

Российский физиологический журнал им. И.М. Сеченова, 2021, T. 107, № 4-5, стр. 518-532
Глюкокортикоидная регуляция глутаматергического синапса: механизмы стресс-зависимой нейропластичности
1 Институт высшей нервной деятельности и нейрофизиологии РАН
Москва, Россия
2 Научно-практический психоневрологический центр им. З.П. Соловьева
Москва, Россия
* E-mail: nata_gul@ihna.ru
Поступила в редакцию 21.01.2021
После доработки 31.01.2021
Принята к публикации 01.02.2021
Аннотация
Глюкокортикоиды, высвобождающиеся из коры надпочечников при действии стрессорных факторов, являются важнейшими медиаторами интегративной регуляции адаптивной пластичности мозга, осуществляемой нейрогуморальной гипоталамо-гипофизарно-надпочечниковой системой. Возбуждающие синапсы рассматриваются как ключевые участники синаптической пластичности и поведенческой адаптации. В обзоре представлены накопленные к настоящему времени данные о механизмах глюкокортикоидной регуляции глутаматергического синапса, в первую очередь, на примерах гиппокампа и префронтальной коры. Глюкокортикоиды, запуская трансдукцию сигнала через минералокортикоидные и глюкокортикоидные рецепторы, локализованные на синаптических мембранах и в цитозоле глутаматергических нейронов, регулируют пластичность синапса на уровне пре- и постсинаптического компартментов. Глюкокортикоиды модулируют возбудимость синапса за счет изменений везикулярного транспорта и высвобождения глутамата, опосредуют изменения экспрессии, состава и свойств ионотропных NMDA- и AMPA- и других глутаматных рецепторов. Представлена подробная схема множественных регуляторных механизмов, реализуемых в глутаматергическом синапсе при связывании глюкокортикоидов со специфическими рецепторами.
ВВЕДЕНИЕ: ГЛЮКОКОРТИКОИДЫ – РЕГУЛЯТОРЫ СТРЕСС-ЗАВИСИМОЙ НЕЙРОПЛАСТИЧНОСТИ
Stress is the spice of life.
Hans Selye
Эмоционально окрашенные переживания в стрессорных ситуациях запоминаются лучше, чем в ситуациях с менее выраженной эмоциональной компонентой, и это адаптивный феномен, поскольку он способствует преодолению сопоставимых ситуаций в будущем. Нейроэндокринный ответ на действие стрессорных факторов, наряду с активацией вегетативной нервной системы, включает реакцию гипоталамо-гипофизарно-надпочечниковой оси (ГГНО): высвобождение гипоталамического кортикотропин-рилизинг гормона (КРГ), который стимулирует высвобождение адренокортикотропного гормона (АКТГ) из гипофиза и, наконец, высвобождение глюкокортикоидных гормонов (ГК) из коры надпочечников (кортикостерона у большинства грызунов; кортизола у человека) [1]. ГК поступают в кровообращение, осуществляя как периферическое, так и центральное действие, достигая однотипных рецепторов практически во всех органах и тканях, включая мозг (ГК свободно проходят гемато-энцефалический барьер). Клеточные эффекты КС опосредованы глюкокортикоидными рецепторами (ГР) и минералокортикоидными рецепторами (МР), которые существуют в цитоплазматической/ядерной и мембранной формах и отвечают соответственно за медленные геномные и быстрые негеномные эффекты ГК. ГР экспрессируются в большинстве структур и регионов мозга, в то время как МР экспрессированы, в основном, в тех регионах мозга, которые имеют решающее значение для формирования памяти и эмоций, таких как гиппокамп, миндалина и префронтальная кора [2]. Высокоаффинные МР связаны с ГК при низком уровне гормонов, а сродство ГР к кортикостерону (КС) на порядок ниже, поэтому активация этих рецепторов происходит главным образом при повышении уровня ГК, в частности, при стрессорной реакции. Преимущественно благодаря трансдукции сигнала через мембранные МР, ГК запускают быстрые негеномные эффекты на возбудимость и активацию нейронов в гиппокампе, гипоталамусе, амигдале и префронтальной коре, таким образом в течение минут оказывая влияние на когнитивные функции, адаптивное поведение и нейроэндокринную систему. Трансдукция сигнала ГК через их рецепторы дает возможность всем клеткам мозга, и в первую очередь, нейронам, адекватно отвечать на действие разнообразных стрессорных факторов. Опосредованный ГК сигналинг регулирует и интегрирует ответы различных систем (нейротрансмиттерных, нейротрофиновой, цитокиновой и др.), обеспечивая их взаимодействие в процессе поддержания и настройки нейропластичности. Это, в частности, обеспечивается сбалансированностью работы системы “один гормон/два рецептора”, способной модулировать широкий спектр активностей ЦНС [1, 2]. В частности, это достигается за счет того, что ГК могут регулировать синаптическую функцию с помощью геномных и негеномных эффектов как через активацию МР, так и ГР [3].
Стрессорные факторы оказывают существенное влияние на обучение и память, в частности, через действие ГК на пластичность мозга на субклеточном, клеточном и сетевом уровне [1, 4]. Исследования последних лет показали, что ГК вместе с катехоламинами очень динамично регулируют синаптическую функцию и синаптическую пластичность, лежащие в основе обучения и памяти [5]. ГК модулируют формирование памяти и кодирование информации, в первую очередь через сигнальные механизмы, опосредованные МР, и способствуют длительной консолидации через активацию ГР. Дисфункция ГГНО и кортикостероидных рецепторов приводит к неадекватной реакции организма на стрессорные факторы и развитие как психопатологий, так и неврологических заболеваний [1].
ВОЗБУЖДАЮЩИЕ СИНАПСЫ ГИППОКАМПА КАК МИШЕНЬ ГЛЮКОКОРТИКОИДНОЙ РЕГУЛЯЦИИ
В этой связи особое внимание привлекает гиппокамп, ключевая структура, участвующая в переработке когнитивной и эмоциональной информации. Селективная чувствительность гиппокампа к действию различных стрессорных факторов рассматривается как основа для развития вызванной стрессом поведенческой дисфункции [6, 7]. При этом результаты многочисленных работ свидетельствуют о том, что хронический избыток ГК, в первую очередь при хроническом стрессе, вызывает специфический синаптический дефицит в гиппокампе. Важнейшую роль в этом играют возбуждающие синапсы, ключевые участники синаптической передачи, синаптической пластичности и поведенческой адаптации [8, 9]. Настройка глутаматергической передачи является важным механизмом коммуникации между нейронами. Накапливается все больше доказательств, что патофизиология нервно-психических расстройств, включая расстройства настроения и памяти, связана с нарушением функции и регуляции глутаматергической системы. Избыточные глутаматергические токи в гиппокампе лежат в основе развития деменции, и их нормализация в экспериментальных исследованиях позволяет предотвращать нарушения памяти [10]. В данном обзоре рассмотрены накопленные к настоящему времени данные о механизмах глюкокортикоидной регуляции глутаматергического синапса, в первую очередь, на примере гиппокампа.
В 1988 г. Halpain и McEwen [11] показали, что КС снижает связывание 3Н-глутамата в гиппокампе (на срезах мозга крыс и в синаптических мембранах). Связывание 3Н-глутамата с областями дорсального гиппокампа было значительно снижено у адреналэктомированных животных после введения КС, который однако не изменял связывание 3Н-глутамата в других областях мозга. Эти результаты были первыми, которые показали, что КС может избирательно изменять сайты связывания возбуждающих аминокислот в ткани гиппокампа. В 1990 г. группа Sapolsky показала, что повреждающее действие КС на нейроны гиппокампа зависит от NMDA-рецептора [12]. ГК снижали способность нейронов гиппокампа выживать в патологических для мозга ситуациях (включая гипоксию–ишемию и судорожную активность), опосредованных избытком возбуждающих нейромедиаторов, в первую очередь, глутамата, активацией NMDA-рецепторов и избыточной мобилизацией цитозольного кальция в постсинапсе. Авторы показали, что повреждающее действие ГК на нейроны гиппокампа опосредовано усилением глутаматергической трансмиссии и/или повышением уязвимости нейронов к избытку глутамата. В качестве возможного объяснения была предложена гипотеза о том, что ГК ингибируют поглощение глюкозы нейронами гиппокампа, а многочисленные этапы рецепторного каскада NMDA усиливаются, когда запасы энергии нейронов уменьшаются. Действительно, это группа далее показала, что ГК ингибируют транспорт глюкозы и захват глутамата астроцитами гиппокампа, и этот феномен может быть одним из факторов нейротоксичности ГК [13]. Позже Sandi [14], обсуждая роль и механизмы участия ГК в хранении памяти, констатировал, что стероидные гормоны надпочечников модулируют процессы обучения и памяти, взаимодействуя со специфическими рецепторами ГК в различных областях мозга, и выдвинул гипотезу о роли ГК, действующих через эти рецепторы, в механизмах консолидации памяти. Были рассмотрены возможные ассоциированные с действием ГК через глутаматергическую передачу и молекулы клеточной адгезии молекулярные механизмы, которые могут опосредовать влияние ГК на синаптическую пластичность, способствующую формированию долговременной памяти. За последние два десятилетия получены многочисленные подтверждения влияния ГК на глутаматергические синапсы и раскрыты некоторые механизмы эффектов ГК.
ГЛЮКОКОРТИКОИДНАЯ РЕГУЛЯЦИЯ AMPA-РЕЦЕПТОРОВ В ГИППОКАМПЕ
Показано, что стресс/ГК динамически модулируют синаптическую пластичность на уровне глутаматергических синапсов взрослого мозга. Karst и Joëls исследовали влияние хронического стресса на синаптические ответы гранулярных нейронов зубчатой извилины крыс на стимуляцию перфорантного пути в срезах гиппокампа [15]. Стресс не влиял на базовые синаптические ответы, однако синаптические ответы хронически стрессированных и контрольных крыс на активацию ГР кортикостероном in vitro различались. В среде без магния с добавлением селективных антагонистов глутаматных рецепторов максимальный ответ на синаптическую активацию нейронов при потенциале –70 мВ был значительно усилен после введения КС в срезах стрессированных, но не контрольных животных. Соответственно амплитуда токов, опосредованных АМРА-, но не NMDA-рецепторами, в присутствии КС была выше у стресссированных крыс во всем диапазоне потенциалов. КС также уменьшал время достижения пика токов АМРА, но этот эффект не зависел от предшествующего стрессорного воздействия. Усиленный под действием ГК синаптический ток может способствовать усиленному возбуждению проекционных областей зубчатой извилины, особенно области СА3 гиппокампа. Принято считать, что в височно-гиппокампальном (temporoammonic) возбуждающем синапсе (TA-CA1) вызванное стрессом повышение уровня ГК запускает развитие синаптической дисфункции. Хронически повышенные уровни КС у крыс приводили к снижению возбуждения, опосредованного AMPA-рецепторами, в синапсах TA-CA1 и снижению экспрессии белка субъединицы 1 AMPA-рецепторов. Эти изменения предотвращались аппликацией ингибитора синтеза КС метирапона, который также предотвращал вызванные хроническим стрессом нарушения консолидации пространственной памяти [16]. Авторы работы заключили, что КС достаточен и необходим для развития глутаматергической дисфункции, лежащей в основе стресс-индуцированных синаптических и поведенческих фенотипов.
АМРА-рецепторы играют критическую роль в зависимой от активности пластичности и гиппокамп-зависимом обучении. Показано, что КС избирательно увеличивает поверхностную экспрессию субъединицы GluR2 AMPA-рецепторов в первичных культурах гиппокампа через активацию ГР и механизм, зависящий от синтеза белка. КС также резко увеличивал долю экспрессируемой на поверхности мембраны субъединицы GluR2, способной к латеральной диффузии [17]. При этом КС способствовал зависимому от NMDA-рецепторов эндоцитозу как синаптической, так и экстрасинаптической GluR2 в условиях, ослабляющих синаптическую передачу. Индуцированное КС повышение мобильности АМРА-рецепторов, обеспечивающее синаптический транспорт AMPA-рецепторов, содержащих GluR2, может как способствовать рекрутированию этих рецепторов, так и, при соответствующих условиях, снижению плотности синаптических АМРА-рецепторов и развитию длительной депрессии. Эти процессы могут лежать в основе как облегчающего, так и подавляющего действия ГК на синаптическую пластичность, обучение и память. Groc и соавт. также показали, что КС через различные рецепторы ГК вызывает зависимое от времени увеличение поверхностной подвижности субъединицы GluR2 АМРА-рецепторов и содержания GluR2 на синаптической поверхности; при этом важно, что КС также потенцирует увеличение содержания GluR2 на синаптической поверхности в условиях длительной потенциации, что указывает на участие ГК в трафике АМРА-рецепторов [18]. В экспериментах с пространственным обучением мышей в условиях умеренного стресса показано облегчение обучения под действием стресса и ассоциация с усиленной синаптической экспрессией субъединицы GluA2 АМРА в гиппокампе. Ингибитор синтеза ГК метирапон блокировал как облегчение обучения, так и усиление трафика GluA2. Внутримозговое введение пептида pep2m, который блокирует синаптический трафик GluA2, нарушало немедленное воспроизведение при обучении, а также долгосрочную память, что свидетельствует о ключевой роли синаптического транспорта GluA2 в вызванном стрессом облегчении обучения и памяти [19]. В опытах in vivo и in vitro показано, что опосредованное ГК сопряжение эндоплазматического ретикулума и митохондрий снижает трафик AMPA-рецепторов и митохондрий в синапсы за счет дестабилизации микротрубочек, ассоциированных с митохондриальными ГР [20].
ГЛЮКОКОРТИКОИДНАЯ РЕГУЛЯЦИЯ NMDA-РЕЦЕПТОРОВ В ГИППОКАМПЕ
Модуляция синаптической пластичности ГК в гиппокампе связана с пластическими изменениями синаптических NMDA-рецепторов, которые, наряду с AMPA-рецепторами, являются критическими для синаптической функции и необходимы для индукции многих форм синаптической пластичности. Наряду с влиянием на синаптическую пластичность путем изменения функциональных свойств АМРА-рецепторов, включая изменения их подвижности в мембране и функционирования, ГК регулируют синаптическую пластичность, динамически модулируя функцию синаптических NMDA-рецепторов. Понимание того, как ГК регулируют функцию NMDA-рецепторов, недостаточно, возможно, из-за принятого мнения, что функция NMDA во взрослом мозге стабильна. Функционирование NMDA-рецепторов во взрослом гиппокампе чувствительно даже к кратковременному воздействию ГК, а события, происходящие в раннем возрасте, могут ре-программировать развитие гиппокампа, повышая его чувствительность к модуляции функции NMDA-рецептора ГК. Модуляция функции NMDA-рецепторов опосредует динамическую регуляцию синаптической пластичности и адаптации при стрессе, однако усиленная функция этих рецепторов может быть вовлечена в механизмы связанных со стрессом психопатологий, включая депрессию [21].
КС через МР регулирует поверхностную динамику субъединицы GluN2B NMDA-рецептора и состав синапса. Показано, что внеклеточный КС увеличивает экспрессию GluN2B в синапсах, при этом функционально потенцирование содержания АМРА-рецепторов в синапсах, вызванное КС, ассоциировано с изменениями поверхностной динамики NMDA-рецепторов [22]. Эти данные, полученные путем визуализации с высоким разрешением, показали, что в гиппокампальных сетях ГК являются естественными, мощными, быстрыми и специфическими регуляторами мембранного трафика GluN2B субъединицы NMDA-рецептора, модулирующими зависимые от NMDA-рецептора адаптации синапсов. В срезах гиппокампа взрослых крыс КС потенцировал опосредованные NMDA-рецепторами быстрые вызванные синаптические ответы, а затем происходило медленное увеличение синаптической экспрессии субъединицы GluN2A NMDA-рецепторов [23]. Параллельно с усилением функции NMDA-рецепторов при аппликации КС происходило преходящее облегчение как длительной потенциации, так и длительной депрессии, свидетельствующее о разнонаправленных изменениях пластичности и прекращающееся, когда синаптическая экспрессия GluN2A повышалась. Опосредованное ГК медленное увеличение GluN2A в синапсах гиппокампа может быть гомеостатическим механизмом нормализации синаптической пластичности после быстрой стресс-индуцированной фасилитации.
Krugers и соавт. [24] исследовали in vitro синаптическое потенцирование в области СА1 срезов гиппокампа мышей с низким базальным уровнем КС. Они показали, что ГК быстро настраивают синаптические NMDA-рецепторы за счет мембранной динамики и сигналинга через МР. Введение КС способствовало синаптическому потенцированию, зависимому от активации потенциал-зависимых кальциевых каналов, одновременно ухудшая синаптическую пластичность, опосредованную активацией NMDA-рецептора. Антагонист ГР RU 38486 блокировал оба эти эффекта КС. По-видимому, окончательный эффект ГР на синаптическую пластичность определяется балансом между различными типами потенцирования, который может быть регион-специфичным и зависит от условий эксперимента. Можно предположить, что эти различные эффекты на синаптическую эффективность участвуют в противоположно направленной модуляции когнитивных функций кортикостероидными гормонами. КС быстро снижал активность NMDA-рецепторов в культивируемых нейронах гиппокампа, а при введении в мозг подавлял длительную потенциацию в области СА1 гиппокампа [25]. При этом ГК действовали на рецептор, зависимый от G белка, активируя несколько сигнальных путей в нейронах гиппокампа. Быстрое подавление активности NMDA-рецепторов ГК зависело от фосфолипазы C и соответствующего пути трансдукции сигнала. В срезах гиппокампа взрослых крыс КС, даже в низких концентрациях, индуцирует быстрое увеличение плотности шипиков пирамидных нейронов СА1 гиппокампа. Аппликация RU486, антагониста ГР, препятствовала действию КС. Блокирование любой из киназ – MAPK, PKA, PKC или PI3K, или NMDA-рецепторов подавляло индуцированное КС усиление образования шипиков [26]. Таким образом, стресс/ГК модулируют обновление шипиков через синаптические ГР и множественные киназные пути.
ГЛЮКОКОРТИКОИДНАЯ РЕГУЛЯЦИЯ ДРУГИХ ТИПОВ ГЛУТАМАТНЫХ РЕЦЕПТОРОВ В ГИППОКАМПЕ
Наряду с АМРА- и NMDA-рецепторами, функция других глутаматных рецепторов также регулируется ГК, хотя такая регуляция остается недостаточно изученной. Острый стресс/КС путем стимуляции ГР облегчал долговременную депрессию, вызванную агонистом метаботропного глутаматного рецептора (mGluR) группы 1 (R,S)-3,5-дигидроксифенилглицина (DHPG) в нейронах CA1 гиппокампа [27]. Поскольку считается, что в основе обучения и хранения памяти лежат процессы, аналогичные долговременной потенциации и долговременной депрессии синаптической эффективности, двум основным формам синаптической пластичности, это указывает на потенциальную вовлеченность mGluR в глюкокортикоидную регуляцию обучения и памяти. Каинатные рецепторы – класс ионотропных глутаматных рецепторов, которые играют важную роль в модуляции высвобождения глутамата и синаптической пластичности в гиппокампе крыс. Адреналэктомия приводила к увеличению экспрессии в гиппокампе мРНК субъединиц KA1, GluR6 и GluR7, и этот эффект блокировался КС в случае KA1 и GluR7 и альдостероном в случае GluR6. Хронический иммобилизационный стресс повышал экспрессию субъединицы KA1, но не оказывал влияния на экспрессию других субъединиц, а хроническое введение умеренных доз КС также увеличивало экспрессию мРНК KA1 в зубчатой извилине, тогда как высокая доза не оказывала эффекта [28]. Таким образом, ГК влияют на состав каинатных рецепторов гиппокампа и селективно модулируют субъединицу KA1 в зубчатой извилине и CA3, принимая тем самым участие в вызванной стрессом адаптивной структурной пластичности.
ГК высвобождаются не только в результате действия стрессорных факторов, но и в ходе ультрадианных и циркадианных импульсов и ассоциированы с ритмическими гипофизарно-надпочечниковыми взаимодействиями. Ритмическая активность появления ГК в крови, контролируемая супрахиазматическим ядром гипоталамуса, связана с специфическими для клеточного типа структурными и функциональными изменениями, которые происходят с циркадианной ритмичностью в нейронах и астроцитах в области CA1 гиппокампа. В пирамидных нейронах изменяется поверхностная экспрессия NMDA-рецепторов, изменяется расстояние астроцитов до синапсов, вместе эти события изменяют клиренс глутамата, активацию рецепторов и интеграцию временно кластеризованных возбуждающих синаптических входов, в конечном счете модифицируя гиппокамп-зависимое обучение in vivo [29]. При этом уровень ГК является ключевым фактором изменения синаптической силы. Эффекты повторных импульсов КС на характеристики глутаматергической трансмиссии в нейронах гиппокампа зависят от предистории поступления КС [30, 31]. Например, первый импульс вызывает преходящее увеличение частоты миниатюрных возбуждающих постсинаптических потенциалов, трафика АМРА-рецепторов и синаптической пластичности, в то время как базальные вызванные полевые потенциалы остаются неизменными. При последующих аппликациях КС реакции становятся более вариабельными, ослабевают или даже меняют знак с течением времени, в зависимости от времени после аппликации.
ГЛЮКОКОРТИКОИДНАЯ РЕГУЛЯЦИЯ ГЛУТАМАТНЫХ РЕЦЕПТОРОВ В ДРУГИХ СТРУКТУРАХ МОЗГА
Эффекты ГК/стресса на синаптическую пластичность продемонстрированы не только в гиппокампе, но и в других структурах мозга, ключевых для когнитивной функции и эмоционального состояния. Ряд исследований влияния ГК/стресса на пластичность глутаматергических синапсов касается префронтальной коры. Это ключевая область мозга, вовлеченная в реализацию обучения и эмоций, чрезвычайно чувствительна к стрессорным воздействиям. В то время как хронический стресс часто оказывает пагубное воздействие, острый стресс может стимулировать обучение и память, и эти эффекты опосредованы эффектами ГК на глутаматергические синапсы префронтальной коры. Было обнаружено, что поведенческие стрессоры in vivo вызывают длительное опосредованное ГР потенцирование синаптических токов через NMDA- и AMPA-рецепторы избирательно в пирамидных нейронах префронтальной коры. Этот эффект сопровождался повышенной экспрессией субъединиц NMDA- и AMPA-рецепторов в острой фазе стресса [32]. Кроме того, поведенческие тесты показали, что рабочая память, ключевая функция, основанная на рекуррентном возбуждении в сетях нейронов префронтальной коры, усиливается при остром стрессе за счет ГР-зависимых механизмов. Иными словами, была выявлена форма ГР-зависимого долговременного потенцирования синаптической передачи в префронтальной коре, индуцированной естественными стимулами in vivo. Исследования последнего десятилетия подтвердили, что стресс, в первую очередь, за счет изменения секреции ГК и ГК-зависимого сигналинга, вызывает сложные изменения в трансдукции глутаматергического сигнала в префронтальной коре, модулируя когнитивные процессы [33]. Модификации глутаматергической системы, вызванные стрессом, в префронтальной коре, по-видимому, являются двухфазными: в то время как быстрая реакция на стресс предполагает увеличение числа возбуждающих синапсов, синаптической передачи и рабочей памяти, долгосрочные адаптационные изменения, в том числе связанные с хроническим стрессом, вызывают противоположные эффекты. Считают, что действие стресса/ГК на глутаматергическую трансмиссию имеет “U-образную форму” в зависимости от продолжительности и тяжести действия стрессора, при этом двухфазная реакция в условиях острого и хронического стресса отражает адаптивные и дезадаптивные реакции на стрессорные стимулы. Как и в гиппокампе, стресс-индуцированная модуляция возбуждающей синаптической передачи включает изменения в пресинаптическом высвобождении глутамата, постсинаптическом трафике и деградации мембранных глутаматных рецепторов, структуре шипиков и сети цитоскелета, а также эпигенетическом контроле экспрессии генов. В префронтальной и лобной коре острый стресс индуцирует усиление высвобождения глутамата и глутаматной нейротрансмиссии, зависящие от активации рецепторов ГК, причем увеличение легко высвобождаемого пула глутаматных везикул опосредовано синаптическим, негеномным действим ГК, в первую очередь, через МР [34]. Более медленные, частично геномные механизмы задействованы в усилении глутаматной нейротрансмиссии, вызванной стрессом. Изменения в высвобождении глутамата и нейротрансмиссии опосредуют ремоделирование дендритов и другие стресс-зависимые морфологические изменения.
Bonini и соавт. [35] обнаружили, что острый неизбегаемый электроболевой стресс (footshock), хотя и не индуцирует никаких транскрипционных изменений субъединиц AMPA- и NMDA-ионотропных рецепторов, оказывает быстрые преходящие эффекты на экспрессию их белков, фосфорилирование (увеличение для Ser(845) GluA1 и Ser(880) GluA2) и локализацию в постсинаптических шипиках префронтальной и лобной коры. В постсинаптических шипиках стресс индуцировал быстрое снижение экспрессии GluA2 вместе с увеличением его фосфорилирования по Ser(880), что свидетельствует об интернализации GluA2-содержащих АМРА-рецепторов. Экспрессия субъединиц NMDA-рецепторов GluN1 и GluN2A была повышена в постсинаптических шипиках. Эти результаты свидетельствуют о раннем и преходящем усилении опосредованных АМРА-рецепторами токов с последующей преходящей активацией NMDA-рецепторов [36]. Острый неизбегаемый электроболевой стресс стимулирует вызванное деполяризацией высвобождение глутамата в префронтальной и лобной коре на фоне повышения циркулирующих уровней КС, который, связываясь с мембранными рецепторами ГК, индуцирует быстрое повышение уровня глутамата в синаптической щели за счет усиления трафика везикул и увеличения пула высвобождаемого глутамата в перфорированных синапсах, плотность которых в префронтальной коре увеличивается [37]. Индуцированный стрессом ex novo синаптогенез асимметричных синапсов обнаруживается уже через 40 мин после начала стрессорного воздействия, а через 1–14 дней наблюдается значительная атрофия и ремоделирование апикальных дендритов. Таким образом, острый стресс может вызвать быстрые структурные/функциональные изменения в глутаматных синапсах префронтальной коры. Интересно, что стресс в раннем возрасте нарушает гомеостаз глутаматергических синапсов при взрослении. На модели раннего стресса материнской депривации у мышей показаны повышение базального уровня КС, концентрации глутамата и нейронной активности в соматосенсорной коре. В отличие от контрольных животных, дополнительная стрессорная нагрузка у депривированных в раннем возрасте крыс, не вызывая нормального дополнительного повышения уровня КС, тем не менее, повышала уровень глутамата, а также вызывала отличные от контрольных изменения экспрессии АМРА- и NMDA-рецепторов [38].
Как было отмечено выше, в гиппокампе высокие уровни ГК, в том числе в результате стресс-реакции, быстро и обратимо усиливают глутаматергическую передачу через негеномные эффекты, зависимые от МР. Затем ГК медленно нормализуют функцию клеток гиппокампа за счет сигнализации через ядерные/цитоплазматические ГР. Метапластические ответы базолатеральной миндалины на КС существенно отличаются от эффектов ГК в гиппокампе. Вначале происходит быстрое МР-зависимое усиление глутаматергической передачи в нейронах. В отличие от гиппокампа это быстрое усиление является длительным, потенциально позволяя расширить окно для кодирования эмоциональных реакций во время стрессорных событий [39]. Важно отметить, что длительное изменение состояния нейронов миндалевидного тела значительно модифицирует ответную реакцию на последующие повышения уровня КС, вызывая быстрое подавление глутаматергической передачи, опосредованное ГР. Таким образом, реакция нейронов базолатеральной миндалины на ГК зависит от предшествующих стрессорных событий. Делеция ГР у мышей специфически в глутаматергических, но не в ГАМКергических нейронах индуцировала гиперактивность ГГНО и снижала проявления поведения, связанного со страхом и тревогой. Это сопровождалось снижением ГР-зависимых электрофизиологических реакций в базолатеральной миндалине [40]. Полученная при помощи вирусной конструкции делеция ГР дополнительно показала, что именно реакция страха, но не тревоги, регулируется ГР в глутаматергических нейронах базолатеральной миндалины. По-видимому, патологическая тревожность является результатом измененной передачи сигналов ГР в глутаматергических цепях нескольких областей переднего мозга, в то время как модуляция поведения, связанного со страхом, в значительной степени может быть опосредована передачей сигналов ГР в глутаматергических нейронах базолатеральной миндалины. Таким образом, при регуляции поведения страха и тревожности вклад ГР значителен в ключевую возбуждающую, но не тормозную нейромедиаторную систему мозга, и этот факт принципиально важен для нашего понимания молекулярных механизмов, лежащих в основе тревожных расстройств.
Глюкокортикоиды, секретируемые в ответ на стрессорную активацию ГГНО, попадают и в гипоталамус, где обеспечивают быстрое подавление нейроэндокринной активации, включая секрецию окситоцина и вазопрессина. Хронический стресс стимулирует экспрессирующие КРГ нейроны в гипоталамусе и приводит к гиперактивности ГГНО. Хронический непредсказуемый стресс у крыс повышал экспрессию субъединицы GluN1 NМDA-рецепторов в паравентрикулярном ядре гипоталамуса и усиливал синаптическую активность NMDA-рецепторов, способствуя гиперактивности экспрессирующих КРГ нейронов паравентрикулярного ядра и активации ГГНО [41].
Как уже упоминалось, стресс вызывает различные, иногда противоположные изменения в глутаматергической системе в разных областях мозга, в том числе в структурах лимбической системы, а именно в префронтальной коре, гиппокампе и миндалевидном теле. АМРА-рецепторы – ионотропные глутаматные рецепторы, опосредующие быструю синаптическую передачу, фосфорилирование специфических сериновых остатков на субъединицах которых (GluA1 и GluA2) считаются критическими посттрансляционными модификациями, регулирующими активность и субклеточный трафик AMPA-рецепторов. Caudal и соавт. [42] исследовали динамику фосфорилирования трех остатков серина субъединиц АМРА-рецепторов (Ser831-GluA1, Ser845-GluA1 и Ser880-GluA2) в четырех областях мозга крысы (миндалевидное тело, медиальная префронтальная кора, дорсальный гиппокамп и вентральный гиппокамп) в течение часа после поведенческого стресса. Оба остатка серина субъединицы GluA1 демонстрировали повышенное фосфорилирование после стресса, однако введение антагонистов кортикостероидных рецепторов снижало этот эффект только для Ser831-GluA1. Напротив, Ser880-GluA2 проявлял зависимую от времени тенденцию к раннему снижению фосфорилирования (которое избирательно усиливалось обработкой антагонистом ГР мифепристоном в миндалевидном теле и медиальной префронтальной коре стрессированных животных) с последующим повышением фосфорилирования. В другой работе на модели острого неизбегаемого стресса у крыс обнаружили противоположные эффекты на фосфорилирование рецепторов AMPA в медиальной префронтальной коре и дорсальном гиппокампе по сравнению с миндалиной и вентральным гиппокампом. После стресса фосфорилирование Ser831-GluA1 было заметно снижено в первых двух областях, тогда как фосфорилирование Ser845-GluA1 было увеличено в миндалевидном теле и вентральном гиппокампе [43]. Стресс вызывал снижение фосфорилирования остатков Tyr876-GluA2 и Ser880-GluA2 в миндалевидном теле и увеличение фосфорилирования Ser880-GluA2 в медиальной фронтальной коре. Эти результаты показывают, что острый стресс вызывает специфические в отношении субъединиц и регион-специфичные изменения глутаматергической трансмиссии, которые, вероятно, приводят к снижению синаптической эффективности в медиальной фронтальной коре и дорсальном гиппокампе и к увеличению активности в миндалевидном теле и вентральном гиппокампе. Модификации фосфорилирования глутаматных рецепторов могут опосредовать патологическое действие стресса на когнитивные функции, а также противоположные эффекты, которые стресс оказывает на синаптическую пластичность в этих областях. Ранний стресс, вызванный выращиванием потомства мышей с ограниченным гнездовым и подстилочным материалом, вызывал нарушения решения гиппокамп-зависимых задач, сопровождающийся снижением соотношения опосредованных NMDA- и AMPA-рецепторами возбуждающих постсинаптических токов и вероятности высвобождения глутамата в нейронах CA1 гиппокампа, но не в базолатеральной миндалине [44].
ЗАКЛЮЧЕНИЕ: ОПОСРЕДОВАННЫЕ РЕЦЕПТОРАМИ ГЛЮКОКОРТИКОИДОВ ПУТИ РЕГУЛЯЦИИ ГЛУТАМАТНОГО СИНАПСА
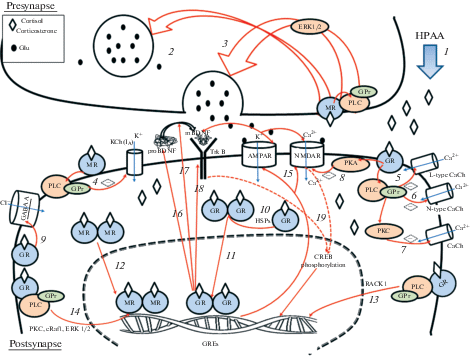
Обзор имеющихся данных показывает, что влияние ГК на глутаматергический синапс зависит от типа и продолжительности стресса (а следовательно, уровня и динамики ГК), оно в определенной степени регион-специфично и зависит от возраста. Высокая пластичность глутаматергических синапсов определятся большим числом точек приложения прямого или опосредованного влияния ГР и в связи с этим множественностью механизмов, вовлеченных в реакцию на ГК в каждой конкретной ситуации. Наличие как мембранных, так и цитоплазматических форм рецепторов ГК (МР и ГР) в глутаматергическом синапсе существенно увеличивает число возможных путей регуляции синаптической пластичности. Такая множественность, несомненно, лежащая в основе экстраординарных пластических свойств глутаматергического синапса, тем не менее, создает существенные трудности для исследования и формирования полноценного представления об этих процессах. На рис. 1 схематически представлены доказанные экспериментальными исследованиями множественные пути регуляции глутаматергического синапса, опосредованные рецепторами ГК, с подробным объяснением и перечислением этих путей. Одной из ключевых систем, вовлеченных в регуляцию синаптической пластичности и тесно связанной различными механизмами с ГГНО, ГК и их рецепторами, является нейротрофиновая система, в первую очередь, связанная с мозговым нейротрофическим фактором (BDNF) [1]. Нейропластичность требует тонко настроенного взаимодействия сосуществующих в ЦНС систем BDNF и ГК на всех уровнях, включая синаптический. Недавно мы проанализировали множественные взаимодействия между системами BDNF и глутамата [48], показав, как ГК изменяют экспрессию и сигнальную трансдукцию BDNF. На приведенной схеме (рис. 1) дополнительно представлены основные пути ГК-зависимой координации функционирования BDNF/TrkB в глутаматных синапсах.
Рис. 1.
Механизмы синаптических эффектов глюкокортикоидов, опосредованные минералокортикоидными и глюкокортикоидными рецепторами, в глутаматергическом синапсе [1, 3, 9, 45–49]. Взаимодействие ГК (Cortisol, Corticosterone) с минералокортикоидными рецепторами (MR) и глюкокортикоидными рецепторами (GR) лежит в основе координации и взаимодействия гипоталамо-гипофизарно-надпочечниковой оси (HPAA) со всеми основными системами, важными для синаптической пластичности. ГК связываются с разным сродством с MR и GR, которые существуют в мембраносвязанной и цитоплазматической/ядерной формах и опосредуют соответственно быстрые и отложенные эффекты ГК. Негеномные механизмы опосредованы мембранными рецепторами ГК. Выброс ГК при активации ГГНО в результате действия стрессорного фактора или циркадианного повышения уровня ГК (1) за счет связывания с высокоаффинными MR в пресинапсе регулирует возбудимость и высвобождение везикул (2), а частота высвобождения глутамата (Glu) регулируется опосредованным G-белком (GPr) сигнальным путем ERK1/2 (внеклеточные киназы, регулируемые сигналом, 1/2; extracellular signal-regulated kinase 1/2) (3). В постсинапсе мембранные MR ингибируют активацию калиевых каналов KCh (IA) (4). Мембранные постсинаптические GR, связанные с GPr, ингибируют кальциевые каналы (CaCh) L-типа (5) и N-типа (6), а также ингибируют протеинкиназу С (РКС), снижая приток кальция в клетку (7). Мембранные постсинаптические GR непрямым образом, через протеинкиназу А (РКА), ингибируют ионотропные N-метил-D-аспартатные рецепторы (NMDAR) (8), а также повышают гиперполяризацию, регулируя ионотропные рецепторы гамма-аминомасляной кислоты (GABA A) (9). Геномные механизмы действия ГК реализуются главным образом в постсинаптическом компартменте и опосредованы, как правило, цитоплазматическими GR и реже MR. ГК, диффундируя через клеточную мембрану, связываются в цитоплазме с MR и GR, которые димеризуются при участии белков теплового шока (HSPs) (10). После диссоциации HSPs гомодимер рецептора транслоцируется в ядро и связывается со специфическими участками ДНК glucocorticoid response elements (GREs) (11, 12). Затем с областью GREs, с которой связан гомодимер рецептора, могут ассоциироваться кофакторы и гистон-модифицирующие элементы (histone-modifying elements), инициируя транскрипцию и синтез белка. В случае связывания с таким участком факторов IRF-3, NF-κB и AP1 транскрипция и экспрессия генов ингибируются. Исключениями, когда геномный эффект ГК осуществляется через мембранный рецептор, являются пути, в которых мембранный GR в комплексе с GPr и RACK1 транслоцируется в ядро и вызывает трансрепрессию (13), а также запускаемая мембранным GR ERK1/2-зависимая транскрипция генов, требующая PKC и cRaf1 (14). Индуцированные ГК геномные эффекты включают модуляцию экспрессии субъединиц NMDAR и ионотропных рецепторов α-амино-3-гидрокси-5-метил-4-изоксазолпропионовой кислоты (AMPAR) (15), а также метаболизма и транспорта глюкозы за счет изменения активности ее мембранных транспортеров (на схеме не указано). Важно, что ГК регулируют также синтез, процессинг, трафик, секрецию и сигналинг мозгового нейротрофического фактора (BDNF), одного из основных регуляторов состояния глутаматергического синапса [45]. Глюкокортикоидная регуляция синтеза и функционирования BDNF происходит на разных уровнях, от регуляции транскрипции определенных экзон-специфичных транскриптов Bdnf до сигнальной трансдукции в постсинаптическом нейроне. Транскрипция Bdnf модулируется GR либо через описанный выше путь связывания с GREs на промотерных областях (16), либо за счет подавления действия других факторов, участвующих в транскрипции Bdnf, например, комплекса AP1 и фактора CREB (cAMP response element binding protein). ГК могут влиять на трансляцию гена Bdnf, модулируя активность трансляционного аппарата. BDNF синтезируется в виде проформы (proBDNF), которая в результате протеолиза превращается в зрелый белок BDNF (mBDNF), и это важное превращение опосредовано многими внутри- и внеклеточными протеазами, модуляция которых глюкокортикоидами в конце концов регулирует уровни доступного mBDNF (17). Геномные эффекты ГК связаны также с регуляцией экспрессии рецептора BDNF тропомиозин киназы В (TrkB) (18). Взаимодействие BDNF с TrkB запускает несколько сигнальных путей, стимулирующих выживание, рост и дифференцировку нейронов, с участием митоген-активируемой протеинкиназы (MAPK), фосфолипазы (PLC) и фосфатидилинозитол-3-киназы (PI3К), результатом которых является функциональная модуляция молекул-мишеней, участвующих в синаптической пластичности, выживании нейрона и клеточной возбудимости. Например, активация пути PLC приводит к изменению транскрипции генов за счет опосредованного Ca2+/кальмодулин-зависимой киназой II (CAMKII) фосфорилирования CREB (19).
Гиппокамп, ассоциированный как с когнитивной функцией, так и с эмоциями, является ярким примером высокопластичного региона мозга. Процессы пластичности на молекулярном, синаптическом, клеточном и сетевом уровнях образуют сложную пирамиду взаимосвязанных как вертикально, так и горизонтально механизмов [1]. Связи и координацию этих уровней осуществляют ГК через сигнализацию, опосредованную МР и ГР, при этом ГК одновременно обеспечивают интегративный нейрогуморальный контроль нейропластичности со стороны ГГНО. Высокая адаптивная пластичность отделов мозга, ответственных за его интегративную функцию, в том числе обучение и память (в частности, гиппокампа), в значительной степени обеспечивается динамичностью и гибкостью регуляции глутаматергического синапса. Однако ценой высокой пластичности является селективная чувствительность таких структур мозга к развитию патологических процессов [50], и это также в полной мере относится к глутаматергическому синапсу, дисфункция которого приводит к гиперглутаматергической трансдукции сигнала и развитию патологических изменений мозга [10].
Список литературы
Gulyaeva N.V. (2019) Biochemical Mechanisms and Translational Relevance of Hippocampal Vulnerability to Distant Focal Brain Injury: The Price of Stress Response. Biochemistry (Mosc). 84(11): 1306–1328. https://doi.org/10.1134/S0006297919110087
de Kloet E.R., Meijer O.C., de Nicola A.F., de Rijk R.H., Joëls M. (2018) Importance of the brain corticosteroid receptor balance in metaplasticity,cognitive performance and neuro-inflammation. Front. Neuroendocrinol. 49: 124–145. https://doi.org/10.1016/j.yfrne.2018.02.003
Prager E.M., Johnson L.R. (2009) Stress at the synapse: signal transduction mechanisms of adrenal steroids at neuronal membranes. Sci. Signal. 2(86): re5. https://doi.org/10.1126/scisignal.286re5
Gulyaeva N.V. (2017) Molecular Mechanisms of Neuroplasticity: An Expanding Universe. Biochemistry (Mosc). 82: 237–242. https://doi.org/10.1134/S0006297917030014
Xiong H., Krugers H.J. (2015) Tuning hippocampal synapses by stress-hormones: Relevance for emotional memory formation. Brain Res. 1621: 114–120. https://doi.org/10.1016/j.brainres.2015.04.010
Gulyaeva N. (2019) Functional Neurochemistry of the Ventral and Dorsal Hippocampus: Stress, Depression, Dementia and Remote Hippocampal Damage. Neurochem. Res. 44: 1306–1322. https://doi.org/10.1007/s11064-018-2662-0
Podgorny O.V., Gulyaeva N.V. (2020) Glucocorticoid-mediated mechanisms of hippocampal damage: Contribution of subgranular neurogenesis. J. Neurochem. https://doi.org/10.1111/jnc.15265
Kessels H.W., Malinow R. (2009) Synaptic AMPA receptor plasticity and behaviour. Neuron. 61: 340–350. https://doi.org/10.1016/j.neuron.2009.01.015
Timmermans W., Xiong H., Hoogenraad C.C., Krugers H.J. (2013) Stress and excitatory synapses: from health to disease. Neuroscience. 248: 626–636. https://doi.org/10.1016/j.neuroscience.2013.05.043
Gulyaeva N.V. (2020) Hippocampal hyperglutamatergic signaling matters: Early targeting glutamate neurotransmission as a preventive strategy in Alzheimer’s disease: An. Editorial. Highlight for “Riluzole attenuates glutamatergic tone and cognitive decline in AβPP/PS1 mice”. J. Neurochem. https://doi.org/10.1111/jnc.15238
Halpain S., McEwen B.S. (1988) Corticosterone decreases 3H-glutamate binding in rat hippocampal formation. Neuroendocrinology. 48: 235–241. https://doi.org/10.1159/000125017
Armanini M.P., Hutchins C., Stein B.A., Sapolsky R.M. (1990) Glucocorticoid endangerment of hippocampal neurons is NMDA-receptor dependent. Brain Res. 532: 7–12. https://doi.org/10.1016/0006-8993(90)91734-x
Virgin C.E. Jr, Ha T.P., Packan D.R., Tombaugh G.C., Yang S.H., Horner H.C., Sapolsky R.M. (1991) Glucocorticoids inhibit glucose transport and glutamate uptake in hippocampal astrocytes: implications for glucocorticoid neurotoxicity. J. Neurochem. 57: 1422–1428. https://doi.org/10.1111/j.1471-4159.1991.tb08309.x
Sandi C. (1998) The role and mechanisms of action of glucocorticoid involvement in memory storage. Neural. Plast. 6: 41–52. https://doi.org/10.1155/NP.1998.41
Karst H., Joëls M. (2003) Effect of chronic stress on synaptic currents in rat hippocampal dentate gyrus neurons. J. Neurophysiol. 89: 625–633. https://doi.org/10.1152/jn.00691.2002
Kvarta M.D., Bradbrook K.E., Dantrassy H.M., Bailey A.M., Thompson S.M. (2015) Corticosterone mediates the synaptic and behavioral effects of chronic stress at rat hippocampal temporoammonic synapses. J. Neurophysiol. 114: 1713–1724. https://doi.org/10.1152/jn.00359.2015
Martin S., Henley J., Holman D., Zhou M., Wiegert O., van Spronsen M., Joëls M., Hoogenraad C.C., Krugers H.J. (2009) Corticosterone alters AMPAR mobility and facilitates bidirectional synaptic plasticity. PLoS One. 4(3): e4714. https://doi.org/10.1371/journal.pone.0004714
Groc L., Choquet D., Chaouloff F. (2008) The stress hormone corticosterone conditions AMPAR surface trafficking and synaptic potentiation. Nat. Neurosci. 11: 868–870. https://doi.org/10.1038/nn.2150
Conboy L., Sandi C. (2010) Stress at learning facilitates memory formation by regulating AMPA receptor trafficking through a glucocorticoid action. Neuropsychopharmacology. 35(3): 674–685.https://doi.org/10.1038/npp.2009.172
Choi G.E., Oh J.Y., Lee H.J., Chae C.W., Kim J.S., Jung Y.H., Han H.J. (2018) Glucocorticoid-mediated ER-mitochondria contacts reduce AMPA receptor and mitochondria trafficking into cell terminus via microtubule destabilization. Cell Death Dis. 9: 1137. https://doi.org/10.1038/s41419-018-1172-y
Tse Y.C., Bagot R.C., Wong T.P. (2012) Dynamic regulation of NMDAR function in the adult brain by the stress hormone corticosterone. Front. Cell Neurosci. 6: 9.https://doi.org/10.3389/fncel.2012.00009
Mikasova L., Xiong H., Kerkhofs A., Bouchet D., Krugers H.J., Groc L. (2017) Stress hormone rapidly tunes synaptic NMDA receptor through membrane dynamics and mineralocorticoid signalling. Sci. Rep. 7: 8053. https://doi.org/10.1038/s41598-017-08695-3
Tse Y.C., Bagot R.C., Hutter J.A., Wong A.S., Wong T.P. (2011) Modulation of synaptic plasticity by stress hormone associates with plastic alteration of synaptic NMDA receptor in the adult hippocampus. PLoS One. 6: e27215. https://doi.org/10.1371/journal.pone.0027215
Krugers H.J., Alfarez D.N., Karst H., Parashkouhi K. van Gemert N., Joëls M. (2005) Corticosterone shifts different forms of synaptic potentiation in opposite directions. Hippocampus. 15: 697–703. https://doi.org/10.1002/hipo.20092
Zhang Y., Sheng H., Qi J., Ma B., Sun J., Li S., Ni X. (2012) Glucocorticoid acts on a putative G protein-coupled receptor to rapidly regulate the activity of NMDA receptors in hippocampal neurons. Am. J. Physiol. Endocrinol. Metab. 302: E747–E58. https://doi.org/10.1152/ajpendo.00302.2011
Komatsuzaki Y., Hatanaka Y., Murakami G., Mukai H., Hojo Y., Saito M., Kimoto T., Kawato S. (2012) Corticosterone induces rapid spinogenesis via synaptic glucocorticoid receptors and kinase networks in hippocampus. PLoS One. 7: e34124. https://doi.org/10.1371/journal.pone.0034124
Chaouloff F., Hémar A., Manzoni O. (2008) Local facilitation of hippocampal metabotropic glutamate receptor-dependent long-term depression by corticosteroneand dexamethasone. Psychoneuroendocrinology. 33: 686–691. https://doi.org/10.1016/j.psyneuen.2007.12.013
Hunter R.G., Bellani R., Bloss E., Costa A., McCarthy K., McEwen B.S. (2009) Regulation of kainate receptor subunit mRNA by stress and corticosteroids in the rat hippocampus. PLoS One. 4: e4328. https://doi.org/10.1371/journal.pone.0004328
McCauley J.P., Petroccione M.A., D’Brant L.Y., Todd G.C., Affinnih N., Wisnoski J.J., Zahid S., Shree S., Sousa A.A., De Guzman R.M., Migliore R., Brazhe A., Leapman R.D., Khmaladze A., Semyanov A., Zuloaga D.G., Migliore M., Scimemi A. (2020) Circadian Modulation of Neurons and Astrocytes Controls Synaptic Plasticity in Hippocampal Area CA1. Cell Rep. 33: 108255. https://doi.org/10.1016/j.celrep.2020.108255
Sarabdjitsingh R.A., Jezequel J., Pasricha N., Mikasova L., Kerkhofs A., Karst H., Groc L., Joëls M. (2014) Ultradian corticosterone pulses balance glutamatergictransmission and synaptic plasticity. Proc. Natl. Acad. Sci. U S A. 111: 14265–14270. https://doi.org/10.1073/pnas.1411216111
Sarabdjitsingh R.A., Pasricha N., Smeets J.A., Kerkhofs A., Mikasova L., Karst H., Groc L., Joëls M. (2016) Hippocampal Fast Glutamatergic Transmission Is Transiently Regulated by Corticosterone Pulsatility. PLoS One. 11: e0145858. https://doi.org/10.1371/journal.pone.0145858
Yuen E.Y., Wei J., Yan Z. (2017) Molecular and Epigenetic Mechanisms for the Complex Effects of Stress on Synaptic Physiology and Cognitive Functions. Int. J. Neuropsychopharmacol. 20(11): 948–955. https://doi.org/10.1093/ijnp/pyx052
Musazzi L., Treccani G., Popoli M. (2015) Functional and structural remodeling of glutamate synapses in prefrontal and frontal cortex induced by behavioral stress. Front. Psychiatry. 6: 60. https://doi.org/10.3389/fpsyt.2015.00060
Bonini D., Mora C., Tornese P., Sala N., Filippini A., La Via L., Milanese M., Calza S., Bonanno G., Racagni G., Gennarelli M., Popoli M., Musazzi L., Barbon A. (2016) Acute Footshock Stress Induces Time-Dependent Modifications of AMPA/NMDA Protein Expression and AMPA Phosphorylation. Neural. Plast. 2016: 7267865. https://doi.org/10.1155/2016/7267865
Nava N., Treccani G., Liebenberg N., Chen F., Popoli M., Wegener G., Nyengaard J.R. (2014) Chronic desipramine prevents acute stress-induced reorganization of medial prefrontal cortex architecture by blocking glutamate vesicle accumulation and excitatory synapse increase. Int. J. Neuropsychopharmacol. 18: pyu085. https://doi.org/10.1093/ijnp/pyu085
Musazzi L., Tornese P., Sala N., Popoli M. (2017) Acute stress is not acute: sustained enhancement of glutamate release after acute stress involves readily releasable pool size and synapsin I activation. Mol. Psychiatry. 22: 1226–1227. https://doi.org/10.1038/mp.2016.175
Toya S., Takatsuru Y., Kokubo M., Amano I., Shimokawa N., Koibuchi N. (2014) Early-life-stress affects the homeostasis of glutamatergic synapses. Eur. J. Neurosci. 40: 3627–3634. https://doi.org/10.1111/ejn.12728
Karst H., Berger S., Erdmann G., Schütz G., Joëls M. (2010) Metaplasticity of amygdalar responses to the stress hormone corticosterone. Proc. Natl. Acad. Sci. U S A. 107: 14449–14454. https://doi.org/10.1073/pnas.0914381107
Hartmann J., Dedic N., Pöhlmann M.L., Häusl A., Karst H., Engelhardt C., Westerholz S., Wagner K.V., Labermaier C., Hoeijmakers L., Kertokarijo M., Chen, Joëls M., Deussing J.M., Schmidt M.V. (2017) Forebrain glutamatergic, but not GABAergic, neurons mediate anxiogenic effects of the glucocorticoid receptor. Mol. Psychiatry. 22: 466–475. https://doi.org/10.1038/mp.2016.87
Zhou J.J., Gao Y., Zhang X., Kosten T.A., Li D.P. (2018) Enhanced Hypothalamic NMDA Receptor Activity Contributes to Hyperactivity of HPA Axis in Chronic Stress in Male Rats. Endocrinology. 159: 1537–1546. https://doi.org/10.1210/en.2017-03176
Caudal D., Rame M., Jay T.M., Godsil B.P. (2016) Dynamic Regulation of AMPAR Phosphorylation In Vivo Following Acute Behavioral Stress. Cell. Mol. Neurobiol. 36: 1331–1342. https://doi.org/10.1007/s10571-016-0332-9
Caudal D., Godsil B.P., Mailliet F., Bergerot D., Jay T.M. (2010) Acute stress induces contrasting changes in AMPA receptor subunit phosphorylation within the prefrontal cortex, amygdala and hippocampus. PLoS One. 5: e15282. https://doi.org/10.1371/journal.pone.0015282
Pillai A.G., Arp M., Velzing E., Lesuis S.L., Schmidt M.V., Holsboer F., Joëls M., Krugers H.J. (2018) Early life stress determines the effects of glucocorticoids and stress on hippocampal function: Electrophysiological and behavioral evidence respectively. Neuropharmacology. 133: 307–318. https://doi.org/10.1016/j.neuropharm.2018.02.001
Gulyaeva N.V. (2017) Interplay between Brain BDNF and Glutamatergic Systems: A Brief State of the Evidence and Association with the Pathogenesis of Depression. Biochemistry (Mosc.). 82: 301–307. https://doi.org/10.1016/j.ejphar.2013.07.015
Joëls M., Pasricha N., Karst H. (2013) The interplay between rapid and slow corticosteroid actions in brain. Eur. J. Pharmacol. 719: 44–52.
Joëls M., de Kloet E.R. (2017) 30 Years of the mineralocorticoid receptor: The brain mineralocorticoid receptor: a saga in three episodes. J. Endocrinol. 234: T49–T66. https://doi.org/10.1530/JOE-16-0660
Le Menuet D., Lombès M. (2014).The neuronal mineralocorticoid receptor: from cell survival to neurogenesis. Steroids. 91: 11–19. https://doi.org/10.3389/fendo.2016.00066
Suri D., Vaidya V.A. (2013).Glucocorticoid regulation of brain-derived neurotrophic factor: relevance to hippocampal structural and functional plasticity. Neuroscience. 239: 196–213.
Гуляева Н.В. (2020) Физиологический континуум пластичности и патологии нервной системы. Интегративная физиология. 1: 294–302. [Gulyaeva N.V. (2020) Physiological continuum of plasticity and pathology of the nervous system. Integrative. Physiol. 1: 294–302. (In Russ)]. https://doi.org/10.33910/2687-1270-2020-1-4-294-302
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова