

Российский физиологический журнал им. И.М. Сеченова, 2021, T. 107, № 4-5, стр. 492-517
Синаптические дисфункции при эпилепсии
А. В. Зайцев 1, *, Д. В. Амахин 1, А. В. Дёмина 1, М. В. Захарова 1, Ю. Л. Ергина 1, Т. Ю. Постникова 1, Г. П. Диеспиров 1, Л. Г. Магазаник 1
1 Институт эволюционной физиологии и биохимии им. И.М. Сеченова РАН
Санкт-Петербург, Россия
* E-mail: aleksey_zaitsev@mail.ru
Поступила в редакцию 20.01.2021
После доработки 08.02.2021
Принята к публикации 09.02.2021
Аннотация
Эпилепсия является одним из распространенных заболеваний мозга, и несмотря на интенсивные многолетние исследования этой патологии современная медицина не может эффективно купировать судорожные проявления почти у трети больных. При эпилепсии происходит реорганизация нейронных сетей, которая обусловлена гибелью части нейронов и формированием новых нейронных связей с измененными свойствами. В этом обзоре мы сфокусировались на анализе изменений свойств ключевого элемента нейронных сетей – химического синапса – сразу после эпилептической активности, во время эпилептогенеза, а также при хронической эпилепсии. Так как синапс включает в себя не только нейрональные пре- и постсинаптическую части, но и глиальные компоненты, то в наше рассмотрение включены изменения свойств астроцитов и микроглии. Эпилептическая активность вызывает многочисленные модификации в работе синапса: меняется вероятность выброса медиатора, трансформируется субъединичный состав и соотношение постсинаптических рецепторов, нарушается синаптическая пластичность, меняется морфология и активность астроцитов и микроглии. Глиальные клетки выделяют ряд глиатрансмиттеров и цитокинов, которые, в свою очередь, модифицируют синаптическую передачу. В некоторых случаях комплекс этих изменений благоприятен и позволяет практически полностью скомпенсировать последствия эпилептической активности для нервной системы. Однако нередко эти изменения, наоборот, запускают цепь процессов, ведущих к эпилептизации и долговременным нарушениям в функционировании нейронных сетей. За последние 10 лет достигнут существенный прогресс в расшифровке этих изменений и их механизмов, который и отражен в нашем обзоре. Однако до сих пор у исследователей не сложилось четкое понимание, какие именно модификации в функционировании синапсов обеспечивают наилучшую компенсацию и способны предотвратить эпилептогенез. Эти знания могли бы стать основой для разработки действенных методов профилактики эпилептогенеза и лечения эпилепсии.
Эпилепсия является одним из наиболее распространенных неврологических расстройств, существенно влияющих на качество жизни пациентов [1]. Эпилепсия не единое заболевание, а целая группа расстройств. Эпилепсия может быть обусловлена как генетической предрасположенностью (идиопатическая эпилепсия), так и вызвана различными травмами мозга, инсультом, гипоксией, инфекциями, опухолями (приобретенная, вторичная, или симптоматическая эпилепсия). Однако общим для всех форм эпилепсии является повторное появление самопроизвольной судорожной активности в связи с гиперактивностью нейронов головного мозга.
На сегодняшний день до 30% случаев эпилепсии остаются фармакорезистентными [2], поэтому важнейшей проблемой как фундаментальной нейробиологии, так и клинической нейрофизиологии является выяснение механизмов эпилептизации мозга и поиск эффективных методов предотвращения и лечения эпилепсии [3]. Современная концепция эпилептогенеза предполагает, что в результате действия неблагоприятных факторов происходит утрата или нарушение механизмов, препятствующих судорожной активности, появляется дисбаланс между возбуждением и торможением в нейронных сетях, где глутаматергические и ГАМКергические нейроны соответственно играют важную роль.
Однажды проявившись, судорожные состояния, как правило, ведут к дальнейшим патологическим перестройкам на молекулярном, клеточном и сетевых уровнях, что сопровождается морфологическими и функциональными нарушениями в работе центральной нервной системы, усиливающими судорожную готовность и нарушающими работу мозга. Таким образом, сам эпилептогенез условно подразделяется на 3 основных фазы: 1) действие провоцирующего фактора, запускающего процесс; 2) латентный период, в ходе которого самопроизвольных судорог нет, но идут перестройки, приводящие к утрате антисудорожных механизмов и превращению нормального мозга в эпилептический; 3) хроническая фаза эпилепсии [4].
Во время эпилептогенеза наблюдается нейродегенерация и нейрогенез, повреждение аксонов и их разрастание, активация глиальных клеток, инвазия периферических иммунных клеток, повреждение сосудов и ангиогенез, трансформации во внеклеточном матриксе, а также изменения в молекулярной структуре различных ионных каналов [3]. Важным фактором, усиливающим эпилептизацию мозга, является формирование многочисленных новых возбуждающих синапсов в тех местах, где они отсутствуют в норме [5]. Появление новых синапсов и изменение свойств существующих синапсов ведет к патологическим формам синаптической пластичности, усиливающей процессы синхронизации в нейронных цепях. Это, в свою очередь, ведет к более легкому возникновению новых эпилептических приступов [6].
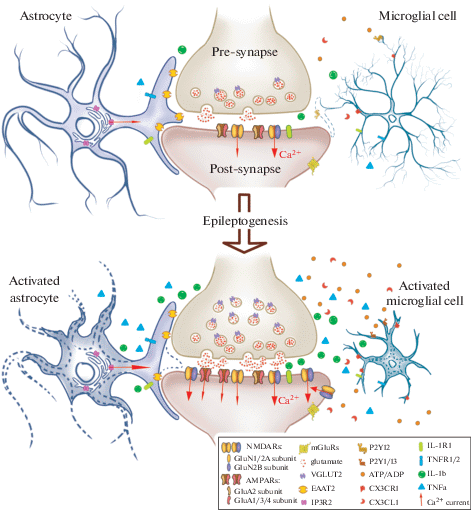
В настоящем обзоре мы не будем обсуждать вклад и значение всех процессов, наблюдаемых при эпилептогенезе, а рассмотрим прежде всего современные данные об изменениях свойств ключевого элемента нейронных сетей – химического синапса – сразу после эпилептической активности, во время эпилептогенеза, а также при хронической эпилепсии. Так как синапс включает в себя не только нейрональные пресинаптическую и постсинаптическую части, но также глиальные компоненты, то в наше рассмотрение также включены изменения свойств астроцитов и микроглии (рис. 1 ).
Рис. 1.
Патологические изменения в синапсе при эпилепсии. На рисунке представлены основные структурные и молекулярные изменения в разных частях глутаматергического синапса, наблюдаемые при эпилепсии (эти изменения могут не совпадать друг с другом по времени возникновения при развитии патологии). При эпилепсии увеличивается площадь пресинаптического бутона и количество глутамат-содержащих везикул. На постсинаптической поверхности наблюдается увеличение NMDA-рецепторов, содержащих GluN2B-субъединицу, а также появляются AMPA-рецепторы, лишенные GluA2-субъединицы, что в целом увеличивает ток ионов Ca2+ внутрь нейрона. Обратите вниманием, что красные стрелки иллюстрируют только ток ионов Ca2+, без учета других ионных токов. Увеличивается внеклеточная концентрация хемокина CX3CL1 и ADP/ATP, что приводит к изменению морфологии микроглиальных клеток и повышению продукции провоспалительных цитокинов, что в свою очередь может модулировать работу NMDA-рецепторов через колокализованные на постсинаптической мембране IL-1R1-рецепторы. Астроциты втягивают свои отростки, из-за чего площадь покрытия синапса астроцитарными лепестками уменьшается. Это ослабляет обратный захват глутамата из синаптической щели. Усиливается высвобождение Ca2+ из внутриклеточных депо астроцита, что может приводить к усилению выброса глиотрансмиттеров. Подробное объяснение всех процессов дано в соответствующих разделах обзора.
1. ПРЕСИНАПТИЧЕСКИЕ ИЗМЕНЕНИЯ
Изучению пресинаптических изменений в ходе эпилептогенеза и при эпилепсии уделяется относительно мало внимания. Значительная доля из проведенных исследований посвящена оценке частоты миниатюрных возбуждающих постсинаптических токов (мВПСТ). Показано увеличение частоты мВПСТ в ходе эпилептогенеза; в частности, увеличение выявлено у нейронов области СА3 в модели постишемической спонтанной эпилептиформной активности [7], проекционных нейронов латеральной амигдалы спустя 14–16 нед. после эпилептического статуса, вызванного пилокарпином [8], нейронов области СА1 в латентный период, спустя 7 дней после эпилептического статуса, вызванного введением каината [9]. Однако во многих случаях остается неясным, связано ли наблюдаемое увеличение частоты мВПСТ именно с увеличением вероятности выброса глутамата или же обусловлено подключением молчащих синапсов. Кроме того, высвобождение глутамата контролируется рядом пресинаптических рецепторов: аденозиновыми, мускариновыми и ГАМКб-рецепторами, действующих по принципу гетерорецепторов и выступающими в роли ауторецепторов ионотропными (NMDA-рецепторы, каинатные рецепторы) и метаботропными рецепторами глутамата [10]. Graebenitz и соавт. продемонстрировали, что различия в частоте мВПСТ у контрольных и пилокарпиновых мышей нивелируются в присутствии антагониста NMDA-рецепторов AP5, что может быть связано с регулирующим влиянием пресинаптических NMDA-ауторецепторов на процесс высвобождения глутамата [8]. Согласно исследованию Thompson и соавт., агонисты ГАМКб-рецепторов, CGP44533 и CGP35024, значительно снижали частоту мВПСТ в энторинальной коре. Ни один из агонистов не оказывал никакого влияния на амплитуду или кинетику миниатюрных ВПСТ. Однако эффект CGP44533 на частоту миниатюрных ВПСТ оказался снижен в срезах пилокарпиновых крыс, что может указывать на снижение функциональной активности пресинаптических ГАМКб-рецепторов в пилокарпиновой модели эпилепсии [11].
Интересно, что в модели судорожных состояний, вызванных применением конвульсанта пентилентетразола, наоборот, выявлено уменьшение вероятности выброса глутамата. Спустя сутки после введения пентилентетразола наблюдается фасилитация полевых ответов в области СА1 гиппокампа при парной стимуляции коллатералей Шаффера, что свидетельствует об уменьшении вероятности выброса глутамата в этих синапсах [12, 13]. Пентилентетразол, в отличие от пилокарпина и каината, не приводит к развитию приобретенной эпилепсии. По-видимому, снижение вероятности выброса глутамата в синапсах гиппокампа в этой модели может быть важным антиэпилептогенным фактором.
Морфологические исследования позволяют подойти к проблеме с другой стороны, отвечая на вопрос о том, что происходит с количеством везикул, а также числом и размерами пресинаптических терминалей. Так, эффект киндлинга на пресинаптические отростки был оценен при помощи иммуномечения – в качестве маркера пресинаптических везикулярных мембран использовался синаптофизин. Значительное увеличение иммунореактивности было отмечено на 28-й день в радиальном слое области CA1, в люцидном и радиальном слоях CA3, в хилусе и во внутренней трети молекулярного слоя зубчатой извилины, а также в II/III слоях пириформной коры [14]. 3D-реконструкция синаптических контактов нейронов СА3–СА1 гиппокампа показала, что эпилептический статус, индуцированный неоднократным введением низких доз каината, приводит к увеличению числа докированных (готовых к высвобождению) везикул в пресинаптических окончаниях уже через 7 дней после его индукции [9]. Согласно работе Murthy и соав, размер пула готовых к высвобождению везикул сильно коррелирует с вероятностью высвобождения медиатора, следовательно, полученные морфологические данные позволяют предположить, что вероятность высвобождения глутамата может быть повышена в исследуемых синапсах [15].
С помощью метода двухфотонной микроскопии было подробно исследовано, как индуцированный пилокарпином эпилептический статус влияет на пресинаптические процессы в терминалях мшистых волокон. В хроническую фазу пилокарпиновой модели значимо увеличились средняя площадь бутонов мшистых волокон и число активных зон, приходящихся на один бутон. Проведенные эксперименты со стимул-индуцированным выбросом медиатора свидетельствовали как об ускорении высвобождения везикул из бутонов мшистых волокон, так и о появлении новой субпопуляции бутонов, характеризующейся более высокой скоростью высвобождения глутамата. Согласно данным электронной микроскопии, у животных, перенесших эпилептический статус, пресинаптические окончания содержали бóльшее число готовых к высвобождению везикул, повышенным было также соотношение среднего числа готовых к высвобождению везикул к протяженности активной зоны [16].
Использование in vivo моделей эпилепсии позволяет учитывать не только внутригиппокампальные связи, но и афференты, приходящие из других областей мозга, а значит и роль, которую они играют в процессах эпилептогенеза. В работе 2015 г. при помощи пилокарпиновой модели эпилепсии была исследована динамика связей между супрамаммиллярным ядром гипоталамуса и зубчатой извилиной и подробно изучена их реорганизация, начинающаяся в ходе латентного периода и продолжающаяся также после появления у животных спонтанных судорог [17]. Помимо нарушения паттерна распределения изучаемых афферентов, весь внутренний молекулярный слой зубчатой извилины получал гораздо более высокое, по сравнению с контролем, число окончаний аксонов, приходящих из латеральной и медиальной областей супрамаммиллярного ядра. В частности, спустя 2 месяца после пилокарпин-индуцированного эпилептического статуса, на всем ростро-каудальном протяжении зубчатой извилины во внутренней трети молекулярного слоя присутствовало множество крупных VGLUT-2-иммунопозитивных терминалей (VGLUT2 – везикулярный транспортер глутамата 2 типа). Количественный анализ показал, что средняя плотность VGLUT2- и VGLUT2/VGAT-иммуномеченных бутонов в молекулярном слое зубчатой извилины была выше у пилокарпиновых животных. Применение антероградного трейсера BDA продемонстрировало схожий паттерн реорганизации афферентов, идущих от супрамаммиллярного ядра, с данными, полученными при иммуномечении окончаний аксонов на содержание VGLUT2 [17].
Таким образом, пресинаптические изменения могут быть одной из важных причин повышенной возбудимости нейронов при эпилепсии.
2. ПОСТСИНАПТИЧЕСКИЕ ИЗМЕНЕНИЯ
2.1.1. Нарушения глутаматергической передачи
Индуцированная эпилептическая активность in vivo и эпилептиформная активность in vitro могут вызвать длительное усиление постсинаптических возбуждающих ответов во всех глутаматергических синапсах гиппокампа [18–21]. Вызванная эпилептической активностью потенциация сопровождается увеличением экспрессии GluA1-субъединицы AMPA-рецепторов в дендритных шипиках, что указывает на происходящее встраивание AMPA-рецепторов в постсинаптическую мембрану [18, 20]. Также имеются свидетельства происходящего увеличения числа NMDA-рецепторов в синапсах [22, 23]. В пирамидных нейронах энторинальной коры уже спустя несколько минут после начала эпилептиформной активности наблюдается схожее по своим свойствам усиление глутаматергических синапсов [24].
Предполагается, что происходящее во время эпилептической активности усиление возбуждающей синаптической передачи может быть одним из механизмов перехода от одиночных судорог к эпилептическому статусу [22, 25, 26]. Подробнее изменения долговременной синаптической пластичности (ДВП) рассмотрено в следующем разделе этого обзора.
2.1.2. Нарушения ГАМК-опосредованного торможения при эпилептической активности
Известно, что концентрация хлорид-ионов внутри клетки достаточно лабильна и может изменяться в широких пределах в зависимости от интенсивности активации ГАМКа-рецепторов и эффективности регулирующих ее гомеостатических механизмов [27–29]. Предполагается, что именно аномальная активность ГАМКергических нейронов приводит к уменьшению градиента хлорид-ионов, что ведет к эпилептическому припадку [28, 30–32]. Исследования с применением моделей эпилептической активности позволяют связать переход от одиночных эпилептических судорог к эпилептическому статусу с необратимым изменением полярности ГАМКа-рецептор-опосредованных ответов. В результате избыточной активации ГАМКа-рецепторов гомеостатические механизмы оказываются не в состоянии восстановить функциональное торможение в нейронной сети, в результате чего генерация эпилептических разрядов не может остановиться [33].
О происходящих вследствие эпилептической активности постсинаптических изменениях ГАМКергической синаптической передачи свидетельствует снижение эффективности противосудорожных препаратов. Например, противосудорожный эффект бензодиазепинов, являющихся положительными модуляторами ГАМКа-рецепторов, снижается, если их применению предшествовал значительный период эпилептической активности [33].
Другим патологическим механизмом, который снижает эффективность ГАМК-опосредованного торможения во время эпилептической активности и способствует усугублению эпилептического состояния, является снижение проводимости ГАМКа-рецепторов [33–35]. Это может быть следствием интернализации ГАМКа-рецепторов в ходе эпилептиформной активности [36, 37]: скорость интернализации коррелирует с нейрональной активностью и, по-видимому, регулируется посредством кальций-зависимых механизмов.
2.2. Постсинаптические изменения, ассоциированные с развитием приобретенной эпилепсией
Даже однократный эпилептический припадок зачастую приводит к отложенным во времени постсинаптическим изменениям, которые могут стать причиной развития приобретенной эпилепсии. Примером наиболее часто описываемых нарушений, влияющих на свойства синаптической передачи, является изменение относительного вклада GluN2B-содержащих NMDA-рецепторов и кальций-проницаемых AMPA-рецепторов в постсинаптический ответ.
2.2.1. Изменение уровня экспрессии GluN2B-содержащих NMDA-рецепторов
GluN2B-содержащие NMDA-рецепторы значительно отличаются по своим свойствам от GluN2A-содержащих: они имеют более медленную кинетику, чем обеспечивают более продолжительный постсинаптический ответ, а также сильно отличаются по аминокислотной последовательности цитоплазматических доменов и характеру взаимодействия с внутриклеточными сигнальными молекулами [38]. Например, показано, что активация GluN2A-содержащих рецепторов увеличивает экспрессию нейротрофического фактора мозга (BDNF), тогда как активация GluN2B-содержащих рецепторов в большей мере усиливает ERK1/2-опосредованное фосфорилирование [39]. В пилокарпиновой и литий-пилокарпиновой моделях эпилепсии у крыс было продемонстрировано увеличение относительного вклада GluN2B-содержащих NMDA-рецепторов в постсинаптический ответ на ранних стадиях эпилептогенеза [40–42]; аналогичные результаты были получены и в модели с пентилентетразоловым киндлингом [43]. Также было показано, что последствием судорог может быть усиление фосфорилирования GluN2B-субъединицы NMDA-рецепторов [44]. Данные, касающиеся эффекта фармакологической блокады GluN2B-содержащих NMDA-рецепторов на этапе эпилептогенеза противоречивы: в ряде исследований продемонстрировано, что применение антагониста этих рецепторов (ифенпродила) препятствует эпилептогенезу после провоцирующего воздействия [43–45], но есть свидетельства отсутствия подобного эффекта [39]. Все вышеперечисленное позволяет рассматривать GluN2B-содержащие NMDA-рецепторы как перспективную мишень для фармакологического воздействия в целях предотвращения развития приобретенной эпилепсии.
2.2.2. Экспрессия кальций-проницаемых AMPA-рецепторов
AMPA-рецепторы, не содержащие GluA2-субъединицы (или в некоторых случаях, содержащие неотредактированную версию GluA2-субъединицы) проницаемы для ионов кальция. Кроме того, такие рецепторы обладают большей проводимостью, иными временными характеристиками открытия и закрытия ионного канала, а также иначе взаимодействуют с внутриклеточными сигнальными и регуляторными молекулами [46, 47]. На раннем этапе постнатального развития кальций-проницаемые AMPA-рецепторы экспрессируются в пирамидных нейронах гиппокампа. По мере взросления организма, они замещаются на кальций-непроницаемые AMPA-рецепторы [48].
Помимо относительно кратковременной экспрессии кальций-проницаемых AMPA-рецепторов в глутаматергических нейронах непосредственно во время эпилептической активности, аномальная экспрессия данных рецепторов может также являться отложенным следствием перенесенных судорожных состояний. В большом числе работ на различных моделях эпилепсии показано снижение относительной экспрессии GluA2-субъединицы AMPA-рецепторов на определенных этапах развития заболевания [49–55], хотя есть и противоположные свидетельства о снижении их экспрессии [56, 57].
В одной из наиболее хорошо воспроизводящей процесс эпилептогенеза моделей – пилокарпиновой, с помощью электрофизиологических методов было продемонстрировано кратковременное появление кальций-проницаемых AMPA-рецепторов на пирамидных нейронах префронтальной коры крыс спустя несколько дней после перенесенного эпилептического статуса [54]. Также в ряде моделей эпилепсии было продемонстрировано, что антагонисты кальций-проницаемых AMPA-рецепторов обладают некоторым непосредственным противосудорожным эффектом [58–61]
Эпилептические приступы, перенесенные в раннем возрасте, могут еще больше увеличить уровень экспрессии кальций-проницаемых AMPA-рецепторов [62], результатом чего является развитие эпилепсии и различных когнитивных расстройств. В ряде случаев подобные последствия перенесенных судорог могут быть предотвращены с помощью терапии антагонистами AMPA-рецепторов [62, 63].
В пользу непосредственного вклада кальций-проницаемых AMPA-рецепторов в развитие эпилепсии свидетельствуют эксперименты с искусственным повышением экспрессии неотредактированной формы GluA2-субъединицы AMPA-рецепторов у мышей, что приводит к повышению количества постсинаптических кальций-проницаемых AMPA-рецепторов. В этом случае наблюдалась повышенная склонность к эпилептическим судорогам, низкая выживаемость подопытных животных после эпилептических приступов, гибель пирамидных нейронов CA1 гиппокампа, сниженная плотность дендритных шипиков, нарушения обучения и памяти [61, 64, 65].
Цитоплазматический белок PICK1, активность которого повышается с увеличением внутриклеточной концентрация кальция, способствует интернализации GluA2-содержащих рецепторов из плазматической мембраны, тем самым увеличивая относительный вклад кальций-проницаемых AMPA-рецепторов в синаптический ответ и еще больше увеличивая вход кальция в клетку. Показано, что экспрессия белка PICK1 снижается вследствие перенесенных судорог, что, вероятно, представляет собой адаптивный механизм, препятствующий слишком сильному снижению количества GluA2-содержащих рецепторов в синапсах [66].
2.3. Постсинаптические изменения при хронической эпилепсии
Постсинаптические изменения при хронической эпилепсии исследуются как на образцах ткани головного мозга человека, полученных в ходе нейрохирургических операций или посмертно, так и с применением моделей эпилепсии на лабораторных животных. Наиболее распространенным экспериментальным подходом к изучению постсинаптических изменений при эпилепсии у человека является выявление изменений экспрессии субъединиц постсинаптических рецепторов молекулярно-биологическими и иммуногистохимическими методами [67–72].
Большинство выявленных изменений субъединичного состава синаптических рецепторов при эпилепсии у человека воспроизводятся в животных моделях, что подтверждает их валидность. Например, часто выявляемое увеличение относительной экспрессии GluN2B-субъединицы NMDA-рецепторов у человека при эпилепсии [69, 73] показано в пилокарпиновой модели хронической эпилепсии у крыс [74]. Однако в каинатной модели хронической эпилепсии выявлено небольшое снижение экспрессии GluN2B-субъединицы [75].
В образцах ткани мозга пациентов с эпилепсией показано снижение экспрессии GluA2-субъединицы AMPA-рецепторов, что ведет к появлению кальций-проницаемых AMPA-рецепторов [69, 76]. Аналогичное снижение экспрессии GluA2-субъединицы наблюдалось в периринальной коре крыс в модели хронической эпилепсии [77]. Другой механизм увеличения числа кальций-проницаемых AMPA-рецепторов при эпилепсии выявлен при изучении образцов ткани гипоталамической гамартомы, вызывающей эпилептические припадки [76]. Предположительно, количество кальций-проницаемых AMPA-рецепторов в этих нейронах увеличивается за счет снижения посттрансляционной модификации РНК GluA2-субъединицы, поскольку у них было снижено количество фермента аденозин-дезаминазы (ADAR2), осуществляющей эту модификацию.
Наиболее характерным изменением субъединичного состава ГАМКа-рецепторов, регистрируемого у пациентов с эпилепсией, является увеличение соотношения субъединиц α2/α1 ГАМКа-рецепторов [68, 69], что характерно для более ранних стадий онтогенеза [78]. Подобные изменения описаны и в животных моделях [79]. В сочетании с ослаблением трансмембранного градиента ионов хлора и повышенной экспрессией кальций-проницаемых AMPA-рецепторов, которые также характерны для синаптической передачи в незрелом мозге, это наблюдение позволяет предполагать, что в ходе хронической эпилепсии может наблюдаться своего рода инверсия процесса созревания синаптической передачи [80].
3. ДОЛГОВРЕМЕННАЯ СИНАПТИЧЕСКАЯ ПЛАСТИЧНОСТЬ ПРИ ЭПИЛЕПСИИ
При эпилепсии нарушается одна из основных функций мозга – способность обучаться и модифицировать поведение, реализуемая благодаря синаптической пластичности [81, 82]. Суть феномена синаптической пластичности заключается в том, что в ответ как на физиологические, так и на патологические повторяющиеся раздражители происходит изменение синаптических связей, эти изменения могут длиться от секунд до часов, дней и даже месяцев [81]. Долговременная потенциация (ДВП) и депрессия вместе с другими формами синаптической пластичности обеспечивают нейронный субстрат для процессов обучения и памяти [83]. Наибольший интерес у исследователей эпилепсии вызывает изучение долговременной пластичности в гиппокампе [84, 85]. Это связано с тем, что гиппокамп играет важную роль в процессах памяти и обучения, моторном контроле и стереотипном поведении [86]. Кроме того, гиппокамп является одним из наиболее уязвимых участков головного мозга при возникновении эпилепсии [87].
Сейчас достаточно распространена гипотеза, что потенциация синаптической передачи, происходящая вследствие эпилептической активности, и классическая ДВП имеют общий механизм реализации [18–20, 24], поэтому дальнейшая потенциация синапсов после судорожных состояний ослаблена в результате окклюзии. Однако недавнее экспериментальное сравнение свойств синаптической пластичности, вызванной эпилептоподобной активностью и классическими индукционными протоколами, выявило ряд различий между этими процессами [88]. Кратковременное встраивание кальций-проницаемых AMPA-рецепторов необходимо для консолидации ДВП в синапсах гиппокампа, и блокада этих рецепторов нарушает экспрессию ДВП [89]. Потенциация синапсов зоны CA1 гиппокампа, вызываемая эпилептиформной активностью, наоборот, более выражена и сохраняется дольше в условиях блокады кальций-проницаемых AMPA-рецепторов [88].
Следует подчеркнуть, что изменения синаптической пластичности после эпилептических припадков очень разнообразны. В экспериментальных моделях на животных было показано как ослабление долговременной потенциации [12, 90–95], так и ее усиление [96–98]. Вероятно, различия в результатах определяются особенностями экспериментальных условий и выбора модели. Нами было показано ослабление ДВП в CA3–CA1 синапсах гиппокампа у крыс через 1, 3 и 7 дней после пентилентетразол (ПТЗ)-индуцированного генерализованного эпилептического припадка [12, 95]. Кроме того, мы обнаружили ослабление долговременной синаптической депрессии [99] и ДВП в латентную [93] и хроническую [94] фазы литий-пилокарпиновой модели височной эпилепсии.
Нарушение ДВП, вероятно, следует связать с повреждением молекулярных механизмов индукции. Известно, что для формирования ДВП в синапсах гиппокампа необходима активация глутаматных NMDA-рецепторов [100]. Как уже подробно обсуждалось выше, эпилептическая активность изменяет не только количество NMDA-рецепторов [23], но и их функциональные свойства, которые напрямую зависят от их субъединичного состава [101].
Ряд генетических и фармакологических исследований показывают, что синаптические GluN2A-содержащие NMDA-рецепторы играют большую роль в индукции долговременной потенциации, в то время как внесинаптические GluN2B-содержащие – в индукции долговременной депрессии [101–107]. Поэтому было высказано предположение, что соотношение продукции субъединиц GluN2A и GluN2B является наиболее значимым фактором при определении знака синаптической пластичности [108] и изменение их соотношения влияет на пластичность. Наши данные согласуются с этим обобщением. Так, ослабление ДВП в синапсах СА3–СА1 гиппокампа крыс через 24 ч после генерализованного припадка, вызванного ПТЗ, сопровождалось увеличением экспрессии мРНК GluN2B субъединицы [12].
Однако другие исследователи отмечают, что как GluN2A-, так и GluN2B-содержащие NMDA-рецепторы необходимы для индукции и ДВП, и долговременной депрессии [109]. Некоторые результаты однако не укладываются в это обобщение. Например, Muller с соавт. обнаружили усиление ДВП в СА3–CA1 синапсах у животных с хронической эпилепсией, которая полностью подавлялась селективным блокатором GluN2B-содержащих NMDA-рецептора (Ro 25-6981, 1 мкМ), в то время блокатор GluN2A-содержащих NMDA-рецепторов (NVP-AAM077, 50 нМ) не оказывал влияния на ДВП [98].
Нарушение пластичности может быть связано с работой метаботропных глутаматных рецепторов. Известно, что mGluR I группы располагаются на постсинаптической мембране перисинаптически [110], участвуют в выработке и длительном поддержании ДВП в синапсах [111], а также оказывают модулирующее действие на NMDA-рецепторы [112, 113]. Временная активация mGluR I группы с помощью агонистов вызывает эпилептиформные разряды в срезах гиппокампа, которые сохраняются в течение нескольких часов после отмывки [114]. В наших исследованиях в модели генерализованных ПТЗ-индуцированных эпилептических припадков мы впервые выявили смену механизма индукции ДВП с NMDAR-зависимого на обусловленный работой mGluR I группы 1 подтипа (mGluR1) [95]. Применение специфического антагониста mGluR1 (FTIDC, 5 мкМ) полностью блокировало выработку ДВП в CA3–CA1 синапсах гиппокампа у крыс через одни сутки после судорог и не влияло на индукцию ДВП в контрольных срезах и срезах через 3 и 7 дней после судорог. Эта форма синаптической пластичности не требовала активации NMDA-рецепторов, и сохранялась в присутствии их конкурентного блокатора AP-5. Ранее такая форма ДВП в поле CA1 гиппокампа, которая обусловлена лишь работой mGluRs, наблюдалась только в синапсах на интернейронах [115, 116]. Временное появление дополнительных функционально активных рецепторов mGluR1 в синапсах CA1 после судорог подтверждается биохимическими исследованиями [117, 118].
4. ГЛИЯ-НЕЙРОНАЛЬНЫЕ ВЗАИМОДЕЙСТВИЯ
Сейчас все большее внимание уделяется роли глия-нейрональных взаимодействий в патогенезе эпилепсии и формировании постсудорожных неврологических нарушений [119]. Эти исследования проводятся в рамках “трехчастной” или “четырехчастной” концепций строения и функционирования синапсов. Идея заключается в том, что в состав синапса помимо пре- и постсинаптических нейронов входят астроцит и микроглиальная клетка [120–122].
4.1. Участие астроцитов в патогенезе эпилепсии
Астроглия быстро реагирует на развитие судорожной активности в ЦНС. Наиболее ярким эффектом является астроглиоз – увеличение числа и размеров астроцитарных клеток, приводящее к изменению функциональных свойств астроглии [121]. Некоторые молекулярные и морфологические особенности реактивных (активированных) астроцитов рассматриваются как отличительные признаки активации астроцитов при эпилепсии [123]. Наиболее заметным признаком является повышенная экспрессия белков промежуточных филаментов, в частности, увеличение продукции глиального фибриллярного кислого белка (GFAP), который является основным компонентом системы промежуточных филаментов взрослых астроцитов. Увеличение продукции GFAP рассматривается в качестве маркера развития астроглиоза [124].
Степень астроглиоза может значительно различаться, сильный астроглиоз приводит к образованию глиального рубца, закрывающего пустое пространство, образованного после гибели нейронов. Формирование астроглиального рубца является частым признаком хронической фокальной эпилепсии [121, 125]. Увеличение содержания GFAP в зонах CA1 и CA3 гиппокампа больных височной эпилепсией найдено с помощью иммуногистохимических методов [126]. Усиление продукции мРНК и/или белка GFAP также было продемонстрированы в различных моделях приобретенной эпилепсии на животных, включая каинатную [127] и литий-пилокарпиновую модель [128].
Увеличение числа астроцитарных клеток может влиять на баланс глутамата за счет участия астроцитов в контроле поглощения, деградации и рециркуляции глутамата, играющего ключевую роль в эпилептогенезе [129]. Глутамат активно удаляется из синаптической щели с помощью нескольких транспортеров возбуждающих аминокислот (EAATs). Основным из них является астроцитарный транспортер EAAT2, опосредующий поглощение астроцитами около 80–90% всего глутамата, он играет одну из ключевых ролей в нейрон-глиальных взаимодействиях [130]. Нарушение обратного захвата глутамата влияет на сдвиг баланса в сторону возбуждения и способствует развитию эпилептиформной активности [131].
В исследовании, проведенном на каинатной модели, было показано, что экспрессия гена Slc1a2, кодирующего белок EAAT2, имеет волновую динамику: она увеличивалась через 1 день после эпилептического статуса, далее через 7 дней отмечалось снижение экспрессии и ее повторное усиление через 30 дней после судорог [127]. Кроме того, исследования на трансгенных мышах с гиперэкспрессией EAAT2 показали, что на острой стадии пилокарпин-индуцированных судорог значительного различия в степени тяжести приступа между мышами EAAT2 и мышами дикого типа не наблюдается, однако в хроническую стадию модели у генно-модифицированных мышей отмечается уменьшение спонтанных рецидивирующих судорог. Также было показано, что у трансгенных животных в острой и хронической фазах пилокарпиновой модели слабее выражена нейродегенерация [132].
Астроциты реагируют на повышение синаптической активности внутриклеточным повышением Ca2+, опосредованным активацией различных метаботропных и ионотропных рецепторов глутамата [129, 133]. В пилокарпиновой модели с использованием двухфотонной микроскопии in vivo было выявлено усиление астроцитарных кальциевых волн в коре через 3 дня после введения конвульсанта, что сопровождалось отсроченной гибелью нейронов [134]. Активация кальциевого сигналинга может способствовать высвобождению химических медиаторов, вовлеченных в эпилептогенез, включая D-серин, АТФ, глутамат, простагландины, цитокины и нейропептиды. Следствием этого является развитие нейронной гиперсинхронизации и аномальной возбудимости, а также нейродегенеративных процессов [129, 135].
Критическим и необходимым компонентом сигнального кальциевого пути в астроцитах являются инозитолтрифосфотные рецепторы второго типа (IP3R2), что было доказано в работах с животными нокаутными по гену соответствующего белка [136, 137]. Нокаутные мыши Itpr2–/– проявляют на 60% меньше эпилептиформной активности по сравнению с мышами дикого типа, что подтверждает проконвульсивную роль повышенного уровня астроцитарного Са2+ [133].
Таким образом, вовлеченность астроцитов в постсудорожные изменения в мозге носит разноплановый характер. С одной стороны, активация астроглии после эпилептического статуса способствует активации обратного захвата глутамата, снижая риск гиперактивации нейронов и развития эпилептиформной активности. С другой стороны, чрезмерный астроглиоз вызывает активацию кальциевого сигналинга в астроцитах, что приводит к нарушению опосредованной глией регуляции ионов и нейромедиаторов и ведет к гипервозбудимости нервной ткани.
Существует альтернативное мнение, что основная роль астроцитов связана с нейровоспалением. Нейровоспаление – патологический процесс в нервной ткани, имеющий сходство с воспалением по основным механизмам, но характеризующийся особенностями, обусловленными иммунологической обособленностью мозга. Считается, что на короткий период нейровоспаление призвано обеспечить сохранность нейронов, однако хроническое воспаление сопровождает нейродегенеративные заболевания и, возможно, является их причиной [138, 139]. Хотя активированные астроциты высвобождают цитокины [140], основную роль в процессах нейровоспаления отводят микроглиальным клеткам и микроглия-нейрональным, микроглия-астроцитарным взаимодействиям, о чем подробнее рассказано в следующем разделе.
4.2. Участие микроглии в патогенезе эпилепсии
Активация микроглии была выявлена как у людей с эпилепсией, так и в моделях эпилепсии у животных; при этом повышенная реактивность микроглии в гиппокампе коррелирует с частотой и длительностью припадков, что может иметь диагностическую значимость [141, 142]. Детальное исследование механизмов участия микроглии в эпилептогенезе началось преимущественно в последние 10 лет, поэтому до сих пор остается открытым вопрос, оказывает ли микроглия в целом проэпилептогенное действие или, наоборот, антиэпилептогенное [143].
4.2.1. Реакция микроглии на эпилептические судороги
В моделях эпилептогенеза на животных выявлена быстрая активация микроглии в ответ на гиперактивацию нейронов [141]. Высвобождаемый во время судорог глутамат активирует нейрональные NMDA-рецепторы, что ведет к кальций-зависимому высвобождению АТФ из нейронов [144]. АТФ активирует микроглию через P2Y12-рецепторы [145], что, проявляется в виде удлинения отростков микроглии в направлении слоя пирамидных клеток гиппокампа. Это, возможно, оказывает противоэпилептическое действие, так как у мышей с нокаутом P2Y12-рецепторов после внутрибрюшинной инъекции каината припадок начинался раньше, и тяжесть и частота приступов увеличивалась, приводя, в том числе, к гибели животных [144].
Через 3 ч после введения пилокарпина наблюдается утолщение точек ветвления и отростков клеток микроглии [144]. Через 24–48 ч после введения каината или пилокарпина наблюдается гибель нейронов и увеличение сомы микроглиальных клеток и укорочение их отростков, данный фенотип больше соответствует реактивной микроглии, способной к фагоцитозу [146]. В этот же период наблюдается повышение продукции хемокина CX3CL1 [147], уровень которого снова снижается через 3 дня после судорог [148]. При этом введение антител к CX3CL1 или его рецептору снижало гибель нейронов в результате эпилептического статуса [149].
Одновременно с морфологическими изменениями микроглии происходит запуск каскадов воспалительных реакций. Во-первых, активация микроглиального рецептора P2X7 [150] способствует процессингу IL-1β и экспрессии TNF-α. Блокада активации этого рецептора снижала микроглиоз и астроглиоз в гиппокампе мышей в каинтатной модели, а также уменьшало тяжесть спонтанных судорог [151]. С другой стороны, предполагается, что комплекс P2X7 с паннексином-1 может играть роль отрицательного модулятора судорожной активности при индукции судорог пилокарпином [152]. Во-вторых, высвобождаемый во время судорог глутамат также может непосредственно способствовать активации микроглии и продукции провоспалительных цитокинов [153]. В-третьих, ДНК погибших нейронов стимулирует микроглиальные Toll-подобные рецепторы (в частности, TLR9), способствуя высвобождению провоспалительных цитокинов TNFα и IL-1b, что может способствовать подавлению аберрантного нейрогенеза [154].
В то же время избыточная активация микроглии и гиперпродукция провоспалительных цитокинов может провоцировать нарушение функций гематоэнцефалического барьера, процессов торможения и возбуждения в мозге и способствовать эпилептогенезу [155]. В частности, TNFα способствует высвобождению глутамата из микроглии, что усиливает нейротоксические эффекты воспаления [156]. Цитокины оказывают модулирующее действие на работу синапсов [157]. Так, IL-1b через рецептор IL-1R1 может усиливать опосредованные NMDA-рецепторами токи Ca2+ [158], способствуя нарушению синаптической пластичности и эксайтотоксическим эффектам. Более того, IL-1b способствует снижению опосредованной ГАМК нейротрансмиссии [159]. Блокирование образования или действия IL-1b на рецептор IL-1R1 способствует снижению гибели нейронов в гиппокампе и уменьшению тяжести спонтанных судорог в хронической фазе литий-пилокарпиновой модели [160, 161]. В то же время для фебрильных судорог, характерных для раннего возраста, показано, что зависимое от микроглии изменение ГАМК-опосредованных токов способствует снижению возбудимости нейронов и смягчению судорог, следовательно, в данной модели острая активация микроглии имеет скорее нейропротекторное действие [162].
В последние годы обсуждается гипотеза наличия двух типов активации микроглии, определяющих исход патологии. Предполагается, что активация микроглии по первому типу (M1) увеличивает продукцию провоспалительных цитокинов (в частности IL-1b, IL-6, TNFα), усиливает воспалительные реакции, в том числе активацию астроцитов [163]. Активация по второму типу (M2) повышает продукцию противовоспалительных факторов и репарацию тканей [164]. Попытка разграничить M1 и M2 маркеры активации микроглии у мышей в пилокарпиновой модели выявила гетерогенную активацию микроглии в мозге после эпилептического статуса [165]. В переднем мозге наблюдалось повышение продукции и тех, и других маркеров, однако в гиппокампе превалировала экспрессия маркеров M1. В каинатной модели, характеризующейся более ранним началом спонтанных судорог, у мышей выявлено преобладание продукции маркеров M1 сразу после эпилептического статуса. Однако в начале хронической фазы в обеих моделях продукция изучаемых маркеров снижалась.
Согласно результатам другой работы, воспалительные изменения микроглии не являются обязательными для эпилептогенеза, невоспалительная активация микроглии вследствие повышенной активации mTOR может тоже запускать эпилептогенез [166]. Однако у нокаутных мышей с mTOR-дефицитарной микроглией наблюдается усиленная нейродегенерация и развитие более тяжелых спонтанных судорог после эпилептического статуса, чем у контроля [167]. Таким образом, только умеренная активация микроглиального mTOR-пути может иметь антиэпилептогенное действие.
Фагоцитарные функции активированной микроглии могут способствовать облегчению последствий судорожного припадка, обеспечивая снижение образования новых нейронов в зубчатой извилине после эпилептического статуса, которые могут способствовать формированию аберрантных гипервозбудимых нервных цепей [168]. Фармакологическое ингибирование миноциклином активации микроглии после эпилептического статуса снижало фагоцитоз вновь образовавшихся и выживших нейронов, часто находящихся в эктопическом положении [169], и в то же время подавляло миграцию вновь образовавшихся аберрантных нейронов [170]. В каинатной модели применение миноциклина подавляло нейродегенерацию в гиппокампе [171], при этом в пилокарпиновой модели было показано не только снижение гибели нейронов в гиппокампе, но и облегчение тяжести спонтанных рецидивирующих судорог в хронической фазе модели, а также уменьшение повышенной продукции микроглией IL-1b и TNFα в гиппокампе [172].
Элиминация синапсов микроглией после судорог затрагивает не только возбуждающие синапсы, но и тормозные ГАМКергические синапсы интернейронов, что ведет к дисбалансу торможения и возбуждения в гиппокампе [173].
Важно отметить, что эпилептический припадок провоцирует значительные изменения в составе сигнальных молекул, опосредующих фагоцитоз, что меняет фагоцитарные функции микроглии [174].
4.2.2. Хроническая активация микроглии в эпилептической ткани мозга человека
Предполагается, что хроническая активация микроглии может иметь проэпилептическое действие [143, 175]. При эпилепсии, ассоциированной с глионейрональной опухолью, плотность активированных клеток микроглии коррелирует с частотой припадков и длительностью эпилепсии [176]. Morin-Brureau с соавт. изучили морфологические характеристики микроглии в гиппокампах пациентов с височной эпилепсией и выявили преобладание амебоидных клеток микроглии в склеротизированном гиппокампе, то есть морфотипа реактивной микроглии. Однако встречаются также и клетки с сильным ветвлением отростков, более соответствующие форме “покоящейся” микроглии [146]. В областях с меньшим количеством нейронов (как правило, это области CA1 и CA3) встречалась преимущественно амебоидная микроглия. Однако число глиальных клеток и их форма не коррелировали с частотой приступов у пациентов. Также все морфотипы микроглиальных клеток демонстрировали отличную от нормальной реакцию на присутствие внеклеточного АДФ: отростки втягивались, в то время как в норме наоборот начинают удлиняться. Однако по другим данным, в эпилептизированных тканях мозга человека такая реакция зависит от концентрации АДФ: низкие концентрации способствуют удлинению отростков в направлении источника АДФ, опосредованное рецепторами P2Y12, а высокие концентрации, напротив, вызывали втягивание отростков, опосредованное рецепторами P2Y1 и P2Y13 [177]. Также в склеротизированной ткани повышена продукция рецептора CX3CR1, что может представлять собой компенсаторную реакцию для восстановления ГАМК-опосредованных токов, которые, как было показано Roseti и соавт., могут моделироваться посредством CX3CL1 [159].
Обобщая вышесказанное, можно отметить, что с одной стороны, временная активация микроглии после судорог снижает образование аберрантных связей, гибель и гиперактивацию нейронов, с другой стороны, избыточная активация микроглии может способствовать нарушению баланса возбуждения и торможения в нейронах, усугубляя нарушения синаптических функций. Остается открытым вопрос, какие из этих процессов преобладают при формировании патологии.
ЗАКЛЮЧЕНИЕ
За последние два десятилетия значительные усилия были приложены для выявления молекулярных механизмов изменений, происходящих в синапсах после эпилептической активности. Подводя итог нашему рассмотрению выявленных изменений в функционировании синапсов в ходе эпилептогенеза, следует подчеркнуть следующее. Во-первых, эпилептическая активность вносит долговременные комплексные изменения в работе всех функциональных частей синапса. Меняется вероятность выброса медиаторов, модифицируются свойства постсинаптических рецепторов, нарушается долговременная пластичность синаптической передачи. Все это сопровождается анатомическими и функциональными изменениями со стороны глиальных клеток. Всеохватность и масштабность таких изменений, к сожалению, не позволяет однозначно выявить причинно-следственные связи между этими событиями и указать, какие изменения изначально носят компенсаторный характер, а появление каких приведет впоследствии к усилению и закреплению патологической активности в нервных сетях. Поэтому выявить одну универсальную мишень для приложения наиболее эффективного терапевтического воздействия с целью остановки эпилептогенеза сейчас представляется маловероятным. Скорее следует думать о комплексном воздействии на нейронные сети, позволяющем максимально снизить их активность, предотвратить избыточную нейровоспалительную реакцию.
Во-вторых, многие из наблюдаемых изменения имеют сложный волновой характер. Например, встраивание кальций-проницаемых AMPA-рецепторов или GluN2B-содержащих NMDA-рецепторов наблюдается неоднократно на разных сроках, как это было описано нами выше. Вероятно, что эффекты от активации этих рецепторов в различные периоды после эпилептической активности могут сильно различаться. Можно предположить, что быстрое кратковременное встраивание кальций-проницаемых AMPA-рецепторов во время судорог может вести к дополнительной активации кальций-зависимой калиевых каналов и на коротком промежутке времени снижать возбудимость нейронов, оказывая таким образом противоэпилептическое действие. Однако более позднее встраивание и длительное присутствие кальций-проницаемых AMPA-рецепторов будет вести к нарушениям кальциевого сигналинга в нейроне, что в свою очередь может стать причиной его гибели.
Волновая природа изменений экспрессии многих значимых для нормального функционирования синапса молекул указывает нам также на то, что фармакологическое воздействие должно производиться в строго определенный период, так как раньше или позднее срока нарушенной экспрессии оно может оказаться бесполезным или даже вредным. Вероятно, что этим объясняются противоречивые данные об антиэпилептогенном эффекте антагонистов кальций-проницаемых AMPA-рецепторов или GluN2B-содержащих NMDA-рецепторов.
Масштабность и сложная временная динамика выявляемых изменений в синапсе в ходе эпилептогенеза делают необходимым проведение дальнейших детальных исследований с анализом активации различных сигнальных путей в нейронах и глиальных клетках на различных моделях эпилептогенеза. Центральными задачами по-прежнему остаются установление причинно-следственных связей между происходящими изменениями в функционировании синапсов во время и после эпилептической активности, а также выявление тех критических механизмов, которые запускают эпилептизацию мозга и ведут к развитию спонтанной судорожной активности.
Список литературы
Chin J.H., Vora N. (2014) The global burden of neurologic diseases. Neurology. 83: 349–351. https://doi.org/10.1212/WNL.0000000000000610
Laxer K.D., Trinka E., Hirsch L.J., Cendes F., Langfitt J., Delanty N., Resnick T., Benbadis S.R. (2014) The consequences of refractory epilepsy and its treatment. Epilepsy. Behav. 37: 59–70. https://doi.org/10.1016/j.yebeh.2014.05.031
Pitkänen A., Lukasiuk K. (2011) Mechanisms of epileptogenesis and potential treatment targets. Lancet Neurol. 10: 173–186. https://doi.org/10.1016/S1474-4422(10)70310-0
Goldberg E.M., Coulter D.A. (2013) Mechanisms of epileptogenesis: a convergence on neural circuit dysfunction. Nat. Rev. Neurosci. 14: 337–349. https://doi.org/10.1038/nrn3482
Ben-Ari Y. (2008) Epilepsies and neuronal plasticity: for better or for worse? Dialogues Clin. Neurosci. 10: 17–27. https://doi.org/10.31887/DCNS.2008.10.1/ybenari
Avoli M., Louvel J., Pumain R., Köhling R. (2005) Cellular and molecular mechanisms of epilepsy in the human brain. Prog. Neurobiol. 77: 166–200. https://doi.org/10.1016/j.pneurobio.2005.09.006
Epsztein J., Milh M., Bihi R.I., Jorquera I., Ben-Ari Y., Represa A., Crépel V. (2006) Ongoing epileptiform activity in the post-ischemic hippocampus is associated with a permanent shift of the excitatory-inhibitory synaptic balance in CA3 pyramidal neurons. J. Neurosci. 26: 7082–7092. https://doi.org/10.1523/JNEUROSCI.1666-06.2006
Graebenitz S., Lesting J., Sosulina L., Seidenbecher T., Pape H.C. (2010) Alteration of NMDA receptor-mediated synaptic interactions in the lateral amygdala associated with seizure activity in a mouse model of chronic temporal lobe epilepsy. Epilepsia. 51: 1754–1762. https://doi.org/10.1111/j.1528-1167.2010.02561.x
Clarkson C., Smeal R.M., Hasenoehrl M.G., White J.A., Rubio M.E., Wilcox K.S. (2020) Ultrastructural and functional changes at the tripartite synapse during epileptogenesis in a model of temporal lobe epilepsy. Exp. Neurol. 326: 113196. https://doi.org/10.1016/j.expneurol.2020.113196
Yang J., Woodhall G.L., Jones R.S.G. (2006) Tonic facilitation of glutamate release by presynaptic NR2B-containing NMDA receptors is increased in the entorhinal cortex of chronically epileptic rats. J. Neurosci. 26: 406–410. https://doi.org/10.1523/JNEUROSCI.4413-05.2006
Thompson S.E., Ayman G., Woodhall G.L., Jones R.S.G. (2006) Depression of Glutamate and GABA Release by Presynaptic GABAB Receptors in the Entorhinal Cortex in Normal and Chronically Epileptic Rats. Neurosignals. 15: 202–215. https://doi.org/10.1159/000098515
Postnikova T.Y., Zubareva O.E., Kovalenko A.A., Kim K.K., Magazanik L.G., Zaitsev A.V. (2017) Status epilepticus impairs synaptic plasticity in rat hippocampus and is followed by changes in expression of NMDA receptors. Biochemistry (Moscow). 82: 282–290. https://doi.org/10.1134/S0006297917030063
Postnikova T.Y, Amakhin D.V., Trofimova A.M., Smolensky I.V., Zaitsev A.V. (2019) Changes in Functional Properties of Rat Hippocampal Neurons Following Pentylenetetrazole-induced Status Epilepticus. Neuroscience. 399: 103–116. https://doi.org/10.1016/j.neuroscience.2018.12.029
Li S., Reinprecht I., Fahnestock M., Racine R. (2002) Activity-dependent changes in synaptophysin immunoreactivity in hippocampus, piriform cortex, and entorhinal cortex of the rat. Neuroscience. 115: 1221–1229. https://doi.org/10.1016/S0306-4522(02)00485-2
Murthy V.N., Sejnowski T.J., Stevens C.F. (1997) Heterogeneous release properties of visualized individual hippocampal synapses. Neuron. 18: 599–612. https://doi.org/10.1016/s0896-6273(00)80301-3
Upreti C., Otero R., Partida C., Skinner F., Thakker R., Pacheco L.F., Zhou Z., Maglakelidze G., Velíšková J., Velíšek L., Romanovicz D., Jones T., Stanton P.K., Garrido-Sanabria E.R (2012) Altered neurotransmitter release, vesicle recycling and presynaptic structure in the pilocarpine model of temporal lobe epilepsy. Brain. 135: 869–885. https://doi.org/10.1093/brain/awr341
Soussi R., Boulland J.L., Bassot E., Bras H., Coulon P., Chaudhry F.A., Storm-Mathisen J., Ferhat L., Esclapez M. (2015) Reorganization of supramammillary–hippocampal pathways in the rat pilocarpine model of temporal lobe epilepsy: evidence for axon terminal sprouting. Brain Struct. Funct. 220: 2449–2468. https://doi.org/10.1007/s00429-014-0800-2
Abegg M.H., Savic N., Ehrengruber M.U., McKinney R.A., Gähwiler B.H. (2004) Epileptiform activity in rat hippocampus strengthens excitatory synapses. J. Physiol. 554: 439–448. https://doi.org/10.1113/jphysiol.2003.052662
Debanne D., Thompson S.M., Gähwiler B.H. (2006) A brief period of epileptiform activity strengthens excitatory synapses in the rat hippocampus in vitro. Epilepsia. 47: 247–256. https://doi.org/10.1111/j.1528-1167.2006.00416.x
Joshi S., Rajasekaran K., Sun H., Williamson J., Kapur J. (2017) Enhanced AMPA receptor-mediated neurotransmission on CA1 pyramidal neurons during status epilepticus. Neurobiol. Dis. 103: 45–53. https://doi.org/10.1016/j.nbd.2017.03.017
Rajasekaran K., Todorovic M., Kapur J. (2012) Calcium-permeable AMPA receptors are expressed in a rodent model of status epilepticus. Ann. Neurol. 72: 91–102. https://doi.org/10.1002/ana.23570
Naylor D.E., Liu H., Niquet J., Wasterlain C.G. (2013) Rapid surface accumulation of NMDA receptors increases glutamatergic excitation during status epilepticus. Neurobiol. Dis. 54: 225–238. https://doi.org/10.1016/j.nbd.2012.12.015
Wasterlain C.G., Naylor D.E., Liu H., Niquet J., Baldwin R. (2013) Trafficking of NMDA receptors during status epilepticus: Therapeutic implications. Epilepsia. 54: 78–80. https://doi.org/10.1111/epi.12285
Amakhin D.V., Soboleva E.B., Ergina J.L., Malkin S.L., Chizhov A.V., Zaitsev A.V. (2018) Seizure-Induced Potentiation of AMPA Receptor-Mediated Synaptic Transmission in the Entorhinal Cortex. Front. Cell Neurosci. 12: 486. https://doi.org/10.3389/fncel.2018.00486
Rajasekaran K., Joshi S., Kozhemyakin M., Todorovic M.S., Kowalski S., Balint C., Kapur J. (2013) Receptor trafficking hypothesis revisited: Plasticity of AMPA receptors during established status epilepticus. Epilepsia. 54: 14–16. https://doi.org/10.1111/epi.12266
Joshi S., Kapur J. (2018) Mechanisms of status epilepticus: α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor hypothesis. Epilepsia. 59: 71–81. https://doi.org/10.1111/epi.14482
Doyon N., Vinay L., Prescott S.A., De Koninck Y. (2016) Chloride Regulation: A Dynamic Equilibrium Crucial for Synaptic Inhibition. Neuron. 89: 1157–1172. https://doi.org/10.1016/j.neuron.2016.02.030
Glykys J., Dzhala V., Egawa K., Balena T., Saponjian Y., Kuchibhotla K.V., Bacskai B.J., Kahle K.T., Zeuthen T., Staley K.J. (2014) Local impermeant anions establish the neuronal chloride concentration. Science. 343: 670–675. https://doi.org/10.1126/science.1245423
Raimondo J.V., Burman R.J., Katz A.A., Akerman C.J. (2015) Ion dynamics during seizures. Front. Cell Neurosci. 9: 1–14. https://doi.org/10.3389/fncel.2015.00419
Lillis K.P., Kramer M.A., Mertz J., Staley K.J., White J.A. (2012) Pyramidal cells accumulate chloride at seizure onset. Neurobiol. Dis. 47: 358–366. https://doi.org/10.1016/j.nbd.2012.05.016
Fujiwara-Tsukamoto Y., Isomura Y., Nambu A., Takada M. (2003) Excitatory GABA input directly drives seizure-like rhythmic synchronization in mature hippocampal CA1 pyramidal cells. Neuroscience. 119: 265–75. https://doi.org/10.1016/s0306-4522(03)00102-7
Librizzi L., Losi G., Marcon I., Sessolo M., Scalmani P., Carmignoto G., de Curtis M. (2017) Interneuronal Network Activity at the Onset of Seizure-Like Events in Entorhinal Cortex Slices. J. Neurosci. 37: 10398–10407. https://doi.org/10.1523/JNEUROSCI.3906-16.2017
Burman R.J., Selfe J.S., Lee J.H., van den Berg M., Calin A., Codadu N.K., Wright R., Newey S.E., Parrish R.R., Katz A.A., Wilmshurst J.M., Akerman C.J., Trevelyan A.J., Raimondo J.V. (2019) Excitatory GABAergic signalling is associated with benzodiazepine resistance in status epilepticus. Brain. 142: 3482–3501. https://doi.org/10.1093/brain/awz283
Gibbs J.W., Sombati S., Delorenzo R.J., Coulter D.A. (1997) Physiological and Pharmacological Alterations in Postsynaptic GABAA Receptor Function in a Hippocampal Culture Model of Chronic Spontaneous Seizures. J. Neurophysiol. 77: 2139–2152. https://doi.org/10.1152/jn.1997.77.4.2139
Joshi S., Rajasekaran K., Hawk K.M., Brar J., Ross B.M., Tran C.A., Chester S.J., Goodkin H.P. (2015) Phosphatase inhibition prevents the activity-dependent trafficking of GABAA receptors during status epilepticus in the young animal. Epilepsia. 56: 1355–1365. https://doi.org/10.1111/epi.13098
Goodkin H.P., Joshi S., Mtchedlishvili Z., Brar J., Kapur J. (2008) Subunit-specific trafficking of GABA(A) receptors during status epilepticus. J. Neurosci. 28: 2527–2538. https://doi.org/10.1523/JNEUROSCI.3426-07.2008
Goodkin H.P. (2005) Status Epilepticus Increases the Intracellular Accumulation of GABAA Receptors. J. Neurosci. 25: 5511–5520. https://doi.org/10.1523/JNEUROSCI.0900-05.2005
Erreger K., Geballe M.T., Kristensen A., Chen P.E., Hansen K.B., Lee C.J., Yuan H., Le P., Lyuboslavsky P.N., Micale N., Jørgensen L., Clausen R.P., Wyllie D.J.A., Snyder J.P., Traynelis S.F. (2007) Subunit-specific agonist activity at NR2A-, NR2B-, NR2C-, and NR2D-containing N-methyl-D-aspartate glutamate receptors. Mol. Pharmacol. 72: 907–920. https://doi.org/10.1124/mol.107.037333
Chen Q., He S., Hu X.L., Yu J., Zhou Y., Zheng J., Zhang S., Zhang C., Duan W.H., Xiong Z.Q. (2007) Differential roles of NR2A- and NR2B-containing NMDA receptors in activity-dependent brain-derived neurotrophic factor gene regulation and limbic epileptogenesis. J. Neurosci. 27: 542–552. https://doi.org/10.1523/JNEUROSCI.3607-06.2007
Amakhin D.V., Malkin S.L., Ergina J.L., Kryukov K.A., Veniaminova E.A., Zubareva O.E., Zaitsev A.V. (2017) Alterations in Properties of Glutamatergic Transmission in the Temporal Cortex and Hippocampus Following Pilocarpine-Induced Acute Seizures in Wistar Rats. Front. Cell Neurosci. 11 :264. https://doi.org/10.3389/fncel.2017.00264
Di Maio R., Mastroberardino P.G., Hu X., Montero L., Greenamyre J.T. (2011) Pilocapine alters NMDA receptor expression and function in hippocampal neurons: NADPH oxidase and ERK1/2 mechanisms. Neurobiol. Dis. 42: 482–495. https://doi.org/10.1016/j.nbd.2011.02.012
Di Maio R., Mastroberardino P.G., Hu X., Montero L.M., Greenamyre J.T. (2013) Thiol oxidation and altered NR2B/NMDA receptor functions in in vitro and in vivo pilocarpine models: Implications for epileptogenesis. Neurobiol. Dis. 49: 87–98. https://doi.org/10.1016/j.nbd.2012.07.013
Zhu X., Dong J., Shen K., Bai Y., Zhang Y., Lv X., Chao J., Yao H. (2015) NMDA receptor NR2B subunits contribute to PTZ-kindling-induced hippocampal astrocytosis and oxidative stress. Brain Res. Bull. 114: 70–78. https://doi.org/10.1016/j.brainresbull.2015.04.002
Chen B., Feng B., Tang Y., You Y., Wang Y., Hou W., Hu W., Chen Z. (2016) Blocking GluN2B subunits reverses the enhanced seizure susceptibility after prolonged febrile seizures with a wide therapeutic time-window. Exp. Neurol. 283: 29–38. https://doi.org/10.1016/j.expneurol.2016.05.034
Frasca A., Aalbers M., Frigerio F., Fiordaliso F., Salio M., Gobbi M., Cagnotto A., Gardoni F., Battaglia G.S., Hoogland G., Di Luca M., Vezzani A. (2011) Misplaced NMDA receptors in epileptogenesis contribute to excitotoxicity. Neurobiol. Dis. 43: 507–515. https://doi.org/10.1016/j.nbd.2011.04.024
Isaac J.T.R., Ashby M.C., McBain C.J. (2007) The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron. 54: 859–871. https://doi.org/10.1016/j.neuron.2007.06.001
Geiger J.R.P., Melcher T., Koh D.S., Sakmann B., Seeburg P.H., Jonas P., Monyer H. (1995) Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron. 15: 193–204. https://doi.org/10.1016/0896-6273(95)90076-4
Kumar S.S., Bacci A., Kharazia V., Huguenard J.R. (2002) A developmental switch of AMPA receptor subunits in neocortical pyramidal neurons. J. Neurosci. 22: 3005–3015. https://doi.org/10.1523/jneurosci.22-08-03005.2002
Henley J.M., Wilkinson K.A. (2016) Synaptic AMPA receptor composition in development, plasticity and disease. Nat. Rev. Neurosci. 17: 337–350. https://doi.org/10.1038/nrn.2016.37
Grooms S.Y. (2000) Status epilepticus decreases glutamate receptor 2 mRNA and protein expression in hippocampal pyramidal cells before neuronal death. Proc. Natl. Acad. Sci. 97: 3631–3636. https://doi.org/10.1073/pnas.050586497
Sommer C., Roth S.U., Kiessling M. (2001) Kainate-induced epilepsy alters protein expression of AMPA receptor subunits GluR1, GluR2 and AMPA receptor binding protein in the rat hippocampus. Acta Neuropathol. 101: 460–468. https://doi.org/10.1007/s004010000310
Sanchez R.M., Koh S., Rio C., Wang C., Lamperti E.D., Sharma D., Corfas G., Jensen F.E. (2001) Decreased glutamate receptor 2 expression and enhanced epileptogenesis in immature rat hippocampus after perinatal hypoxia-induced seizures. J. Neurosci. 21: 8154–8163. https://doi.org/10.1523/jneurosci.21-20-08154.2001
Friedman L.K. (1998) Selective reduction of GluR2 protein in adult hippocampal CA3 neurons following status epilepticus but prior to cell loss. Hippocampus. 8: 511–525. https://doi.org/10.1002/(SICI)1098-1063(1998)8:5<511::AID-HIPO9>3.0.CO;2-W
Malkin S.L., Amakhin D.V., Veniaminova E.A., Kim K.K., Zubareva O.E., Magazanik L.G., Zaitsev A.V. (2016) Changes of ampa receptor properties in the neocortex and hippocampus following pilocarpine-induced status epilepticus in rats. Neuroscience. 327: 146–155. https://doi.org/10.1016/j.neuroscience.2016.04.024
Pellegrini-Giampietro D.E., Gorter J.A., Bennett M.V.L., Zukin R.S. (1997) The GluR2 (GluR-B) hypothesis: Ca2+-permeable AMPA receptors in neurological disorders. Trends Neurosci. 20: 464–470. https://doi.org/10.1016/S0166-2236(97)01100-4
Hu Y., Jiang L., Chen H., Zhang X.P. (2012) Expression of AMPA receptor subunits in hippocampus after status convulsion. Child’s Nerv. Syst. 28: 911–918. https://doi.org/10.1007/s00381-012-1747-3
Russo I., Bonini D., Via L.La, Barlati S., Barbon A. (2013) AMPA receptor properties are modulated in the early stages following pilocarpine-induced status epilepticus. Neuromol. Med. 15: 324–338. https://doi.org/10.1007/s12017-013-8221-6
Zaitsev A.V., Kim K.K, Frolova E.V., Lavrent’eva V.V., Lukomskaya N.Y., Magazanik L.G. (2014) Anticonvulsant activities of antagonists of NMDA and calcium-permeable AMPA receptors in a model of maximum electroshock in rats. Neurochem. J. 8: 301–305. https://doi.org/10.1134/S1819712414040138
Szczurowska E., Mares P. (2015) An antagonist of calcium permeable AMPA receptors, IEM1460: Anticonvulsant action in immature rats? Epilepsy Res. 109: 106–113. https://doi.org/10.1016/j.eplepsyres.2014.10.020
Szczurowska E., Mareš P. (2013) NMDA and AMPA receptors: Development and status epilepticus. Physiol. Res. 62: 21–38. https://doi.org/10.33549/physiolres.932662
Konen L.M., Wright A.L., Royle G.A., Morris G.P., Lau B.K., Seow P.W., Zinn R., Milham L.T., Vaughan C.W., Vissel B. (2020) A new mouse line with reduced GluA2 Q/R site RNA editing exhibits loss of dendritic spines, hippocampal CA1-neuron loss, learning and memory impairments and NMDA receptor-independent seizure vulnerability. Mol. Brain. 13: 27. https://doi.org/10.1186/s13041-020-0545-1
Rakhade S.N., Zhou C., Aujla P.K., Fishman R., Sucher N.J., Jensen F.E. (2008) Early Alterations of AMPA Receptors Mediate Synaptic Potentiation Induced by Neonatal Seizures. J. Neurosci. 28: 7979–7990. https://doi.org/10.1523/JNEUROSCI.1734-08.2008
Lippman-Bell J.J., Rakhade S.N., Klein P.M., Obeid M., Jackson M.C., Joseph A., Jensen F.E. (2013) AMPA Receptor antagonist NBQX attenuates later-life epileptic seizures and autistic-like social deficits following neonatal seizures. Epilepsia. 54: 1922–1932. https://doi.org/10.1111/epi.12378
Krestel H.E., Shimshek D.R., Jensen V., Nevian T., Kim J., Geng Y., Bast T., Depaulis A., Schonig K., Schwenk F., Bujard H., Hvalby Ø., Sprengel R., Seeburg P.H. (2004) A Genetic Switch for Epilepsy in Adult Mice. J. Neurosci. 24: 10568–10578. https://doi.org/10.1523/JNEUROSCI.4579-03.2004
Brusa R., Zimmermann F., Koh D.-S., Feldmeyer D., Gass P., Seeburg P.H., Sprengel R. (1995) Early-Onset Epilepsy and Postnatal Lethality Associated with an Editing-Deficient GluR-B Allele in Mice. Science. 270: 1677–1680. https://doi.org/10.1126/science.270.5242.1677
Lorgen J.-Ø., Egbenya D.L., Hammer J., Davanger S. (2017) PICK1 facilitates lasting reduction in GluA2 concentration in the hippocampus during chronic epilepsy. Epilepsy Res. 137: 25–32. https://doi.org/10.1016/j.eplepsyres.2017.08.012
Pirker S., Schwarzer C., Czech T., Baumgartner C., Pockberger H., Maier H., Hauer B., Sieghart W., Furtinger S., Sperk G. (2003) Increased Expression of GABA A Receptor β-Subunits in the Hippocampus of Patients with Temporal Lobe Epilepsy. J. Neuropathol. Exp. Neurol. 62: 820–834. https://doi.org/10.1093/jnen/62.8.820
Loup F., Wieser H.-G., Yonekawa Y., Aguzzi A., Fritschy J.-M. (2000) Selective Alterations in GABA A Receptor Subtypes in Human Temporal Lobe Epilepsy. J. Neurosci. 20: 5401–5419. https://doi.org/10.1523/JNEUROSCI.20-14-05401.2000
Loddenkemper T., Talos D.M., Cleary R.T., Joseph A., Sánchez Fernández I., Alexopoulos A., Kotagal P., Najm I., Jensen F.E. (2014) Subunit composition of glutamate and gamma-aminobutyric acid receptors in status epilepticus. Epilepsy Res. 108: 605–615. https://doi.org/10.1016/j.eplepsyres.2014.01.015
Mathern G.W., Pretorius J.K., Kornblum H.I., Mendoza D., Lozada A., Leite J.P., Chimelli L.M.C., Fried I., Sakamoto A.C., Assirati J.A., Lévesque M.F., Adelson P.D., Peacock W.J. (1997) Human hippocampal AMPA and NMDA mRNA levels in temporal lobe epilepsy patients. Brain. 120: 1937–1959. https://doi.org/10.1093/brain/120.11.1937
Talos D.M., Kwiatkowski D.J., Cordero K., Black P.M., Jensen F.E. (2008) Cell-specific alterations of glutamate receptor expression in tuberous sclerosis complex cortical tubers. Ann. Neurol. 63 :454–465. https://doi.org/10.1002/ana.21342
Sánchez Fernández I., Loddenkemper T. (2014) Subunit Composition of Neurotransmitter Receptors in the Immature and in the Epileptic Brain. Biomed. Res. Int. 2014: 1–11. https://doi.org/10.1155/2014/301950
Möddel G., Jacobson B., Ying Z., Janigro D., Bingaman W., González-Martínez J., Kellinghaus C., Prayson R.A., Najm I.M. (2005) The NMDA receptor NR2B subunit contributes to epileptogenesis in human cortical dysplasia. Brain Res. 1046: 10–23. https://doi.org/10.1016/j.brainres.2005.03.042
Klatte K., Kirschstein T., Otte D., Pothmann L., Müller L., Tokay T., Kober M., Uebachs M., Zimmer A., Beck H. (2013) Impaired D-serine-mediated cotransmission mediates cognitive dysfunction in epilepsy. J. Neurosci. 33: 13066–13080. https://doi.org/10.1523/JNEUROSCI.5423-12.2013
Egbenya D.L., Hussain S., Lai Y.-C., Xia J., Anderson A.E., Davanger S. (2018) Changes in synaptic AMPA receptor concentration and composition in chronic temporal lobe epilepsy. Mol. Cell Neurosci. 92: 93–103. https://doi.org/10.1016/j.mcn.2018.07.004
Kitaura H., Sonoda M., Teramoto S., Shirozu H., Shimizu H., Kimura T., Masuda H., Ito Y., Takahashi H., Kwak S., Kameyama S., Kakita A. (2017) Ca 2+ -permeable AMPA receptors associated with epileptogenesis of hypothalamic hamartoma. Epilepsia. 58: e59–e63. https://doi.org/10.1111/epi.13700
Prince H.C., Tzingounis A.V., Levey A.I., Conn P.J. (2000) Functional downregulation of GluR2 in piriform cortex of kindled animals. Synapse. 38: 489–498. https://doi.org/10.1002/1098-2396(20001215)38:4<489::AID-SYN15>3.0.CO;2-N
Brooks-Kayal A.R., Pritchett D.B. (1993) Developmental changes in human gamma-aminobutyric acidA receptor subunit composition. Ann. Neurol. 34: 687–693. https://doi.org/10.1002/ana.410340511
Poulter M.O., Brown L.A., Tynan S., Willick G., William R., McIntyre D.C. (1999) Differential Expression of α 1, α 2, α 3, and α 5 GABA A Receptor Subunits in Seizure-Prone and Seizure-Resistant Rat Models of Temporal Lobe Epilepsy. J. Neurosci. 19: 4654–4661. https://doi.org/10.1523/JNEUROSCI.19-11-04654.1999
Galanopoulou A.S., Moshé S.L. (2014) Does Epilepsy Cause a Reversion to Immature Function? Adv. Exp. Med. Biol. 813: 195–209. https://doi.org/10.1007/978-94-017-8914-1_16
Citri A., Malenka R.C. (2008) Synaptic Plasticity: Multiple Forms, Functions, and Mechanisms. Neuropsychopharmacology. 33: 18–41. https://doi.org/10.1038/sj.npp.1301559
Lepeta K., Lourenco M.V., Schweitzer B.C., Martino Adami P.V., Banerjee P., Catuara-Solarz S., de La Fuente Revenga M., Guillem A.M., Haidar M., Ijomone O.M., Nadorp B., Qi L., Perera N.D., Refsgaard L.K., Reid K.M., Sabbar M., Sahoo A, Schaefer N., Sheean R.K., Suska A., Verma R., Vicidomini C., Wright D., Zhang X.-D., Seidenbecher C. (2016) Synaptopathies: synaptic dysfunction in neurological disorders – A review from students to students. J. Neurochem. 138: 785–805. https://doi.org/10.1111/jnc.13713
Bliss T.V.P., Collingridge G.L. (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 361: 31–39. https://doi.org/10.1038/361031a0
Neves G., Cooke S.F., Bliss T.V.P. (2008) Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat. Rev. Neurosci. 9: 65–75. https://doi.org/10.1038/nrn2303
Brown T.H., Chapman P.F., Kairiss E.W., Keenan C.L. (1988) Long-term synaptic potentiation. Science. 242: 724–728. https://doi.org/10.1126/science.2903551
Shah P., Bassett D.S., Wisse L.E.M., Detre J.A., Stein J.M., Yushkevich P.A., Shinohara R.T., Pluta J.B., Valenciano E., Daffner M., Wolk D.A., Elliott M.A., Litt B., Davis K.A., Das S.R. (2018) Mapping the structural and functional network architecture of the medial temporal lobe using 7T MRI. Hum. Brain Mapp. 39: 851–865. https://doi.org/10.1002/hbm.23887
Mathern G.W., Adelson P.D., Cahan L.D, Leite J.P. (2002) Hippocampal neuron damage in human epilepsy: Meyer’s hypothesis revisited. Prog. Brain. Res. 135: 237–51. https://doi.org/10.1016/s0079-6123(02)35023-4
Postnikova T.Y., Amakhin D.V., Trofimova A.M., Zaitsev A.V. (2020) Calcium-permeable AMPA receptors are essential to the synaptic plasticity induced by epileptiform activity in rat hippocampal slices. Biochem. Biophys. Res. Commun. 529: 1145–1150. https://doi.org/10.1016/j.bbrc.2020.06.121
Plant K., Pelkey K.A., Bortolotto Z.A., Morita D., Terashima A., McBain C.J., Collingridge G.L., Isaac J.T.R. (2006) Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat. Neurosci. 9: 602–604. https://doi.org/10.1038/nn1678
Zhou J.-L., Shatskikh T.N., Liu X., Holmes G.L. (2007) Impaired single cell firing and long-term potentiation parallels memory impairment following recurrent seizures. Eur. J. Neurosci. 25: 3667–77. https://doi.org/10.1111/j.1460-9568.2007.05598.x
Suárez L.M., Cid E., Gal B., Inostroza M., Brotons-Mas J.R., Gómez-Domínguez D., de la Prida L.M., Solís J.M. (2012) Systemic Injection of Kainic Acid Differently Affects LTP Magnitude Depending on its Epileptogenic Efficiency. PLoS One. 7: e48128. https://doi.org/10.1371/journal.pone.0048128
Cunha A.O.S., de Oliveira J.A.C., Almeida S.S., Garcia-Cairasco N., Leão R.M. (2015) Inhibition of long-term potentiation in the schaffer-CA1 pathway by repetitive high-intensity sound stimulation. Neuroscience. 310: 114–127. https://doi.org/10.1016/j.neuroscience.2015.09.040
Kryukov K.A., Kim K.K., Magazanik L.G., Zaitsev A.V. (2016) Status epilepticus alters hippocampal long-term synaptic potentiation in a rat lithium-pilocarpine model. Neuroreport. 27: 1191–1195. https://doi.org/10.1097/WNR.0000000000000656
Plata A., Lebedeva A., Denisov P., Nosova O., Postnikova T.Y., Pimashkin A., Brazhe A., Zaitsev A.V., Rusakov D.A., Semyanov A. (2018) Astrocytic Atrophy Following Status Epilepticus Parallels Reduced Ca2+ Activity and Impaired Synaptic Plasticity in the Rat. Hippocampus. Front. Mol. Neurosci. 11: 215. https://doi.org/10.3389/fnmol.2018.00215
Postnikova T.Y., Trofimova A.M., Ergina J.L., Zubareva O.E., Kalemenev S.V., Zaitsev A.V. (2019) Transient Switching of NMDA-Dependent Long-Term Synaptic Potentiation in CA3–CA1 Hippocampal Synapses to mGluR1-Dependent Potentiation After Pentylenetetrazole-Induced Acute Seizures in Young Rats. Cell Mol. Neurobiol. 39: 287–300. https://doi.org/10.1007/s10571-018-00647-3
Guli X., Tokay T., Kirschstein T., Köhling R. (2016) Status Epilepticus Enhances Depotentiation after Fully Established LTP in an NMDAR-Dependent but GluN2B-Independent Manner. Neural. Plast. 2016: 6592038. https://doi.org/10.1155/2016/6592038
O’Leary H., Bernard P.B., Castano A.M., Benke T.A. (2016) Enhanced long term potentiation and decreased AMPA receptor desensitization in the acute period following a single kainate induced early life seizure. Neurobiol. Dis. 87: 134–144. https://doi.org/10.1016/j.nbd.2015.12.005
Müller L., Tokay T., Porath K., Köhling R., Kirschstein T. (2013) Enhanced NMDA receptor-dependent LTP in the epileptic CA1 area via upregulation of NR2B. Neurobiol. Dis. 54: 183–193. https://doi.org/10.1016/j.nbd.2012.12.011
Ivanov A.D., Zaitsev A.V. (2017) NMDAR-independent hippocampal long-term depression impairment after status epilepticus in a lithium-pilocarpine model of temporal lobe epilepsy. Synapse. 71: e21982. https://doi.org/10.1002/syn.21982
Barria A., Malinow R. (2005) NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron. 48: 289–301. https://doi.org/10.1016/j.neuron.2005.08.034
Paoletti P., Bellone C., Zhou Q. (2013) NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 14: 383–400. https://doi.org/10.1038/nrn3504
Parsons M.P., Raymond L.A. (2014) Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron. 82: 279–93. https://doi.org/10.1016/j.neuron.2014.03.030
Franchini L., Carrano N., Di Luca M., Gardoni F. (2020) Synaptic GluN2A-Containing NMDA Receptors: From Physiology to Pathological Synaptic Plasticity. Int. J. Mol. Sci. 21: 1538. https://doi.org/10.3390/ijms21041538
Li R., Huang F.S., Abbas A.K., Wigström H. (2007) Role of NMDA receptor subtypes in different forms of NMDA-dependent synaptic plasticity. BMC Neurosci. 8: 55. https://doi.org/10.1186/1471-2202-8-55
Liu L., Wong T.P., Pozza M.F., Lingenhoehl K., Wang Y.T., Sheng M., Auberson Y.P., Wang Y.T. (2004) Role of NMDA Receptor Subtypes in Governing the Direction of Hippocampal Synaptic Plasticity. Science. 304: 1021–1024. https://doi.org/10.1126/science.1096615
Massey P.V., Johnson B.E., Moult P.R., Auberson Y.P., Brown M.W., Molnar E., Collingridge G.L., Bashir Z.I. (2004) Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J. Neurosci. 24: 7821–7828. https://doi.org/10.1523/JNEUROSCI.1697-04.2004
Bartlett T.E., Bannister N.J., Collett V.J., Dargan S.L., Massey P.V., Bortolotto Z.A., Fitzjohn S.M., Bashir Z.I., Collingridge G.L., Lodge D. (2007) Differential roles of NR2A and NR2B-containing NMDA receptors in LTP and LTD in the CA1 region of two-week old rat hippocampus. Neuropharmacology. 52: 60–70. https://doi.org/10.1016/j.neuropharm.2006.07.013
Xu Z., Chen R.Q., Gu Q.H., Yan J.Z., Wang S.H., Liu S.Y., Lu W. (2009) Metaplastic regulation of long-term potentiation/long-term depression threshold by activity-dependent changes of NR2A/NR2B ratio. J. Neurosci. 29: 8764–8773. https://doi.org/10.1523/JNEUROSCI.1014-09.2009
Fox C.J., Russell K.I., Wang Y.T., Christie B.R. (2006) Contribution of NR2A and NR2B NMDA subunits to bidirectional synaptic plasticity in the hippocampus in vivo. Hippocampus. 16: 907–915. https://doi.org/10.1002/hipo.20230
Scheefhals N., MacGillavry H.D. (2018) Functional organization of postsynaptic glutamate receptors. Mol.Cell Neurosci. 91: 82–94. https://doi.org/10.1016/j.mcn.2018.05.002
Anwyl R. (2009) Metabotropic glutamate receptor-dependent long-term potentiation. Neuropharmacology. 56: 735–740. https://doi.org/10.1016/j.neuropharm.2009.01.002
O’neill N., McLaughlin C., Komiyama N., Sylantyev S. (2018) Biphasic modulation of NMDA receptor function by metabotropic glutamate receptors. J. Neurosci. 38: 9840–9855. https://doi.org/10.1523/JNEUROSCI.1000-18.2018
Lai T.K.Y., Zhai D., Su P., Jiang A., Boychuk J., Liu F. (2019) The receptor-receptor interaction between mGluR1 receptor and NMDA receptor: a potential therapeutic target for protection against ischemic stroke. FASEB J. 33: 14423–14439. https://doi.org/10.1096/fj.201900417R
Merlin L.R., Wong R.K.S. (1997) Role of group I metabotropic glutamate receptors in the patterning of epileptiform activities in vitro. J. Neurophysiol. 78: 539–544. https://doi.org/10.1152/jn.1997.78.1.539
Lapointe V., Morin F., Ratté S., Croce A., Conquet F., Lacaille J.C. (2004) Synapse-specific mGluR1-dependent long-term potentiation in interneurones regulates mouse hippocampal inhibition. J. Physiol. 555: 125–135. https://doi.org/10.1113/jphysiol.2003.053603
Perez Y., Morin F., Lacaille J.C. (2001) A hebbian form of long-term potentiation dependent on mGluR1a in hippocampal inhibitory interneurons. Proc. Natl. Acad. Sci. USA. 98: 9401–9406. https://doi.org/10.1073/pnas.161493498
Akbar M.T., Rattray M., Powell J.F., Meldrum B.S. (1996) Altered expression of group I metabotropic glutamate receptors in the hippocampus of amygdala-kindled rats. Mol. Brain. Res. 43: 105–116. https://doi.org/10.1016/S0169-328X(96)00162-3
Blümcke I., Suter B., Behle K., Kuhn R., Schramm J., Elger C.E., Wiestler O.D. (2000) Loss of hilar mossy cells in Ammon’s horn sclerosis. Epilepsia. 41. Suppl 6: S174–180. https://doi.org/10.1111/j.1528-1157.2000.tb01577.x
Vizuete A.F.K., Hansen F., Negri E., Leite M.C., de Oliveira D.L, Gonçalves C.-A. (2018) Effects of dexamethasone on the Li-pilocarpine model of epilepsy: protection against hippocampal inflammation and astrogliosis. J. Neuroinflam. 15: 68. https://doi.org/10.1186/s12974-018-1109-5
Stogsdill J.A., Eroglu C. (2017) The interplay between neurons and glia in synapse development and plasticity. Curr. Opin. Neurobiol. 42: 1–8. https://doi.org/10.1016/j.conb.2016.09.016
Patel D.C., Tewari B.P., Chaunsali L., Sontheimer H. (2019) Neuron–glia interactions in the pathophysiology of epilepsy. Nat. Rev. Neurosci. 20: 282–297. https://doi.org/10.1038/s41583-019-0126-4
Croft W., Dobson K.L., Bellamy T.C. (2015) Plasticity of Neuron-Glial Transmission: Equipping Glia for Long-Term Integration of Network Activity. Neural. Plast. 2015: 765792. https://doi.org/10.1155/2015/765792
Hol E.M., Pekny M. (2015) Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the central nervous system. Curr. Opin. Cell Biol. 32: 121–130. https://doi.org/10.1016/j.ceb.2015.02.004
Sofroniew M.V., Vinters H.V. (2010) Astrocytes: biology and pathology. Acta Neuropathol. 119: 7–35. https://doi.org/10.1007/s00401-009-0619-8
Pekny M., Wilhelmsson U., Pekna M. (2014) The dual role of astrocyte activation and reactive gliosis. Neurosci. Lett 565: 30–38. https://doi.org/10.1016/j.neulet.2013.12.071
Kandratavicius L., Peixoto-Santos J.E., Monteiro M.R., Scandiuzzi R.C., Carlotti C.G., Assirati J.A., Hallak J.E., Leite J.P. (2015) Mesial temporal lobe epilepsy with psychiatric comorbidities: A place for differential neuroinflammatory interplay. J. Neuroinflam. 12: 1–12. https://doi.org/10.1186/s12974-015-0266-z
Hubbard J.A., Szu J.I., Yonan J.M., Binder D.K. (2016) Regulation of astrocyte glutamate transporter-1 (GLT1) and aquaporin-4 (AQP4) expression in a model of epilepsy. Exp. Neurol. 283: 85–96. https://doi.org/10.1016/j.expneurol.2016.05.003
Rossi A.R., Angelo M.F., Villarreal A., Lukin J., Ramos A.J. (2013) Gabapentin Administration Reduces Reactive Gliosis and Neurodegeneration after Pilocarpine-Induced Status Epilepticus. PLoS One. 8: e78516. https://doi.org/10.1371/journal.pone.0078516
Steinhäuser C., Grunnet M., Carmignoto G. (2016) Crucial role of astrocytes in temporal lobe epilepsy. Neuroscience. 323: 157–169. https://doi.org/10.1016/j.neuroscience.2014.12.047
Zaitsev A.V., Smolensky I.V., Jorratt P., Ovsepian S.V. (2020) Neurobiology, Functions, and Relevance of Excitatory AminoAcid Transporters (EAATs) to Treatment of Refractory Epilepsy. CNS Drugs. 34: 1089–1103. https://doi.org/10.1007/s40263-020-00764-y
Boison D., Steinhäuser C. (2018) Epilepsy and astrocyte energy metabolism. Glia 66: 1235–1243. https://doi.org/10.1002/glia.23247
Kong Q., Takahashi K., Schulte D., Stouffer N., Lin Y., Lin C.-L.G. (2012) Increased glial glutamate transporter EAAT2 expression reduces epileptogenic processes following pilocarpine-induced status epilepticus. Neurobiol. Dis. 47: 145–154. https://doi.org/10.1016/j.nbd.2012.03.032
Heuser K., Nome C.G., Pettersen K.H., Åbjørsbråten K.S., Jensen V., Tang W., Sprengel R., Taubøll E., Nagelhus E.A., Enger R. (2018) Ca2+ Signals in Astrocytes Facilitate Spread of Epileptiform Activity. Cereb. Cortex 28: 4036–4048. https://doi.org/10.1093/cercor/bhy196
Ding S., Fellin T., Zhu Y., Lee S.-Y., Auberson Y.P., Meaney D.F., Coulter D.A., Carmignoto G., Haydon P.G. (2007) Enhanced Astrocytic Ca2+ Signals Contribute to Neuronal Excitotoxicity after Status Epilepticus. J. Neurosci. 27: 10674–10684. https://doi.org/10.1523/JNEUROSCI.2001-07.2007
Wetherington J., Serrano G., Dingledine R. (2008) Astrocytes in the Epileptic Brain. Neuron
Eilert-Olsen M., Hjukse J.B., Thoren A.E., Tang W., Enger R., Jensen V., Pettersen K.H., Nagelhus E.A. (2019) Astroglial endfeet exhibit distinct Ca2+ signals during hypoosmotic conditions. Glia. 67: 2399–2409. https://doi.org/10.1002/glia.23692
Petravicz J., Boyt K.M., McCarthy K.D. (2014) Astrocyte IP3R2-dependent Ca2+ signaling is not a major modulator of neuronal pathways governing behavior. Front. Behav. Neurosci. 8: 384. https://doi.org/10.3389/fnbeh.2014.00384
Bazan N.G. (2012) Neuroinflammation and proteostasis are modulated by endogenously biosynthesized neuroprotectin D1. Mol. Neurobiol. 46: 221–226. https://doi.org/10.1007/s12035-012-8322-5
Skaper S.D. (2007) The brain as a target for inflammatory processes and neuroprotective strategies. Ann. N Y Acad. Sci. 1122: 23–34. https://doi.org/10.1196/annals.1403.002
Devinsky O., Vezzani A., Najjar S., De Lanerolle N.C., Rogawski M.A. (2013) Glia and epilepsy: excitability and inflammation. Trends Neurosci. 36: 174–184. https://doi.org/10.1016/j.tins.2012.11.008
Hiragi T., Ikegaya Y., Koyama R. (2018) Microglia after Seizures and in Epilepsy. Cells. 7: 26. https://doi.org/10.3390/cells7040026
Kagitani-Shimono K., Kato H., Kuwayama R., Tominaga K., Nabatame S., Kishima H., Hatazawa J., Taniike M. (2021) Clinical evaluation of neuroinflammation in child-onset focal epilepsy: a translocator protein PET study. J. Neuroinflam. 18: 8. https://doi.org/10.1186/s12974-020-02055-1
Kinoshita S., Koyama R. (2021) Pro- and anti-epileptic roles of microglia. Neural Regen. Res. 16: 1369. https://doi.org/10.4103/1673-5374.300976
Eyo U.B., Peng J., Swiatkowski P., Mukherjee A., Bispo A., Wu L.-J. (2014) Neuronal Hyperactivity Recruits Microglial Processes via Neuronal NMDA Receptors and Microglial P2Y12 Receptors after Status Epilepticus. J. Neurosci. 34: 10528–10540. https://doi.org/10.1523/JNEUROSCI.0416-14.2014
Kumaria A., Tolias C.M., Burnstock G. (2008) ATP signalling in epilepsy. Purinergic Signal. 4: 339–346. https://doi.org/10.1007/s11302-008-9115-1
Morin-Brureau M., Milior G., Royer J., Chali F., Le Duigou C., Savary E., Blugeon C., Jourdren L., Akbar D., Dupont S., Navarro V., Baulac M., Bielle F., Mathon B., Clemenceau S., Miles R. (2018) Microglial phenotypes in the human epileptic temporal lobe. Brain. 141: 3343–3360. https://doi.org/10.1093/brain/awy276
Ransohoff R.M.(2009) Chemokines and Chemokine Receptors: Standing at the Crossroads of Immunobiology and Neurobiology. Immunity. 31: 711–721. https://doi.org/10.1016/j.immuni.2009.09.010
Yeo S.-I., Kim J.-E., Ryu H.J., Seo C.H., Lee B.C., Choi I.-G., Kim D.-S., Kang T.-C. (2011) The roles of fractalkine/CX3CR1 system in neuronal death following pilocarpine-induced status epilepticus. J. Neuroimmunol. 234:93–102. https://doi.org/10.1016/j.jneuroim.2011.03.005
Ueno M., Fujita Y., Tanaka T., Nakamura Y., Kikuta J., Ishii M., Yamashita T. (2013) Layer V cortical neurons require microglial support for survival during postnatal development. Nat. Neurosci. 16: 543–551. https://doi.org/10.1038/nn.3358
Rappold P.M., Lynd-Balta E., Joseph S.A. (2006) P2X7 receptor immunoreactive profile confined to resting and activated microglia in the epileptic brain. Brain Res. 1089: 171–178. https://doi.org/10.1016/j.brainres.2006.03.040
Jimenez-Pacheco A., Diaz-Hernandez M., Arribas-Blázquez M., Sanz-Rodriguez A., Olivos-Oré L.A., Artalejo A.R., Alves M., Letavic M., Miras-Portugal M.T., Conroy R.M., Delanty N., Farrell M.A., O’Brien D.F., Bhattacharya A., Engel T., Henshall D.C. (2016) Transient P2X7 Receptor Antagonism Produces Lasting Reductions in Spontaneous Seizures and Gliosis in Experimental Temporal Lobe Epilepsy. J. Neurosci. 36: 5920–5932. https://doi.org/10.1523/JNEUROSCI.4009-15.2016
Kim J.-E., Kang T.-C. (2011) The P2X7 receptor–pannexin-1 complex decreases muscarinic acetylcholine receptor–mediated seizure susceptibility in mice. J. Clin. Invest. 121: 2037–2047. https://doi.org/10.1172/JCI44818
Raghunatha P., Vosoughi A., Kauppinen T.M., Jackson M.F. (2020) Microglial NMDA receptors drive pro-inflammatory responses via PARP-1/TRMP2 signaling. Glia. 68: 1421–1434. https://doi.org/10.1002/glia.23790
Matsuda T., Murao N., Katano Y., Juliandi B., Kohyama J., Akira S., Kawai T., Nakashima K. (2015) TLR9 signalling in microglia attenuates seizure-induced aberrant neurogenesis in the adult hippocampus. Nat. Commun. 6: 6514. https://doi.org/10.1038/ncomms7514
Klement W., Garbelli R., Zub E., Rossini L., Tassi L., Girard B., Blaquiere M., Bertaso F., Perroy J., de Bock F., Marchi N. (2018) Seizure progression and inflammatory mediators promote pericytosis and pericyte-microglia clustering at the cerebrovasculature. Neurobiol. Dis. 113: 70–81. https://doi.org/10.1016/j.nbd.2018.02.002
Takeuchi H., Jin S., Wang J., Zhang G., Kawanokuchi J., Kuno R., Sonobe Y., Mizuno T., Suzumura A. (2006) Tumor Necrosis Factor-α Induces Neurotoxicity via Glutamate Release from Hemichannels of Activated Microglia in an Autocrine Manner. J. Biol. Chem. 21: 21362–21368. https://doi.org/10.1074/jbc.M600504200
Stellwagen D., Beattie E.C, Seo J.Y., Malenka R.C. (2005) Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-α. J. Neurosci. 25: 3219–3228. https://doi.org/10.1523/JNEUROSCI.4486-04.2005
Viviani B., Bartesaghi S., Gardoni F., Vezzani A., Behrens M.M., Bartfai T., Binaglia M., Corsini E., Di Luca M., Galli C.L., Marinovich M. (2003) Interleukin-1β enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J. Neurosci. 23: 8692–8700. https://doi.org/10.1523/jneurosci.23-25-08692.2003
Roseti C., van Vliet E.A., Cifelli P., Ruffolo G., Baayen J.C., Di Castro M.A., Bertollini C., Limatola C., Aronica E., Vezzani A., Palma E. (2015) GABAA currents are decreased by IL-1β in epileptogenic tissue of patients with temporal lobe epilepsy: Implications for ictogenesis. Neurobiol. Dis. 82: 311–320. https://doi.org/10.1016/j.nbd.2015.07.003
Dyomina A.V., Zubareva O.E., Smolensky I.V., Vasilev D.S., Zakharova M.V., Kovalenko A.A., Schwarz A.P., Ischenko A.M., Zaitsev A.V. (2020) Anakinra Reduces Epileptogenesis, Provides Neuroprotection, and Attenuates Behavioral Impairments in Rats in the Lithium–Pilocarpine Model of Epilepsy. Pharmaceuticals. 13: 340. https://doi.org/10.3390/ph13110340
Noe F.M., Polascheck N., Frigerio F., Bankstahl M., Ravizza T., Marchini S., Beltrame L., Banderó C.R., Löscher W., Vezzani A. (2013) Pharmacological blockade of IL-1β/IL-1 receptor type 1 axis during epileptogenesis provides neuroprotection in two rat models of temporal lobe epilepsy. Neurobiol. Dis. 59: 183–193. https://doi.org/10.1016/j.nbd.2013.07.015
Wan Y., Feng B., You Y., Yu J., Xu C., Dai H., Trapp B.D., Shi P., Chen Z., Hu W. (2020) Microglial Displacement of GABAergic Synapses Is a Protective Event during Complex Febrile Seizures. Cell Rep. 33: 108346. https://doi.org/10.1016/j.celrep.2020.108346
Mukherjee S., Arisi G.M., Mims K., Hollingsworth G., O’Neil K., Shapiro L.A. (2020) Neuroinflammatory mechanisms of post-traumatic epilepsy. J. Neuroinflam. 17: 193. https://doi.org/10.1186/s12974-020-01854-w
Boche D., Perry V.H., Nicoll J.A.R. (2013) Review: activation patterns of microglia and their identification in the human brain. Neuropathol Appl. Neurobiol. 39: 3–18. https://doi.org/10.1111/nan.12011
Benson M.J., Manzanero S., Borges K. (2015) Complex alterations in microglial M1/M2 markers during the development of epilepsy in two mouse models. Epilepsia. 56: 895–905. https://doi.org/10.1111/epi.12960
Zhao X., Liao Y., Morgan S., Mathur R., Feustel P., Mazurkiewicz J., Qian J., Chang J., Mathern G.W., Adamo M.A., Ritaccio A.L., Gruenthal M., Zhu X., Huang Y. (2018) Noninflammatory Changes of Microglia Are Sufficient to Cause Epilepsy. Cell. Rep. 22: 2080–2093. https://doi.org/10.1016/j.celrep.2018.02.004
Zhao X.-F., Liao Y., Alam M.M., Mathur R., Feustel P., Mazurkiewicz J.E., Adamo M.A., Zhu X.C., Huang Y. (2020) Microglial mTOR is Neuronal Protective and Antiepileptogenic in the Pilocarpine Model of Temporal Lobe Epilepsy. J. Neurosci. 40: 7593–7608. https://doi.org/10.1523/JNEUROSCI.2754-19.2020
Zhou Q.-G., Nemes A.D., Lee D., Ro E.J., Zhang J., Nowacki A.S., Dymecki S.M., Najm I.M., Suh H. (2018) Chemogenetic silencing of hippocampal neurons suppresses epileptic neural circuits. J. Clin. Invest. 129: 310–323. https://doi.org/10.1172/JCI95731
Luo C., Koyama R., Ikegaya Y. (2016) Microglia engulf viable newborn cells in the epileptic dentate gyrus. Glia. 64: 1508–1517. https://doi.org/10.1002/glia.23018
Yang F., Liu Z.-R., Chen J., Zhang S.-J., Quan Q.-Y., Huang Y.-G., Jiang W. (2010) Roles of astrocytes and microglia in seizure-induced aberrant neurogenesis in the hippocampus of adult rats. J. Neurosci. Res. 88: 519–529. https://doi.org/10.1002/jnr.22224
Heo K., Cho Y.-J., Cho K.-J., Kim H.-W., Kim H.-J., Shin H.Y., Lee B.I., Kim G.W. (2006) Minocycline inhibits caspase-dependent and -independent cell death pathways and is neuroprotective against hippocampal damage after treatment with kainic acid in mice. Neurosci. Lett. 398: 195–200. https://doi.org/10.1016/j.neulet.2006.01.027
Wang N., Mi X., Gao B., Gu J., Wang W., Zhang Y., Wang X. (2015) Minocycline inhibits brain inflammation and attenuates spontaneous recurrent seizures following pilocarpine-induced status epilepticus. Neuroscience. 287: 144–156. https://doi.org/10.1016/j.neuroscience.2014.12.021
Andoh M., Ikegaya Y., Koyama R. (2019) Synaptic Pruning by Microglia in Epilepsy. J. Clin. Med. 8: 2170. https://doi.org/10.3390/jcm8122170
Wyatt-Johnson S.K., Brewster A.L. (2020) Emerging Roles for Microglial Phagocytic Signaling in Epilepsy. Epilepsy Curr. 20: 33–38. https://doi.org/10.1177/1535759719890336
Najjar S., Pearlman D., Miller D.C., Devinsky O. (2011) Refractory Epilepsy Associated With Microglial Activation. Neurologist. 17:.249–254. https://doi.org/10.1097/NRL.0b013e31822aad04
Aronica E., Gorter J.A., Redeker S., Ramkema M., Spliet W.G.M., van Rijen P.C., Leenstra S., Troost D. (2005) Distribution, characterization and clinical significance of microglia in glioneuronal tumours from patients with chronic intractable epilepsy. Neuropathol. Appl. Neurobiol. 31: 280–291. https://doi.org/10.1111/j.1365-2990.2004.00636.x
Milior G., Morin-Brureau M., Chali F. Le Duigou C., Savary E., Huberfeld G., Rouach N., Pallud J., Capelle L., Navarro V., Mathon B., Clemenceau S., Miles R. (2020) Distinct P2Y Receptors Mediate Extension and Retraction of Microglial Processes in Epileptic and Peritumoral Human Tissue. J. Neurosci. 40: 1373–1388. https://doi.org/10.1523/JNEUROSCI.0218-19.2019
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова