режимов питания практически отсутствуют.
Комплексный подход в изучении эффектов диеты на работу мозга также встречается редко. Следует учитывать, что диета может действовать через несколько одновременных механизмов, которые различаются в зависимости от типа клеток и области мозга. Исследование влияния диеты на работу мозга – активно развивающаяся область знаний, однако ряд научных вопросов остается не до конца решенным. Например, как изменяются свойства нейронов (глутаматергических, ГАМКергических) в случае использования ими в качестве источника энергии преимущественно кетоновых тел или глюкозы [156, 157]? Как различные метаболические состояния влияют на активность астроцитов и определенных подтипов нейронов [158]? Есть ли разница между глутаматергическими и ГАМКергическими нейронами в их реакции на метаболические изменения, вызванные диетами [159]? В какой степени гликолиз и окислительное фосфорилирование способствуют поддержанию активности нейронов в состоянии покоя и во время активации [160]? Влияет ли сдвиг метаболизма в сторону увеличения окислительного фосфорилирования в митохондриях и уменьшения гликолиза при кетогенной диете и ограничении калорий на возбудимость нейронов [161, 162]? Каковы хронические эффекты кетогенной диеты, ограничения калорий и западной диеты [21, 156]? Какие модели эпилепсии in vivo и in vitro лучше всего подходят для оценки эффектов и механизмов кетогенной диеты и ограничения калорий [163, 164]? За счет каких механизмов может отличаться эффективность применения различных диет при лечении лекарственно-резистентных форм эпилепсии [165, 166]?
Клинический потенциал использования диет в качестве поддерживающей терапии уже был показан для ряда заболеваний, таких как эпилепсия [85, 99], нейродегенерация [83, 115, 118], глиома [167]. Отсутствие дополнительной фармакологической нагрузки и простота применения делают специальные режимы питания идеальными кандидатами в качестве сопутствующей, поддерживающей, превентивной и облегчающей течение болезни терапии (например, в случае высокого генетического фактора риска развития нейродегенеративных заболеваний).
Список литературы
Браже А.Р., Доронин М.С., Попов А.В., Денисов П.А., Семьянов А.В. (2019) Исследование паттернов кальциевой динамики в сетях астроцитов головного мозга. Росс. физиол. журн. им. И.М. Сеченова 105: 1436–1451. [Brazhe A.R., Doronin M.S., Popov A.V., Denisov P.A., Semyanov A.V. (2019) Issledovanie patternov kal’cievoj dinamiki v setyah astrocitov golovnogo mozga. Russ. J. Physiol. 105: 1436–1451 (In Russ)]. https://doi.org/10.1134/s0869813919110037
Wild C.P. (2005) Complementing the genome with an “exposome”: The outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer. Epidemiol. Biomark and Prevention. 14: 1847–1850. https://doi.org/10.1158/1055-9965.EPI-05-0456
Wild C.P. (2012) The exposome: From concept to utility. International. J. Epidemiol. 41:24–32. https://doi.org/10.1093/ije/dyr236
Guloksuz S., van Os J., Rutten B.P.F. (2018) The Exposome Paradigm and the Complexities of Environmental Research in Psychiatry. JAMA Psychiatry. 75: 985–986. https://doi.org/10.1001/jamapsychiatry.2018.1211
Pries L.K., Dal Ferro G.A., van Os J., Delespaul P., Kenis G., Lin B.D., Luykx J.J., Richards A.L., Akdede B., Binbay T., Altlnyazar V., Yallnçetin B., Gümüş-Akay G, Cihan B., Soygür H., Ulaş H., Åžahin Cankurtaran E., Ulusoy Kaymak S. Mihaljevic M.M., Andric Petrovic S., Mirjanic T., Bernardo M., Mezquida G., Amoretti S., Bobes J., Saiz P.A., Garciá-Portilla M.P., Sanjuan J., Aguilar E.J., Santos J.L., Jiménez-López E., Arrojo M., Carracedo A., López G., González-Penãs J., Parellada M., Maric N.P., AtbaşoÄalu C., Ucok A., Alptekin K., Can Saka M., Arango C., O’Donovan M., Tosato S., Rutten B.P.F., Guloksuz S. (2020) Examining the independent and joint effects of genomic and exposomic liabilities for schizophrenia across the psychosis spectrum. Epidemiol. and Psych. Sci. 29: 1–10. https://doi.org/10.1017/S2045796020000943
Colomina M.T., Sánchez-Santed F., Conejo N.M., Collado P., Salvador A., Gallo M., Pinos H., Salas C., Navarro J.F., Adán A., Azpiroz A., Arias J.L. (2018) The psychoexposome: A holistic perspective beyond health and disease. Psicothema. 30: 5–7. https://doi.org/10.7334/psicothema2017.244
Finch C.E., Kulminski A.M. (2019) The Alzheimer’s Disease Exposome. Alzheimer’s and Dementia. 15: 1123–1132. https://doi.org/10.1016/j.jalz.2019.06.3914
Moon Y. (2016) Microbiome-linked crosstalk in the gastrointestinal exposome towards host health and disease. Pediatric Gastroenterol, Hepatol. and Nutrition. 19: 221–228. https://doi.org/10.5223/pghn.2016.19.4.221
Wang J., Jia H. (2016) Metagenome-wide association studies: fine-mining the microbiome. Nat. Rev. Microbiol. 14: 508–522. https://doi.org/10.1038/nrmicro.2016.83
Sanchis-Gomar F., Lavie C.J., Mehra M.R., Henry B.M., Lippi G. (2020) Obesity and Outcomes in COVID-19: When an Epidemic and Pandemic Collide. Mayo. Clinic. Proc. 95: 1445–1453. https://doi.org/10.1016/j.mayocp.2020.05.006
Semyanov A., Henneberger C., Agarwal A. (2020) Making sense of astrocytic calcium signals — from acquisition to interpretation. Nature. Rev. Neurosci. 21: 551–564. https://doi.org/10.1038/s41583-020-0361-8
Domingues H.S., Portugal C.C., Socodato R., Relvas J.B. (2016) Oligodendrocyte, astrocyte, and microglia crosstalk in myelin development, damage, and repair. Front. Cell Develop. Biol. 4: 71. https://doi.org/10.3389/fcell.2016.00071
Nelson D.L., Cox M.M. (2017) Lehninger Principles of Biochemistry 7th. WH Freeman and Company.
Magistretti P.J., Allaman I. (2013) Brain energy metabolism. In: Neuroscience in the 21st Century: From Basic to Clinical. 1591–1620.
Camberos-Luna L., Massieu L. (2020) Therapeutic strategies for ketosis induction and their potential efficacy for the treatment of acute brain injury and neurodegenerative diseases. Neurochem Internat. 133: 104614. https://doi.org/10.1016/j.neuint.2019.104614
Hall K.D., Chen K.Y., Guo J., Lam Y., Leibel R.L., Mayer L., Reitman M.L., Rosenbaum M., Smith S.R., Walsh B.T., Ravussin E. (2016) Energy expenditure and body composition changes after an isocaloric ketogenic diet in overweight and obese men. Am. J. Clin. Nutrit. 104: 324–333.https://doi.org/10.3945/ajcn.116.133561.Dietary
Wilson J.R., Levine S.Z., Rivkin H. (1926) the respiratory metabolism in infancy and in childhood: II. Ketosis and the respiratory exchange in children. Am. J. Diseas. Childr. 31: 335–356. https://doi.org/10.1001/archpedi.1926.04130030022003
Woodyatt R.T. (1910) The action of glycol aldehyd and glycerin aldehyd in diabetes mellitus and the nature of antiketogenesis. J. Am. Med. Assoc. 55: 2109–2112. https://doi.org/10.1001/jama.1910.04330250005003
Schutz Y. (2011) Protein turnover, ureagenesis and gluconeogenesis. Bern. Int. J. Vitam. Nutr. Res. 81: 101–107. https://doi.org/10.1024/0300-9831/a000064
Speakman J.R., Mitchell S.E. (2011) Caloric restriction. Mol. Aspects. Med. 32: 159–221. https://doi.org/10.1016/j.mam.2011.07.001
Maalouf M., Rho J.M., Mattson M.P. (2009) The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain. Res. Rev. 59: 293–315. https://doi.org/10.1016/j.brainresrev.2008.09.002
Masoro E.J. (2010) History of Caloric Restriction, Aging and Longevity. In: Everitt A.V., Rattan S.I.S., le Couteur D.G., de Cabo R. (eds). Springer Netherlands, Dordrecht, 3–14.
Shively C.A., Appt S.E., Vitolins M.Z., Uberseder B., Michalson K.T., Silverstein-Metzler M.G., Register T.C. (2019) Mediterranean versus Western Diet Effects on Caloric Intake, Obesity, Metabolism, and Hepatosteatosis in Nonhuman Primates. Obesity. 27: 777–784. https://doi.org/10.1002/oby.22436
Cordain L., Eaton S.B., Sebastian A., Mann N., Lindeberg S., Watkins B.A., O’Keefe J.H., Brand-Miller J. (2005) Origins and evolution of the Western diet: Health implications for the 21st century. Am. J. Clin. Nutr. 81: 341–354. https://doi.org/10.1093/ajcn.81.2.341
Schönfeld P., Reiser G. (2013) Why does brain metabolism not favor burning of fatty acids to provide energy-Reflections on disadvantages of the use of free fatty acids as fuel for brain. J. Cereb. Blood. Flow. Metab. 33: 1493–1499. https://doi.org/10.1038/jcbfm.2013.128
Pifferi F., Cunnane S.C., Guesnet P. (2020) Evidence of the role of omega-3 polyunsaturated fatty acids in brain glucose metabolism. Nutrients. 12: 1–11. https://doi.org/10.3390/nu12051382
Rose J., Brian C., Pappa A., Panayiotidis M.I., Franco R. (2020) Mitochondrial Metabolism in Astrocytes Regulates Brain Bioenergetics, Neurotransmission and Redox Balance. Front. Neurosci. 14: 1–20. https://doi.org/10.3389/fnins.2020.536682
Cunnane S.C., Courchesne-Loyer A., Vandenberghe C., St-Pierre V., Fortier M., Hennebelle M., Croteau E., Bocti C., Fulop T., Castellano C.A. (2016) Can ketones help rescue brain fuel supply in later life? Implications for cognitive health during aging and the treatment of alzheimer’s disease. Front. Mol. Neurosci. 9: 53. https://doi.org/10.3389/fnmol.2016.00053
Pierre K., Pellerin L. (2005) Monocarboxylate transporters in the central nervous system: Distribution, regulation and function. J. Neurochem. 94: 1–14. https://doi.org/10.1111/j.1471-4159.2005.03168.x
Achanta L.B., Rae C.D. (2017) β-Hydroxybutyrate in the Brain: One Molecule, Multiple Mechanisms. Neurochem. Res. 42: 35–49. https://doi.org/10.1007/s11064-016-2099-2
Pierre K., Magistretti P.J., Pellerin L. (2002) MCT2 is a major neuronal monocarboxylate transporter in the adult mouse brain. J. Cereb. Blood. Flow. Metab. 22: 586–595. https://doi.org/10.1097/00004647-200205000-00010
Chiry O., Pellerin L., Monnet-Tschudi F., Fishbein W.N., Merezhinskaya N., Magistretti P.J., Clarke S. (2006) Expression of the monocarboxylate transporter MCT1 in the adult human brain cortex. Brain. Res. 1070: 65–70. https://doi.org/10.1016/j.brainres.2005.11.064
Magistretti P.J., Allaman I. (2018) Lactate in the brain: From metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 19: 235–249. https://doi.org/10.1038/nrn.2018.19
Chi C.P., Roberts E.L. (2003) Energy Substrates for Neurons during Neural Activity: A Critical Review of the Astrocyte-Neuron Lactate Shuttle Hypothesis. J. Cereb. Blood. Flow. Metab. 23: 1263–1281. https://doi.org/10.1097/01.wcb.0000081369.51727.6f
Larrabee M.G. (1996) Partitioning of CO2 production between glucose and lactate in excised sympathetic ganglia, with implications for brain. J. Neurochem. 67: 1726–1734. https://doi.org/10.1046/j.1471-4159.1996.67041726.x
Dienel G.A., Rothman D.L (2020) Reevaluation of Astrocyte-Neuron Energy Metabolism with Astrocyte Volume Fraction Correction: Impact on Cellular Glucose Oxidation Rates, Glutamate–Glutamine Cycle Energetics, Glycogen Levels and Utilization Rates vs. Exercising Muscle, and Na+/K+ Pumping. Neurochem. Res. 45: 2607–2630. https://doi.org/10.1007/s11064-020-03125-9
Yellen G. (2018) Fueling thought: Management of glycolysis and oxidative phosphorylation in neuronal metabolism. J. Cell. Biol. 217:2235–2246. https://doi.org/10.1083/jcb.201803152
Pellerin L., Magistretti P.J. (1994) Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA. 9:10625–10629. https://doi.org/10.1073/pnas.91.22.10625
Bingul D., Kalra K., Murata E.M., Belser A., Dash M.B. (2020) Persistent changes in extracellular lactate dynamics following synaptic potentiation. Neurobiol. Learn. Mem. 175: 107314. https://doi.org/https://doi.org/10.1016/j.nlm.2020.107314
Dienel G.A. (2019) Brain glucose metabolism: Integration of energetics with function. Physiol. Rev. 99: 949–1045. https://doi.org/10.1152/physrev.00062.2017
Dienel G.A. (2017) Lack of appropriate stoichiometry: Strong evidence against an energetically important astrocyte–neuron lactate shuttle in brain. J. Neurosci. Res. 95: 2103–2125. https://doi.org/10.1002/jnr.24015
Magistretti P.J., Allaman I. (2015) A Cellular Perspective on Brain Energy Metabolism and Functional Imaging. Neuron. 86: 883–901. https://doi.org/10.1016/j.neuron.2015.03.035
Saez I., Duran J., Sinadinos C., Beltran A., Yanes O., Tevy M.F., Martínez-Pons C., Milán M., Guinovart J.J. (2014) Neurons have an active glycogen metabolism that contributes to tolerance to hypoxia. J. Cereb. Blood Flow. Metab. 34: 945–955. https://doi.org/10.1038/jcbfm.2014.33
Gentry M.S., Guinovart J.J., Minassian B.A., Roach P.J., Serratosa J.M. (2018) Lafora disease offers a unique window into neuronal glycogen metabolism. J. Biol. Chem. 293: 7117–7125. https://doi.org/10.1074/jbc.R117.803064
Vilchez D., Ros S., Cifuentes D., Pujadas L., Vallès J., García-Fojeda B., Criado-García O., Fernández-Sánchez E., Medrão-Fernández I., Domínguez J., García-Rocha M., Soriano E., Rodríguez De Córdoba S., Guinovart J.J. (2007) Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat. Neurosci. 10: 1407–1413. https://doi.org/10.1038/nn1998
DiNuzzo M., Schousboe A. (2019) Brain Glycogen Metabolism. Springer.
Duran J., Gruart A., García-Rocha M., Delgado-García J.M., Guinovart J.J. (2014) Glycogen accumulation underlies neurodegeneration and autophagy impairment in lafora disease. Hum. Mol. Genet. 23: 3147–3156. https://doi.org/10.1093/hmg/ddu024
Лебедева А.В., Дембицкая Ю.В., Пимашкин А.С., Журавлева З.Д., Шишкова Е.А., Семьянов А.В. (2015) Роль энергетических субстратов в кальциевой активности астроцитов гиппокампа крыс раннего постнатального период. Соврем. технол. мед. [Lebedeva A.V., Dembitskaya Y.V., Pimashkin A.S., Zhuravleva Z.D., Shishkova E.A. (2015) The Role of Energy Substrates in Astrocyte Calcium Activity of Rat Hippocampus in Early Postnatal Ontogenesis. Sovrem. Tekhnologii. Med. 7: 14–19 (In Russ)]. https://doi.org/10.17691/stm2015.7.3.02
Steiner P. (2019) Brain Fuel Utilization in the Developing Brain. Ann. Nutr. Metab. 75(suppl 1): 8–18. https://doi.org/10.1159/000508054
Turner D.A., Adamson D.C. (2011) Neuronal-astrocyte metabolic interactions: Understanding the transition into abnormal astrocytoma metabolism. J. Neuropathol. Exp. Neurol. 70: 167–176. https://doi.org/10.1097/NEN.0b013e31820e1152
Zilberter Y., Zilberter T. (2020) Glucose-Sparing Action of Ketones Boosts Functions Exclusive to Glucose in the Brain. eneuro 7: ENEURO.0303-20.2020. https://doi.org/10.1523/ENEURO.0303-20.2020
Castellano C.-A., Nugent S., Paquet N., Tremblay S., Bocti C., Lacombe G., Imbeault H., Turcotte É., Fulop T., Cunnane S.C. (2015) Lower Brain 18F-Fluorodeoxyglucose Uptake But Normal 11C-Acetoacetate Metabolism in Mild Alzheimer’s Disease Dementia. J. Alzheimer’s Dis. 43: 1343–1353. https://doi.org/10.3233/JAD-141074
Croteau E., Castellano C.A., Fortier M., Bocti C., Fulop T., Paquet N., Cunnane S.C. (2018) A cross-sectional comparison of brain glucose and ketone metabolism in cognitively healthy older adults, mild cognitive impairment and early Alzheimer’s disease. Exp. Gerontol. 107: 18–26. https://doi.org/https://doi.org/10.1016/j.exger.2017.07.004
Croteau E., Castellano C.-.A., Richard M.A., Fortier M., Nugent S., Lepage M., Duchesne S., Whittingstall K., Turcotte É.E., Bocti C., Fülöp T., Cunnane S.C. (2018) Ketogenic Medium Chain Triglycerides Increase Brain Energy Metabolism in Alzheimer’s Disease. J. Alzheimer’s Dis. 64: 551–561. https://doi.org/10.3233/JAD-180202
Sharp F.R. (1976) Relative cerebral glucose uptake of neuronal perikarya and neuropil determined with 2-deoxyglucose in resting and swimming rat. Brain. Res. 110: 127–139. https://doi.org/10.1016/0006-8993(76)90213-4
Araque A., Parpura V., Sanzgiri R.P., Haydon P.G. (1999) Tripartite synapses: Glia, the unacknowledged partner. Trends. Neurosci. 22: 208–215. https://doi.org/10.1016/S0166-2236(98)01349-6
Tabernero A., Medina J.M., Giaume C. (2006) Glucose metabolism and proliferation in glia: Role of astrocytic gap junctions. J. Neurochem. 99: 1049–1061. https://doi.org/10.1111/j.1471-4159.2006.04088.x
Devivo D.C., Leckie M.P., Ferrendelli J.S., McDougal D.B. (1978) Chronic ketosis and cerebral metabolism. Ann. Neurol. 3: 331–337. https://doi.org/10.1002/ana.410030410
Ma W., Berg J., Yellen G. (2007) Ketogenic diet metabolites reduce firing in central neurons by opening KATP channels. J. Neurosci. 27: 3618–3625. https://doi.org/10.1523/JNEUROSCI.0132-07.2007
Chan O., Lawson M., Zhu W., Beverly J.L., Sherwin R.S. (2007) ATP-sensitive K+ channels regulate the release of GABA in the ventromedial hypothalamus during hypoglycemia. Diabetes. 56: 1120–1126. https://doi.org/10.2337/db06-1102
Tanner G.R., Lutas A., Martínez-François J.R., Yellen G. (2011) Single KATP channel opening in response to action potential firing in mouse dentate granule neurons. J. Neurosci. 31: 8689–8696. https://doi.org/10.1523/JNEUROSCI.5951-10.2011
Mercer R.W., Dunham P.B. (1981) Membrane-bound ATP fuels the Na/K pump: Studies on membrane-bound glycolytic enzymes on inside-out vesicles from human red cell membranes. J. Gen. Physiol. 78: 547–568. https://doi.org/10.1085/jgp.78.5.547
Haller M., Mironov S.L., Karschin A., Richter D.W. (2001) Dynamic activation of K ATP channels in rhythmically active neurons. J. Physiol. 537: 69–81. https://doi.org/10.1111/j.1469-7793.2001.0069k.x
Lipton J.O., Sahin M. (2014) The Neurology of mTOR. Neuron. 84: 275–291. https://doi.org/10.1016/j.neuron.2014.09.034
Sengupta S., Peterson T.R., Laplante M., Oh S., Sabatini D.M. (2010) mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature. 468: 1100–1104. https://doi.org/10.1038/nature09584
Ma D., Wang A.C., Parikh I., Green S.J., Hoffman J.D., Chlipala G., Murphy M.P., Sokola B.S., Bauer B., Hartz A.M.S., Lin A.-L. (2018) Ketogenic diet enhances neurovascular function with altered gut microbiome in young healthy mice. Sci. Rep. 8: 6670. https://doi.org/10.1038/s41598-018-25190-5
Laplante M., Sabatini D.M. (2012) MTOR signaling in growth control and disease. Cell. 149: 274–293. https://doi.org/10.1016/j.cell.2012.03.017
Martuscello R.T., Vedam-Mai V., McCarthy D.J., Schmoll M.E., Jundi M.A., Louviere C.D., Griffith B.G., Skinner C.L., Suslov O., Deleyrolle L.P., Reynolds B.A. (2016) A Supplemented High-Fat Low-Carbohydrate Diet for the Treatment of Glioblastoma. Clin. Cancer. Res. 22: 2482–2495. https://doi.org/10.1158/1078-0432.CCR-15-0916
Rojas-Morales P., Pedraza-Chaverri J., Tapia E. (2020) Ketone bodies, stress response, and redox homeostasis. Redox Biol. 29:101395. https://doi.org/10.1016/j.redox.2019.101395
Haces M.L., Hernández-Fonseca K., Medina-Campos O.N., Montiel T., Pedraza-Chaverri J., Massieu L. (2008) Antioxidant capacity contributes to protection of ketone bodies against oxidative damage induced during hypoglycemic conditions. Exp. Neurol. 211: 85–96. https://doi.org/10.1016/j.expneurol.2007.12.029
Lu Y., Yang Y.Y., Zhou M.W., Liu N., Xing H.Y., Liu X.X., Li F. (2018) Ketogenic diet attenuates oxidative stress and inflammation after spinal cord injury by activating Nrf2 and suppressing the NF-κB signaling pathways. Neurosci. Lett. 683: 13–18. https://doi.org/10.1016/j.neulet.2018.06.016
Maalouf M., Sullivan P.G., Davis L., Kim D.Y., Rho J.M. (2007) Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience. 145(1): 256–264. https://doi.org/10.1016/j.neuroscience.2006.11.065
Cheng C.M., Hicks K., Wang J., Eagles D.A., Bondy C.A. (2004) Caloric restriction augments brain glutamic acid decarboxylase-65 and -67 expression. J. Neurosci. Res. 77: 270–276. https://doi.org/10.1002/jnr.20144
Juge N., Gray J.A., Omote H., Miyaji T., Inoue T., Hara C., Uneyama H., Edwards R.H., Nicoll R.A., Moriyama Y. (2010) Metabolic Control of Vesicular Glutamate Transport and Release. Neuron. 68: 99–112. https://doi.org/10.1016/j.neuron.2010.09.002
Ruskin D.N., Kawamura M., Masino S.A. (2020) Adenosine and Ketogenic Treatments. J. Caffeine Adenosine Res. 10: 104–109. https://doi.org/10.1089/caff.2020.0011
Fredholm B.B., Chen J.F., Cunha R.A., Svenningsson P., Vaugeois J.M. (2005) Adenosine and Brain Function. Int. Rev. Neurobiol. 63: 191–270. https://doi.org/10.1016/S0074-7742(05)63007-3
Kawamura M., Ruskin D.N., Masino S.A. (2010) Metabolic autocrine regulation of neurons involves cooperation among pannexin hemichannels, adenosine receptors, and KATP channels. J. Neurosci. 30: 3886–3895. https://doi.org/10.1523/JNEUROSCI.0055-10.2010
Gzielo K., Soltys Z., Rajfur Z., Setkowicz Z.K. (2019) The Impact of the Ketogenic Diet on Glial Cells Morphology. A Quantitative Morphological Analysis. Neuroscience. 412: 239–251. https://doi.org/10.1016/j.neuroscience.2019.06.009
Zhao Z., Lange D.J., Voustianiouk A., MacGrogan D., Ho L., Suh J., Humala N., Thiyagarajan M., Wang J., Pasinetti G.M. (2006) A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci. 7: 29. https://doi.org/10.1186/1471-2202-7-29
Tai K.K., Truong D.D. (2007) Ketogenic diet prevents seizure and reduces myoclonic jerks in rats with cardiac arrest-induced cerebral hypoxia. Neurosci. Lett. 425: 34–38. https://doi.org/10.1016/j.neulet.2007.08.007
Tai K.K., Nguyen N., Pham L., Truong D.D. (2008) Ketogenic diet prevents cardiac arrest-induced cerebral ischemic neurodegeneration. J. Neural. Transm. 115: 1011–1017. https://doi.org/10.1007/s00702-008-0050-7
Yang Q., Guo M., Wang X., Zhao Y., Zhao Q., Ding H., Dong Q., Cui M. (2017) Ischemic preconditioning with a ketogenic diet improves brain ischemic tolerance through increased extracellular adenosine levels and hypoxia-inducible factors. Brain. Res. 1667: 11–18. https://doi.org/10.1016/j.brainres.2017.04.010
Hertz L., Chen Y., Waagepetersen H.S. (2015) Effects of ketone bodies in Alzheimer’s disease in relation to neural hypometabolism, β-amyloid toxicity, and astrocyte function. J. Neurochem. 134: 7–20. https://doi.org/10.1111/jnc.13107
Keene D.L. (2006) A systematic review of the use of the ketogenic diet in childhood epilepsy. Pediatr. Neurol. 35: 1–5. https://doi.org/10.1016/j.pediatrneurol.2006.01.005
Hallböök T., Ji S., Maudsley S., Martin B. (2012) The effects of the ketogenic diet on behavior and cognition. Epilepsy. Res. 100: 304–309. https://doi.org/10.1016/j.eplepsyres.2011.04.017
Samoilova M., Weisspapir M., Abdelmalik P., Velumian A.A., Carlen P.L. (2010) Chronic in vitro ketosis is neuroprotective but not anti-convulsant. J. Neurochem. 113: 826–835. https://doi.org/10.1111/j.1471-4159.2010.06645.x
Phillis J.W., Wu P.H. (1981) The role of adenosine and its nucleotides in central synaptic transmission. Prog. Neurobiol. 16: 187–239. https://doi.org/10.1016/0301-0082(81)90014-9
Masino S.A., Li T., Theofilas P., Sandau U.S., Ruskin D.N, Fredholm B.B., Geiger J.D., Aronica E., Boison D. (2011) A ketogenic diet suppresses seizures in mice through adenosine A 1 receptors. J. Clin. Invest. 121: 2679–2683. https://doi.org/10.1172/JCI57813
Plata A., Lebedeva A., Denisov P., Nosova O., Postnikova T.Y., Pimashkin A., Brazhe A., Zaitsev A.V., Rusakov D.A., Semyanov A. (2018) Astrocytic Atrophy Following Status Epilepticus Parallels Reduced Ca2+ Activity and Impaired Synaptic Plasticity in the Rat Hippocampus. Front. Mol. Neurosci. 11: 215. https://doi.org/10.3389/fnmol.2018.00215
Neal E.G., Chaffe H., Schwartz R.H. Lawson M.S., Edwards N., Fitzsimmons G., Whitney A., Cross J.H. (2008) The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet. Neurol. 7: 500–506. https://doi.org/10.1016/S1474-4422(08)70092-9
Patel A., Pyzik P.L., Turner Z., Rubenstein J.E., Kossoff E.H.(2010) Long-term outcomes of children treated with the ketogenic diet in the past. Epilepsia. 51: 1277–1282. https://doi.org/10.1111/j.1528-1167.2009.02488.x
Jensen N.J. Wodschow H.Z., Nilsson M., Rungby J. (2020) Effects of ketone bodies on brain metabolism and function in neurodegenerative diseases. Int. J. Mol. Sci. 21: 8767. https://doi.org/10.3390/ijms21228767
VanItallie T.B., Nonas C., di Rocco A., Boyar K., Hyams K., Heymsfield S.B. (2005) Treatment of Parkinson disease with diet-induced hyperketonemia: A feasibility study. Neurology. 64: 728–730. https://doi.org/10.1212/01.WNL.0000152046.11390.45
Tieu K., Perier C., Caspersen C., Teismann P., Wu D.C., Yan S. du, Naini A., Vila M., Jackson-Lewis V., Ramasamy R., Przedborski S. (2003) D-β-Hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J. Clin. Invest. 112: 892–901. https://doi.org/10.1172/JCI200318797
Kashiwaya Y., Takeshima T., Mori N., Nakashima K., Clarke K., Veech R.L. (2000) D-β-hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proc. Natl. Acad. Sci. U S A. 97: 5440–5444. https://doi.org/10.1073/pnas.97.10.5440
Evangeliou A., Vlachonikolis I., Mihailidou H., Spilioti M., Skarpalezou A. Makaronas N., Prokopiou A., Christodoulou P., Liapi-Adamidou G., Helidonis E., Sbyrakis S., Smeitink J. (2003) Application of a ketogenic diet in children with autistic behavior: Pilot study. J. Child. Neurol. 18: 113–118. https://doi.org/10.1177/08830738030180020501
Zhao Q., Stafstrom C.E., Fu D.D., Hu Y., Holmes G.L. (2004) Detrimental Effects of the Ketogenic Diet on Cognitive Function in Rats. Pediatr. Res. 55: 498–506. https://doi.org/10.1203/01.PDR.0000112032.47575.D1
Rubio C., Luna R., Rosiles A., Rubio-Osornio M. (2020) Caloric Restriction and Ketogenic Diet Therapy for Epilepsy: A Molecular Approach Involving Wnt Pathway and KATP Channels. Front. Neurol. 11: 584298. https://doi.org/10.3389/fneur.2020.584298
Phillips-Farfán B.V., Rubio Osornio M del C., Custodio Ramírez V., Paz Tres C., Carvajal Aguilera K.G. (2015) Caloric restriction protects against electrical kindling of the amygdala by inhibiting the mTOR signaling pathway. Front. Cell Neurosci. 9: 90. https://doi.org/10.3389/fncel.2015.00090
Ma L., Wang R., Dong W., Zhao Z. (2018) Caloric restriction can improve learning and memory in C57/BL mice probably via regulation of the AMPK signaling pathway. Exp. Gerontol. 102: 28–35. https://doi.org/10.1016/j.exger.2017.11.013
Hadem I.K.H., Sharma R. (2017) Differential Regulation of Hippocampal IGF-1-Associated Signaling Proteins by Dietary Restriction in Aging Mouse. Cell. Mol. Neurobiol. 37: 985–993. https://doi.org/10.1007/s10571-016-0431-7
Ciobanu O., Elena Sandu R., Tudor Balseanu A., Zavaleanu A., Gresita A., Petcu E.B., Uzoni A., Popa-Wagner A. (2017) Caloric restriction stabilizes body weight and accelerates behavioral recovery in aged rats after focal ischemia. Aging Cell 16: 1394–1403. https://doi.org/10.1111/acel.12678
Julien C., Tremblay C., Émond V., Lebbadi M., Salem N., Bennett D.A., Calon F. (2009) Sirtuin 1 reduction parallels the accumulation of tau in alzheimer disease. J. Neuropathol. Exp. Neurol. 68: 48–58. https://doi.org/10.1097/NEN.0b013e3181922348
Morris K.C., Lin H.W., Thompson J.W., Perez-Pinzon M.A. (2011) Pathways for ischemic cytoprotection: Role of sirtuins in caloric restriction, resveratrol, and ischemic preconditioning. J. Cereb. Blood Flow Metab. 31: 1003–1019. https://doi.org/10.1038/jcbfm.2010.229
Bagherniya M., Butler A.E., Barreto G.E., Sahebkar A. (2018) The effect of fasting or calorie restriction on autophagy induction: A review of the literature. Ageing. Res. Rev. 47: 183–197. https://doi.org/10.1016/j.arr.2018.08.004
Schmeisser K., Parker J.A. (2019) Pleiotropic effects of mTOR and autophagy during development and aging. Front. Cell Dev. Biol. 7: 192. https://doi.org/10.3389/fcell.2019.00192
Dong W., Wang R., Ma L.N., Xu B.L., Zhang J.S., Zhao Z.W., Wang Y.L., Zhang X. (2015) Autophagy involving age-related cognitive behavior and hippocampus injury is modulated by different caloric intake in mice. Int. J. Clin. Exp. Med. 8: 11843–11853.
Liu Y., Wang R., Zhao Z., Dong W., Zhang X., Chen X., Ma L. (2017) Short-term caloric restriction exerts neuroprotective effects following mild traumatic brain injury by promoting autophagy and inhibiting astrocyte activation. Behav. Brain. Res. 331: 135–142. https://doi.org/10.1016/j.bbr.2017.04.024
Popov A., Denisov P., Bychkov M., Brazhe A., Lyukmanova E., Shenkarev Z., Lazareva N., Verkhratsky A., Semyanov A. (2020) Caloric restriction triggers morphofunctional remodeling of astrocytes and enhances synaptic plasticity in the mouse hippocampus. Cell. Death. Dis. 11: 208. https://doi.org/10.1038/s41419-020-2406-3
Rühlmann C., Wölk T., Blümel T., Stahn L., Vollmar B., Kuhla A. (2016) Long-term caloric restriction in ApoE-deficient mice results in neuroprotection via Fgf21-induced AMPK/mTOR pathway. Aging. 8: 2777–2789. https://doi.org/10.18632/aging.101086
Matyi S., Jackson J., Garrett K., Deepa S.S., Unnikrishnan A. (2018) The effect of different levels of dietary restriction on glucose homeostasis and metabolic memory. Geroscience. 40: 139–149. https://doi.org/10.1007/s11357-018-0011-5
Speakman J.R., Mitchell S.E., Mazidi M. (2016) Calories or protein? The effect of dietary restriction on lifespan in rodents is explained by calories alone. Exp. Gerontol. 86: 28–38. https://doi.org/https://doi.org/10.1016/j.exger.2016.03.011
Greene A.E., Todorova M.T., McGowan R., Seyfried T.N. (2001) Caloric restriction inhibits seizure susceptibility in epileptic EL mice by reducing blood glucose. Epilepsia. 42: 1371–1378. https://doi.org/10.1046/j.1528-1157.2001.17601.x
McCay C.M., Crowell M.F., Maynard L.A. (1989) The effect of retarded growth upon the length of life span and upon the ultimate body size. Nutrition. 10: 63–79. https://doi.org/10.1093/jn/10.1.63
Colman R.J., Anderson R.M., Johnson S.C., Kastman E.K., Kosmatka K.J., Beasley T.M., Allison D.B., Cruzen C., Simmons H.A., Kemnitz J.W., Weindruch R. (2009) Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 325: 201–204. https://doi.org/10.1126/science.1173635
Mattison J.A., Colman R.J., Beasley T.M., Allison D.B., Kemnitz J.W., Roth G.S., Ingram D.K., Weindruch R., de Cabo R., Anderson R.M. (2017) Caloric restriction improves health and survival of rhesus monkeys. Nat. Commun. 8: 14063. https://doi.org/10.1038/ncomms14063
Kuhla A., Lange S., Holzmann C., Maass F., Petersen J. Vollmar B., Wree A. (2013) Lifelong Caloric Restriction Increases Working Memory in Mice. PLoS One. 8: e68778. https://doi.org/10.1371/journal.pone.0068778
Villeda S.A., Horowitz A.M. (2017) Therapeutic potential of systemic brain rejuvenation strategies for neurodegenerative disease. F1000Research. 6: 1291. https://doi.org/10.12688/f1000research.11437.1
Aguilera K.G.C., Farfán B.V.P. (2016) Caloric Restriction and Dietary Treatments of Epilepsy: Mechanistic Insights for Drug Discovery. 163–180.
Beilharz J.E., Maniam J., Morris M.J. (2014) Short exposure to a diet rich in both fat and sugar or sugar alone impairs place, but not object recognition memory in rats. Brain. Behav Immun. 37: 134–141. https://doi.org/10.1016/j.bbi.2013.11.016
Molteni R., Barnard R.J., Ying Z,. Roberts C.K., Gómez-Pinilla F. (2002) A high-fat, refined sugar diet reduces hippocampal brain-derived neurotrophic factor, neuronal plasticity, and learning. Neuroscience. 112: 803–814. https://doi.org/10.1016/S0306-4522(02)00123-9
Beilharz J.E., Maniam J., Morris M.J. (2015) Diet-induced cognitive deficits: The role of fat and sugar, potential mechanisms and nutritional interventions. Nutrients. 7: 6719–6738. https://doi.org/10.3390/nu7085307
Kanoski S.E., Zhang Y., Zheng W., Davidson T.L. (2010) The effects of a high-energy diet on hippocampal function and blood-brain barrier integrity in the rat. J. Alzheimer’s Dis. 21: 207–217. https://doi.org/10.3233/JAD-2010-091414
Davidson T.L., Monnot A., Neal A.U., Martin A.A., Horton J.J., Zheng W. (2012) The effects of a high-energy diet on hippocampal-dependent discrimination performance and blood-brain barrier integrity differ for diet-induced obese and diet-resistant rats. Physiol. Behav. 107: 26–33. https://doi.org/10.1016/j.physbeh.2012.05.015
Davidson T.L., Hargrave S.L., Swithers S.E., Sample C.H., Fu X., Kinzig K.P., Zheng W. (2013) Inter-relationships among diet, obesity and hippocampal-dependent cognitive function. Neuroscience. 253: 110–122. https://doi.org/10.1016/j.neuroscience.2013.08.044
Banks W.A., Burney B.O., Robinson S.M. (2008) Effects of triglycerides, obesity, and starvation on ghrelin transport across the blood-brain barrier. Peptides. 29: 2061–2065. https://doi.org/10.1016/j.peptides.2008.07.001
Banks W.A., Coon A.B., Robinson S.M., Moinuddin A., Shultz J.M., Nakaoke R., Morley J.E. (2004) Triglycerides Induce Leptin Resistance at the Blood-Brain Barrier. Diabetes. 53: 1253–1260. https://doi.org/10.2337/diabetes.53.5.1253
Hsu T.M., Kanoski S.E. (2014) Blood-brain barrier disruption: Mechanistic links between western diet consumption and dementia. Front. Aging Neurosci. 6: 88. https://doi.org/10.3389/fnagi.2014.00088
Mulder M., Blokland A., van den Berg D.J., Schulten H., Bakker A.H.F., Terwel D., Honig W., de Kloet E.R., Havekes L.M., Steinbusch H.W.M., de Lange E.C.M. (2001) Apolipoprotein E protects against neuropathology induced by a high-fat diet and maintains the integrity of the blood-brain barrier during aging. Lab. Investig. 81: 953–960. https://doi.org/10.1038/labinvest.3780307
Abbott N.J. (2000) Inflammatory mediators and modulation of blood-brain barrier permeability. Cell. Mol. Neurobiol. 20: 131–147. https://doi.org/10.1023/A:1007074420772
Abbott N.J., Rönnbäck L., Hansson E.(2006) Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 7: 41–53. https://doi.org/10.1038/nrn1824
Ek M., Engblom D., Saha S., Blomqvist A., Jakobsson P.-J., Ericsson-Dahlstrand A. (2001) Pathway across the blood–brain barrier. Nature. 410: 430–431. https://doi.org/10.1038/35068632
Jena P.K., Sheng L., di Lucente J., Jin L.W., Maezawa I., Wan Y.J.Y. (2018) Dysregulated bile acid synthesis and dysbiosis are implicated in Western diet-induced systemic inflammation, microglial activation, and reduced neuroplasticity. FASEB J. 32: 2866–2877. https://doi.org/10.1096/fj.201700984RR
Granholm A.C, Bimonte-Nelson H.A., Moore A.B., Nelson M.E., Freeman L.R., Sambamurti K. (2008) Effects of a saturated fat and high cholesterol diet on memory and hippocampal morphology in the middle-aged rat. J. Alzheimer’s Dis. 14: 133–145. https://doi.org/10.3233/JAD-2008-14202
Biessels G.J., Reagan L.P. (2015) Hippocampal insulin resistance and cognitive dysfunction. Nat. Rev. Neurosci. 16: 660–671. https://doi.org/10.1038/nrn4019
Kothari V., Luo Y., Tornabene T., O’Neill A.M., Greene M.W., Geetha T., Babu J.R. (2017) High fat diet induces brain insulin resistance and cognitive impairment in mice. Biochim. Biophys. Acta – Mol. Basis. Dis. 1863: 499–508. https://doi.org/10.1016/j.bbadis.2016.10.006
Tsai S.F., Wu H.T., Chen P.C., Chen Y.W., Yu M., Wang T.F., Wu S.Y., Tzeng S.F., Kuo Y.M. (2018) High-fat diet suppresses the astrocytic process arborization and downregulates the glial glutamate transporters in the hippocampus of mice. Brain. Res. 1700: 66–77. https://doi.org/10.1016/j.brainres.2018.07.017
Zhao W.Q., Alkon D.L. (2001) Role of insulin and insulin receptor in learning and memory. Mol. Cell. Endocrinol. 177: 125–134. https://doi.org/10.1016/S0303-7207(01)00455-5
Greenwood C.E., Winocur G. (2005) High-fat diets, insulin resistance and declining cognitive function. Neurobiol. Aging. 26 Suppl: 42–45. https://doi.org/10.1016/j.neurobiolaging.2005.08.017
Lizarbe B., Soares A.F., Larsson S. Duarte J.M.N. (2019) Neurochemical modifications in the hippocampus, cortex and hypothalamus of mice exposed to long-term high-fat diet. Front. Neurosci. 12: 985. https://doi.org/10.3389/fnins.2018.00985
Huang P.L. (2009) A comprehensive definition for metabolic syndrome. Dis. Model Mech. 2: 231–237.https://doi.org/10.1242/dmm.001180
Mittra S., Bansal V.S., Bhatnagar P.K. (2008) From a glucocentric to a lipocentric approach towards metabolic syndrome. Drug. Discov. Today. 13: 211–218. https://doi.org/https://doi.org/10.1016/j.drudis.2008.01.006
Avena N.M., Gold M.S. (2011) Food and addiction - sugars, fats and hedonic overeating. Addiction. 106: 1214–1215. https://doi.org/10.1111/j.1360-0443.2011.03373.x
Ebbeling C.B., Swain J.F., Feldman H.A., Wong W.W., Hachey D.L., Garcia-Lago E., Ludwig D.S. (2012) Effects of dietary composition on energy expenditure during weight-loss maintenance. JAMA. 307: 2627–2634. https://doi.org/10.1001/jama.2012.6607
Graham L.C., Harder J.M., Soto I., de Vries W.N., John S.W.M., Howell G.R. (2016) Chronic consumption of a western diet induces robust glial activation in aging mice and in a mouse model of Alzheimer’s disease. Sci. Rep. 6: 21568. https://doi.org/10.1038/srep21568
Escartin C., Galea E., Lakatos A., O’Callaghan J.P., Petzold G.C., Serrano-Pozo A., Steinhäuser C., Volterra A., Carmignoto G., Agarwal A., Allen N.J., Araque A., Barbeito L., Barzilai A., Bergles D.E., Bonvento G., Butt A.M., Chen W.-T., Cohen-Salmon M., Cunningham C., Deneen B., de Strooper B., Díaz-Castro B., Farina C., Freeman M., Gallo V., Goldman J.E., Goldman S.A., Götz M., Gutiérrez A., Haydon P.G., Heiland D.H., Hol E.M., Holt M.G., Iino M., Kastanenka K.V., Kettenmann H., Khakh B.S., Koizumi S., Lee C.J., Liddelow S.A., MacVicar B.A., Magistretti P., Messing A., Mishra A., Molofsky A. v, Murai K.K., Norris C.M., Okada S., Oliet S.H.R. Oliveira J.F., Panatier A., Parpura V, Pekna M. Pekny M., Pellerin L., Perea G., Pérez-Nievas B.G., Pfrieger F.W., Poskanzer K.E., Quintana F.J., Ransohoff R.M., Riquelme-Perez M., Robel S., Rose C.R., Rothstein J.D. Rouach N., Rowitch D.H., Semyanov A., Sirko S., Sontheimer H., Swanson R.A., Vitorica J., Wanner I.-B., Wood L.B., Wu J., Zheng B., Zimmer E.R., Zorec R., Sofroniew M.V., Verkhratsky A. (2021) Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. https://doi.org/10.1038/s41593-020-00783-4
Thériault P., ElAli A., Rivest S. (2016) High fat diet exacerbates Alzheimer’s disease-related pathology in APPswe/PS1 mice. Oncotarget. 7: 67808–67827. https://doi.org/10.18632/ONCOTARGET.12179
Kim H.N., Langley M.R., Simon W.L., Yoon H., Kleppe L. Lanza I.R., LeBrasseur N.K., Matveyenko A., Scarisbrick I.A. (2020) A Western diet impairs CNS energy homeostasis and recovery after spinal cord injury: Link to astrocyte metabolism. Neurobiol. Dis. 141: 104934. https://doi.org/10.1016/j.nbd.2020.104934
Cordner Z.A., Tamashiro K.L.K. (2015) Effects of high-fat diet exposure on learning & memory. Physiol. Behav. 152: 363–371. https://doi.org/10.1016/j.physbeh.2015.06.008
Cinquina V., Calvigioni D., Farlik M., Halbritter F., Fife-Gernedl V., Shirran S.L., Fuszard M.A., Botting C.H., Poullet P., Piscitelli F., Máté Z., Szabó G., Yanagawa Y., Kasper S., di Marzo V., Mackie K., McBain C.J., Bock C., Keimpema E. Harkany T. (2020) Life-long epigenetic programming of cortical architecture by maternal ‘Western’ diet during pregnancy. Mol. Psychiatry. 25: 22–36. https://doi.org/10.1038/s41380-019-0580-4
Buffington S.A., di Prisco G.V., Auchtung T.A., Ajami N.J., Petrosino J.F., Costa-Mattioli M. (2016) Microbial Reconstitution Reverses Maternal Diet-Induced Social and Synaptic Deficits in Offspring. Cell. 165: 1762–1775. https://doi.org/10.1016/j.cell.2016.06.001
Winocur G., Greenwood C.E. (2005) Studies of the effects of high fat diets on cognitive function in a rat model. Neurobiol. Aging. 26 Suppl: 46–49. https://doi.org/10.1016/j.neurobiolaging.2005.09.003
López-Taboada I., González-Pardo H. Conejo N.M. (2020) Western Diet: Implications for Brain Function and Behavior. Front. Psychol. 11: 564413. https://doi.org/10.3389/fpsyg.2020.564413
Hirschberg S., Gisevius B., Duscha A., Haghikia A. (2019) Implications of diet and the gut microbiome in neuroinflammatory and neurodegenerative diseases. Int. J. Mol. Sci. 20: 3109. https://doi.org/10.3390/ijms20123109
Marrone M.C., Coccurello R. (2020) Dietary fatty acids and microbiota-brain communication in neuropsychiatric diseases. Biomolecules. 10: 12. https://doi.org/10.3390/biom10010012
Li R.J., Liu Y., Liu H.Q., Li J. (2020) Ketogenic diets and protective mechanisms in epilepsy, metabolic disorders, cancer, neuronal loss, and muscle and nerve degeneration. J. Food Biochem. 44: 1–14. https://doi.org/10.1111/jfbc.13140
Guo Q., Liu S., Wang S., Wu M., Li Z., Wang Y. (2019) Beta-hydroxybutyric acid attenuates neuronal damage in epileptic mice. Acta Histochem. 121: 455–459. https://doi.org/10.1016/j.acthis.2019.03.009
Boison D., Steinhäuser C. (2018) Epilepsy and astrocyte energy metabolism. Glia. 66: 1235–1243. https://doi.org/10.1002/glia.23247
Yudkoff M., Daikhin Y., Horyn O., Nissim I., Nissim I. (2008) Ketosis and brain handling of glutamate, glutamine, and GABA. Epilepsia. 49: 73–75. https://doi.org/10.1111/j.1528-1167.2008.01841.x
Koppel S.J., Swerdlow R.H. (2018) Neuroketotherapeutics: A modern review of a century-old therapy. Neurochem. Int. 117: 114–125. https://doi.org/10.1016/j.neuint.2017.05.019
Barzegar M., Afghan M., Tarmahi V., Behtari M., Rahimi Khamaneh S., Raeisi S. (2019) Ketogenic diet: overview, types, and possible anti-seizure mechanisms. Nutr. Neurosci. 1–10. https://doi.org/10.1080/1028415X.2019.1627769
McNally M.A., Hartman A.L. (2012) Ketone bodies in epilepsy. J. Neurochem. 121: 28–35. https://doi.org/10.1111/j.1471-4159.2012.07670.x
Masino S.A., Rho J.M. (2012) Mechanisms of ketogenic diet action. Jasper’s Basic Mechanisms of the Epilepsies [Internet] 4th edition.
Minlebaev M., Khazipov R. (2011) Antiepileptic effects of endogenous beta-hydroxybutyrate in suckling infant rats. Epilepsy Res. 95: 100–109. https://doi.org/10.1016/j.eplepsyres.2011.03.003
Yuen A., Sander L. (2014) Rationale for using intermittent calorie restriction as a dietary treatment for drug resistant epilepsy. Epilepsy Behav. 33: 110–114. https://doi.org/10.1016/j.yebeh.2014.02.026
Martin-McGill K.J., Jackson C.F., Bresnahan R., Levy R.G., Cooper P.N. (2018) Ketogenic diets for drug-resistant epilepsy. Cochrane database Syst. Rev.: CD001903–CD001903. https://doi.org/10.1002/14651858.CD001903.pub4
Seyfried T.N., Sanderson T.M., El-Abbadi M.M., McGowan R. Mukherjee P. (2003) Role of glucose and ketone bodies in the metabolic control of experimental brain cancer. Br. J. Cancer. 89: 1375–1382. https://doi.org/10.1038/sj.bjc.6601269
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова