

Российский физиологический журнал им. И.М. Сеченова, 2021, T. 107, № 6-7, стр. 695-716
Молекулярная и функциональная гетерогенность Na,K-АТФазы в скелетной мышце
В. В. Кравцова 1, И. И. Кривой 1, *
1 Санкт-Петербургский государственный университет
Санкт-Петербург, Россия
* E-mail: iikrivoi@gmail.com
Поступила в редакцию 12.03.2021
После доработки 21.03.2021
Принята к публикации 28.03.2021
Аннотация
Активность Na,K-АТФазы критически важна для поддержания электрогенеза, сократительной функции и работоспособности скелетных мышц. Данный обзор посвящен анализу результатов исследований последних лет в области молекулярного и функционального разнообразия Na,K-АТФазы в скелетных мышцах, ко-экспрессирующих α1- и α2-изоформы каталитической и транспортной α-субъединицы Na,K-АТФазы. Рассмотрены проблемы, которые представляются наиболее перспективными с точки зрения их дальнейшего развития. Накопленные факты свидетельствуют, что в отличие от α1-изоформы, демонстрирующей функциональную стабильность, α2-изоформа отличается высокой степенью пластичности, которая обусловлена ее специфической мембранной локализацией, функциональными и молекулярными взаимодействиями с белковым и липидным окружением, а также особенностями регуляции различными факторами. Функциональные нарушения α2-изоформы Na,K-АТФазы относятся к наиболее общим признакам, характерным как для хронических, так и для кратковременных форм двигательной дисфункции.
I. ПРОБЛЕМЫ ИЗУЧЕНИЯ Na,K-АТФазы СКЕЛЕТНЫХ МЫШЦ
При интенсивной двигательной активности, обеспечиваемой скелетной мускулатурой, происходит снижение трансмембранных градиентов Na+ и К+. Наиболее функционально значимо снижение калиевого градиента, поскольку сопутствующая деполяризация плазматической мембраны способна привести к инактивации потенциал-зависимых Na+-каналов и снижению возбудимости, и, как следствие, к нарушению механизма генерации потенциалов действия и функционирования системы электромеханического сопряжения [1–5]. Накопление К+ в синаптических щелях с ограниченной диффузией может привести также к нарушениям в пресинаптическом механизме квантовой секреции медиатора [6]. Среди механизмов поддержания электрогенеза, сократительной функции и работоспособности скелетных мышц важнейшую роль играет активность Na,K-АТФазы. Эта транспортная система, открытая Skou [7], представляет собой интегральный белок, поддерживающий трансмембранные градиенты Na+ и K+ за счет их активного транспорта. Активность Na,K-АТФазы обеспечивает не только электрогенез и возбудимость, но также ряд сопряженных транспортных механизмов клетки [1–4]. Необходимо отметить, что плотность распределения Na,K-АТФазы в сарколемме составляет порядка 1000/мкм2, благодаря чему в скелетных мышцах содержится один из основных пулов Na,K-АТФазы, что важно для поддержания калиевого гомеостазиса всего организма в целом [2].
Na,K-АТФаза состоит из двух основных субъединиц α и β. Каталитическую и транспортную функции выполняет α-субъединица, тогда как β-субъединица в основном функционирует в качестве шаперона. С функциональным комплексом α/β ассоциирован также белок семейства FXYD, являющийся модулятором активности фермента [8–12]. Важной особенностью α-субъединицы Na,K-ATФазы является наличие внеклеточных участков, которые формируют специфический сайт связывания (рецептор) для кардиотонических стероидов (КТС). Эти лиганды растительного и животного происхождения в высоких концентрациях являются ингибиторами Na,K-АТФазы и имеют эндогенные циркулирующие структурные аналоги [9, 10, 13–15]. Na,K-АТФаза, как и многие другие функциональные белки, экспрессируется в различных молекулярных формах. Для млекопитающих известны четыре изоформы α-субъединицы (α1–α4), три изоформы β-субъединицы Na,K-АТФазы, а также семь белков семейства FXYD [8, 9, 11, 12, 16]. Экспрессия этих изоформ носит тканеспецифический характер, а различные их сочетания обеспечивают широкое молекулярное и функциональное разнообразие Na,K-АТФазы. Изоформа α1, экспрессируемая клетками всех тканей, предположительно выполняет основную функцию поддержания ионных градиентов. В большинстве случаев (кроме эритроцитов, почки и некоторых других клеток и тканей) α1-изоформа ко-экспрессируется с одной из изоформ α2–α4, которые выполняют специфические для данной клетки функции [8, 9, 15, 16].
Важно, что помимо молекулярного разнообразия, на функциональную гетерогенность Na,K-АТФазы важное влияние оказывают и другие факторы. Прежде всего, это факторы, обусловленные специфической мембранной локализацией, особенностями регуляции, а также молекулярным и функциональным взаимодействием с белковым и липидным окружением [15, 16]. В частности, наличие у Na,K-АТФазы ряда структурных доменов позволяет этому белку участвовать в качестве скаффолда в формировании функциональных мультимолекулярных комплексов. Благодаря этим особенностям достигается не только модуляция самой Na,K-АТФазы, но и ее вовлечение в реализацию разнообразных сигнальных внутриклеточных путей [17–21].
В скелетных мышцах коэкспрессируются α1- и α2-изоформы Na,K-АТФазы [22, 23], в мышцах человека экспрессируется также α3-изоформа [24]. Физиологическая роль, особенности локализации и функционирования различных молекулярных форм Na,K-АТФазы являются предметом интенсивного изучения [5, 22, 23, 25–27].
Данный обзор посвящен анализу результатов исследований последних лет, в которых авторы обзора принимали непосредственное участие и которые представляются наиболее перспективными с точки зрения их дальнейшего развития.
Одна из проблем обусловлена неоднородностью распределения α1- и α2-изоформ Na,K-АТФазы в сарколемме. Изоформа α1 распределена относительно равномерно. Фракция α2-изоформы доминирует и достигает 87% от общего количества Na,K-АТФазы в скелетной мышце [22, 23]. Вместе с тем, эта изоформа в покое малоактивна и базовую насосную функцию в основном выполняет α1-изоформа [28–30]. Важно, что α2-изоформа Na,K-АТФазы распределена в сарколемме в виде двух основных пулов. Большая часть α2-изоформы локализована в Т-системе, где участвует в поддержании работоспособности мышцы при накоплении К+ в Т-системе в условиях интенсивной двигательной нагрузки [5, 25]. Меньший пул α2-изоформы локализован в постсинаптической области мембраны (концевая пластинка) [26, 30]. Особенности локализации, межмолекулярных взаимодействий, функционирования и регуляции этого пула α2-изоформы исследованы в меньшей степени. Лишь отдельные работы свидетельствуют о существенных различиях в функционировании и регуляции этих двух пулов α2-изоформы Na,K-АТФазы [26, 31]. Одна из причин такого различия может заключаться в функциональном и молекулярном взаимодействии между α2-изоформой и никотиновыми холинорецепторами (нХР) в области концевой пластинки, механизмы и изоформ-специфичность которых во многом остаются неясными [16, 30]. Учитывая существенное влияние, которое оказывает холестерин на функционирование Na,K-АТФазы [32] и ионных каналов [33], роль липидного окружения во взаимодействии нХР/Na,K-АТФаза также заслуживает особого изучения [34].
Другая проблема обусловлена немногочисленностью данных об изоформ-специфичности влияния двигательной активности в отношении Na,K-АТФазы скелетных мышц. Вместе с тем данные о серьезных нарушениях функционирования Na,K-АТФазы с возрастом и при травмах [3, 35], при различных видах миодистрофии [36], в скелетных мышцах гибернирующих животных [37], а также в условиях гравитационной разгрузки [38, 39], настоятельно требуют подобных исследований. Необходимо отметить, что особый интерес представляет начальный этап двигательной разгрузки, который важен для понимания механизмов ранних сигнальных событий, предшествующих развитию атрофии, и который изучен недостаточно [39, 40].
Наконец, достаточно хорошо изучена специфичность регуляции α2-изоформы Na,K-АТФазы сердечной и гладкой мышц КТС, включая их циркулирующие эндогенные аналоги [15, 41]. Однако лишь относительно немногие исследования посвящены изучению эффектов КТС в скелетных мышцах [31, 42–45].
II. МОЛЕКУЛЯРНЫЙ КОМПЛЕКС НИКОТИНОВОГО ХОЛИНОРЕЦЕПТОРА И Na,K-АТФазы
То, что функциональные и молекулярные взаимодействия между Na,K-АТФазой и разнообразными белками (рецепторы нейромедиаторов, транспортеры и др.) вовлечены в регуляцию синаптической функции, уже не вызывает сомнений [46–50].
Одной из первых была выявлена функциональная и молекулярная связь между Na,K-АТФазой и нХР [16, 30, 51]. В основе этих исследований лежали следующие предпосылки. Общеизвестно, что активация нХР ацетилхолином (АХ), освобождающимся в виде квантов (в микромолярных концентрациях), приводит к открыванию ионных каналов рецепторов и кратковременной деполяризации мембраны концевой пластинки. Однако АХ в наномолярных концентрациях, соответствующих уровню негидролизованного АХ в синаптической щели (преимущественно освобождаемый в неквантовой форме), способен вызывать противоположный эффект – гиперполяризацию мембраны [28, 51–54]. Работами акад. Е.Е. Никольского и соавт. было установлено, что этот эффект обусловлен активацией электрогенного Na,K-насоса неквантовым АХ и лежит в основе локальной гиперполяризации мембраны в области концевой пластинки (surplus polarization) [52–54]. Обсуждается физиологическое значение этой локальной гиперполяризации, в частности, при формировании синаптического контакта, а также с точки зрения поддержания постсинаптического электрогенеза [54].
Последующие опыты на мембранных препаратах электрического органа Torpedo показали существование реципрокного функционального и молекулярного взаимодействия между нХР и α-субъединицей Na,K-АТФазы [30, 51]. Было установлено также, что АХ в наномолярных концентрациях вызывает гиперполяризацию мембраны за счет специфического увеличения электрогенной активности именно α2-изоформы Na,K-АТФазы [28, 51]. Наконец, было установлено, что в скелетной мышце часть пула α2-изоформы Na,K-АТФазы сконцентрирована в области концевой пластинки, где ко-локализована с нХР (рис. 1a) и продемонстрирована коиммунопреципитация между нХР, Na,K-АТФазой, фосфолемманом (субъединица FXYD1) и кавеолином-3 (маркер кавеол в мышечных клетках) [30, 55]. Участие холестерина в качестве молекулярного компонента этого комплекса [34, 56] (рис. 1b–d) рассмотрено ниже в части III данного обзора. Эти данные позволили предположить, что некоторая фракция нХР и α2-изоформы Na,K-АТФазы образуют функциональный комплекс нХР/α2Na,K-АТФаза/FXYD1/кавеолин-3 (рис. 1e), участвующий в модуляции и поддержании постсинаптического электрогенеза и нервно-мышечной передачи [16, 30, 34]. Предположительно, этот комплекс локализован в кавеолах, что подтверждается также данными об участии кавеолина-3 в агрин-индуцируемой кластеризации нХР [57].
Рис. 1.
Гипотетическая схема молекулярного комплекса никотинового холинорецептора (нХР) и Na,K-АТФазы. (a) – распределение нХР, меченных α-бунгаротоксином (α-ВТХ, красный канал), и α2-изоформы Na,K-АТФазы, меченной Bodipy-Ouabain (Bodipy-OUA, зеленый канал), в концевых пластинках диафрагмы мыши C57Bl/6. (b) – распределение нХР и липидных плотиков, меченных субъединицей B холерного токсина (СТхВ, зеленый канал), в концевых пластинках камбаловидной мышцы крысы. (c) и (d) – дестабилизация липидных плотиков в концевой пластинке камбаловидной мышцы крысы уабаином (1 мкМ) и их восстановление с помощью комплекса МβЦД/холестерин. Показаны изображения (c) и уровень флуоресценции (d) после начального окрашивания СТхВ (СТхВ), через 15 мин действия уабаина (Oua); через 15 мин после добавления комплекса МβЦД/холестерин (Oua + Chol) и после повторного окрашивания СТхВ (Oua + CTxB). Масштаб: 10 мкМ. (e) – гипотетическая схема мультимолекулярного комплекса нХР/α2Na,K-АТФаза/FXYD1/кавеолин-3/холестерин. Пояснения в тексте. Данные авторов [34, 36, 56] с изменениями.
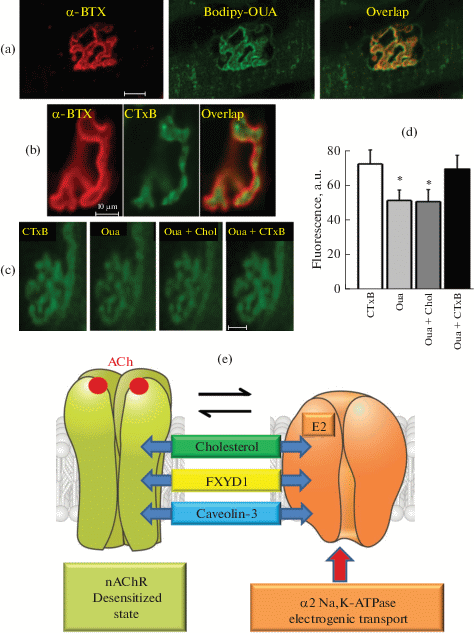
Участие белков цитоскелета дистрофина, спектринов, анкиринов и других возможных молекулярных партнеров в организации комплекса нХР/α2Na,K-АТФаза остается во многом неясным и нуждается в дальнейшем анализе.
Как и другие лиганд-управляемые каналы, нХР осуществляют постоянные конформационные переходы между закрытым, открытым и десенситизированным состояниями. Здесь важно отметить, что в состоянии десенситизации аффинность нХР к АХ лежит в наномолярном диапазоне, то есть на несколько порядков выше по сравнению с другими конформационными состояниями [58, 59]. Опыты с применением лигандов, сдвигающих равновесие между закрытым и десенситизированным состояниями, позволили предположить, что модуляция α2-изоформы Na,K-АТФазы обусловлена связыванием АХ с фракцией нХР, находящихся в состоянии десенситизации. Сами по себе конформационные сдвиги при переходе нХР в десенситизированное состояние также могут служить модулирующим сигналом [30]. Для Na,K-АТФазы принципиальным для осуществления взаимодействия с нХР, предположительно, является конформационный переход Е1–Е2 [16, 30].
Данные о функциональной связи между нХР и α2-изоформой Na,K-АТФазы предполагают возможность модуляции Na,K-АТФазы в скелетной мышце циркулирующими холинергическими лигандами. В частности, никотином, концентрация которого в крови при курении табака составляет порядка 100 нМ [60, 61]. Лишь в сравнительно немногих работах изучается влияние циркулирующего никотина на скелетную мускулатуру [62–64], хотя известно, что в этих условиях происходит переход части нХР в состояние десенситизации [65]. Перераспределение между различными конформационными состояниями нХР может действовать как модулирующий Na,K-АТФазу сигнал. Это подтверждается изменением функционирования α2-изоформы Na,K-АТФазы в сосудах гемато-энцефалического барьера и в мозге [66], а также в диафрагмальной мышце [55] крыс в условиях хронического повышения уровня циркулирующего никотина в крови.
Действует ли подобный механизм в условиях применения ингибиторов ацетилхолинэстеразы, когда повышается уровень циркулирующего негидролизованного эндогенного АХ, неизвестно, и это требует специального анализа. Учитывая применение антихолинэстеразных препаратов в клинике, проведение такого анализа может быть полезным для более глубокого понимания механизмов их побочного действия, а также при отравлении подобными веществами.
Необходимо отметить, что сравнительно недавно функциональная и молекулярная реципрокная связь с Na,K-АТФазой была также показана для нХР нейронального типа у насекомых [67], что позволяет предположить возможность подобного принципа регуляции не только в скелетной мышце, но и в ЦНС.
III. Na,K-АТФаза И ЛИПИДНОЕ ОКРУЖЕНИЕ
Если участие липидов мембраны и холестерина в регуляции пресинаптической квантовой секреции медиатора из моторных нервных окончаний изучено достаточно подробно [34], то данные о роли холестерина в структурно-функциональной организации концевой пластинки сравнительно немногочисленны. Хорошо известно, что все мембранные белки взаимодействуют с липидным окружением [33]. В частности, нХР имеют многочисленные сайты связывания с холестерином, который необходим для нормального функционирования нХР и их встраивания в мембрану концевой пластинки. Установлено, что липидные плотики являются платформой для кластеризации нХР, которая нарушается в случае частичного удаления мембранного холестерина с помощью метил-β-циклодекстрина (МβЦД) [68–70].
Хорошо известно, что липидное окружение оказывает существенное влияние на функционирование Na,K-АТФазы [32, 70–73]. Однако данные об изоформ-специфичности взаимодействия между Na,K-АТФазой и холестерином весьма противоречивы. Так, в опытах на клеточных линиях и клеточных культурах показаны реципрокные взаимодействия между холестерином и Na,K-АТФазой, в которых сама Na,K-АТФаза может влиять на синтез холестерина и его трафик [74]. Ряд данных свидетельствует об уникальности в этой регуляции α1-изоформы Na,K-АТФазы, которая осуществляется с участием молекулярного комплекса Na,K-АТФаза/Src/кавеолин-1 [73].
Однако в работах с использованием Pichia pastoris в качестве экспрессионной системы в условиях применения таких дестабилизирующих факторов, как нагревание и детергенты, было показано, что липиды, включая холестерин, специфически отвечают за встраивание в плазматическую мембрану α2-изоформы Na,K-АТФазы [75, 76]. Предположительно, специфика взаимодействия α2-изоформы с фосфолипидами мембраны обусловлена особенностями ее трансмембранных доменов М8, М9 и М10, а также более слабой ассоциацией с белком FXYD1 [76].
Перечисленные выше противоречивые данные были получены при исследовании клеточных линий и клеточных культур. Данные об изоформ-специфичности взаимодействия между Na,K-АТФазой и холестерином в скелетной мышце весьма немногочисленны.
В частности, установлено, что удаление части мембранного холестерина с помощью МβЦД специфически снижает электрогенную активность α2-изоформы Na,K-АТФазы в диафрагме крысы [77]. Другая серия экспериментов [56] показала, что липидные плотики, локализованные в мембране концевой пластинки (рис. 1b), отвечают дестабилизацией при блокировании α2-изоформы Na,K-АТФазы с помощью 1 мкМ уабаина (рис. 1c), причем эта дестабилизация имеет такой же характер, как и при действии МβЦД. Дестабилизация обусловлена снижением уровня мембранного холестерина, поскольку была обратимой при встраивании в плазматическую мембрану экзогенного холестерина (рис. 1d). Эти данные позволяют предположить реципрокность взаимодействия между холестерином и α2-изоформой Na,K-АТФазы. Поскольку эффекты МβЦД и уабаина были наиболее выражены в постсинаптической области сарколеммы, взаимодействие между холестерином и α2-изоформой может быть наиболее функционально значимым для области концевой пластинки. В совокупности полученные данные легли в основу гипотетической схемы мультимолекулярного комплекса нХР/α2Na,K-АТФаза/FXYD1/кавеолин-3/холестерин (рис. 1e), локализованного в мембране концевой пластинки и служащего физиологическим механизмом поддержания мышечного электрогенеза и гарантийного фактора нервно-мышечной передачи [34, 56]. Полностью этот механизм остается неизвестным, однако наиболее вероятным с точки зрения взаимодействия с холестерином является переход Na,K-АТФазы в конформационное состояние E2 [56, 72].
IV. Na,K-АТФаза И ДВИГАТЕЛЬНАЯ АКТИВНОСТЬ
Хорошо известно, что не только двигательная активность зависит от функционирования Na,K-АТФазы, но существует и обратная зависимость. Усиление двигательной активности характеризуется увеличением общего количества Na,K-АТФазы в сарколемме, длительное ослабление активности вызывает противоположный эффект [3]. Изоформ-специфичность этих изменений Na,K-АТФазы изучена недостаточно, и имеющиеся данные получены в основном при изучении нарушений хронического характера при травмах [78, 79] и возрастных изменениях [35]. В опытах на мышах линий mdx (модель миодистрофии Дюшенна) и Bla/J (модель дисферлинопатии) выявлено, что эти различные по молекулярным механизмам и степени деструкции концевых пластинок модели миодистрофии характеризуются схожими нарушениями электрогенной активности и мембранной локализации α2-изоформы Na,K-АТФазы. Эти нарушения не зависят от функциональной специализации мышц (диафрагмальная или камбаловидная) и, предположительно, являются результатом их адаптивного ремоделирования при хронической моторной дисфункции [36, 80, 81].
Длительная функциональная разгрузка приводит к потере мышечной массы, ослаблению сократительной функции и, в конечном счете, к атрофии скелетной мышцы. Обсуждаемые механизмы атрофических процессов, обусловленных двигательной дисфункцией, весьма многочисленны и во многом остаются неясными [39, 40, 82–84]. При этом с точки зрения поиска ключевых сигнальных событий, запускающих атрофическую программу, наиболее интересны механизмы ранних этапов, предшествующих развитию мышечной атрофии. Это относится и к вопросу изоформ-специфичности нарушений Na,K-АТФазы в таких условиях.
Изучение ранних (6–72 ч) этапов двигательной разгрузки камбаловидной мышцы с использованием метода антиортостатического вывешивания задних конечностей крысы (hindlimb suspension, HS) выявило деполяризацию и снижение возбудимости сарколеммы за счет специфического снижения электрогенной активности α2-изоформы Na,K-АТФазы [26, 85] (рис. 2). При этом был установлен адаптационный характер этих изменений, а также изменений экспрессии мРНК и количества α2-изоформы Na,K-АТФазы. Более того, функционирование α2-изоформы по-разному изменялось в разных районах сарколеммы. В период 12–24 ч HS наблюдалось усиление экспрессии мРНК и количества α2-изоформы; при этом в области концевой пластинки наблюдалось восстановление активности постсинаптического пула α2-изоформы и соответственно величины мембранного потенциала покоя. Напротив, во внесинаптическом районе сарколеммы, где локализован основной пул α2-изоформы, ее электрогенная активность и мембранный потенциал покоя были устойчиво снижены в течение всего периода HS (рис. 2). Механизм этого снижения неясен, но предположительно может быть обусловлен увеличением количества белка FXYD1 и усилением его ассоциации с α2-субъединицей Na,K-АТФазы, что должно тормозить каталитическую активность фермента [26]. Важно, что даже при кратковременной стимуляции двигательного нерва наблюдалось восстановление электрогенной активности α2-изоформы Na,K-АТФазы в обоих районах сарколеммы [26], что показывает прямую зависимость функционирования α2-изоформы от двигательной активности независимо от локализации.
Рис. 2.
Вывешивание задних конечностей (hindlimb suspension, HS) в течение 6–72 ч специфически влияет на α2-изоформу Na,K-АТФазы в камбаловидной мышце крысы. (a) и (b) – изменение величины мембранного потенциала покоя (RMP). (c) и (d) – изменение электрогенной активности α1- и α2-изоформ Na,K-АТФазы. (a, c) и (b, d) – постсинаптический и внесинаптический районы сарколеммы соответственно. (e) – количество белка, (f) – уровень мРНК α1- и α2-изоформ Na,K-АТФазы в общем гомогенате мышц. Пояснения в тексте. Данные авторов [26, 85] с изменениями.

Было установлено также, что даже кратковременная (6 ч) двигательная разгрузка в условиях HS сопровождается такими же нарушениями липид-упорядоченной фазы сарколеммы, как и в случае блокирования α2-изоформы Na,K-АТФазы с помощью 1 мкМ уабаина [56]. Дестабилизация липидных плотиков в обоих случаях была наиболее выражена в постсинаптическом районе мембраны, и кратковременная двигательная нагрузка, вызванная стимуляцией двигательного нерва, приводила к частичному восстановлению плотиков. Нарушения плотиков были также обратимы при встраивании в мембрану экзогенного холестерина, что указывает на частичную потерю мембранного холестерина в условиях HS.
В связи с этими наблюдениями важно отметить, что специфическое снижение электрогенной активности и плотности распределения α2-изоформы Na,K-АТФазы в сарколемме мышей линий mdx и Bla/J, различающихся молекулярными механизмами двигательной дисфункции, также сопровождалось перераспределением мембранного холестерина [36]. Помимо холестерина, в механизмах ранних нарушений, вызываемых двигательной дисфункцией, показано участие церамидов (основная молекула сфинголипидов), которые накапливаются в мышце при двигательной разгрузке [86].
Ультраструктура нервно-мышечного соединения весьма пластична и существенно зависит от двигательной активности. Такие нарушения структуры концевой пластинки, как изменение ее площади и усиление фрагментации в распределении нХР, наблюдаются при снижении двигательной активности: при возрастных изменениях, после денервации, при миодистрофии и других формах мышечной патологии. Молекулярные механизмы, лежащие в основе таких нарушений, интенсивно исследуются, но во многом продолжают оставаться неясными [87–89].
Важно отметить, что даже кратковременная двигательная разгрузка (6–12 ч HS), вызывающая нарушения функционирования α2-изоформы Na,K-АТФазы и дестабилизацию липидных плотиков, сопровождается также изменением структуры концевой пластинки [90]. Перечисленные структурно-функциональные нарушения, вызванные двигательной дисфункцией, относятся к наиболее ранним событиям, предшествующим развитию мышечной атрофии. Механизмы этих нарушений остаются предметом дальнейших исследований, однако рад фактов позволяет предположить, что одним из ключевых факторов является АМФ-активируемая протеинкиназа (AMPK) [39, 40, 83, 91].
В скелетной мышце АМРК является метаболическим сенсором, зависящим от внутриклеточного соотношения АМФ/АТФ. При возрастании потребления энергии клеткой в условиях интенсивной сократительной деятельности, когда увеличивается соотношение АМФ/АТФ, наблюдается активация АМРК. Напротив, при снижении двигательной активности должен наблюдаться противоположный эффект, что подтверждается данными о снижении уровня фосфорилирования AMРK в камбаловидной мышце крысы через 6–24 ч HS [83, 90]. При этом наблюдается также усиление аутофагии [90], которая вовлечена в регуляцию структуры нервно-мышечного соединения и является AMPK-зависимым фактором [92].
К настоящему времени накоплены многочисленные доказательства того, что AMРK в скелетных мышцах является не только регулятором процессов метаболизма, транскрипции, аутофагии и др., но играет также важную роль в поддержания структурно-функциональной организации нервно-мышечного соединения [92–94]. Эксперименты с превентивным введением AICAR (5-aminoimidazole-4-carboxamide ribonucleotide, активатор АМРК) позволяют рассматривать АМРК в качестве фармакологической мишени, обладающей терапевтическим потенциалом и препятствующей структурным нарушениям концевой пластинки и развитию симптомов мышечной атрофии [83, 93, 94]. AMPK известен также как важный фактор липидного метаболизма, участвующий в скелетной мышце в регуляции уровня мембранного холестерина [95].
Кроме того, по ряду данных, АМРК обладает способностью активировать Na,K-АТФазу в скелетной мышце [91]. Показано, что при превентивном введении AICAR отсутствует деполяризация сарколеммы камбаловидной мышцы крысы, характерная для HS и обусловленная дисфункцией Na,K-АТФазы [96]. Помимо АМРК, можно рассматривать и другие потенциальные регуляторы Na,K-АТФазы, в частности, оксид азота (NO) [11, 97], являющийся важным сигнальным фактором в условиях функциональной разгрузки скелетной мышцы [98, 99]. Наконец, можно рассматривать β-катенин, ключевой компонент Wnt-зависимого сигнального пути, который играет важную роль в формировании нервно-мышечного соединения и за счет взаимодействия с α2-изоформой Na,K-АТФазы модулирует ее количество, активность и электрогенез скелетных мышечных клеток [100]. Функциональное значение перечисленных регуляторов Na,K-АТФазы в условиях сниженной двигательной активности еще предстоит выяснить.
V. Na,K-АТФаза И КАРДИОТОНИЧЕСКИЕ СТЕРОИДЫ
Внеклеточные участки α-субъединицы Na,K-АТФазы формируют специфический рецептор для ряда лигандов растительного (уабаин, строфантин, дигоксин, дигитоксин и др.), и животного (маринобуфагенин) происхождения [9, 10, 13–15, 18, 101]. Эти лиганды являются специфическими ингибиторами Na,K-АТФазы и в высоких дозах чрезвычайно токсичны. Однако в низких дозах эти лиганды (строфантин, дигоксин) оказывают положительное инотропное действие, благодаря чему широко применяются в клинике сердечно-сосудистых заболеваний. В настоящее время, учитывая сходную стероидную структуру, эти лиганды обычно называют кардиотоническими стероидами (КТС).
По ряду данных, основную роль в реализации положительного инотропного действия КТС в сердечной и гладкой мышцах играет α2-изоформа Na,K-АТФазы. Предполагается, что это обусловлено особенностями локализации α2-изоформы в местах тесного прилегания плазматической мембраны к саркоплазматическому ретикулуму, где формируются субклеточные компартменты (микродомены) с ограниченной диффузией [41, 102]. В этих участках α2-изоформа Na,K-АТФазы тесно компартментализована с Na+,Ca2+-обменником, различными Ca2+-каналами и Ca2+-АТФазами. Благодаря этому, при связывании молекул КТС с этим пулом α2-изоформы за счет ингибирования фермента происходит локальное накопление внутриклеточного Na+ и сопряженное с ним накопление Ca2+ в результате изменения работы Na+,Ca2+-обменника. За счет работы SERCA происходит увеличение концентрации Ca2+ в саркоплазматическом ретикулуме, что приводит к усилению выброса Ca2+ из ретикулума в ответ на приходящий потенциал действия и соответственно к усилению мышечного сокращения. Помимо SERCA в регуляции кальциевого баланса в этих компартментах также могут принимать участие потенциал- и депо-зависимые Са2+-каналы плазматической мембраны, рианодиновые и IP3 рецепторы саркоплазматического ретикулума [18, 41, 102].
Хотя способность КТС усиливать сокращения скелетной мышцы также установлена [23, 42], механизм этого эффекта до сих пор остается неизвестным. Однако есть основания предполагать некоторую аналогию с механизмом положительного инотропного действия КТС в сердечной и гладкой мышцах. Действительно, в скелетной мышце триадное соединение между Т-тубулами и концевыми цистернами саркоплазматического ретикулума, где локализованы дигидропиридиновые и рианодиновые рецепторы, Na+,Ca2+-обменник, SERCA [103, 104], а также значительный пул α2-изоформы Na,K-АТФазы [5], можно считать определенным аналогом “PLasmERosome”.
Важно особо отметить, что хотя уабаин является специфическим ингибитором Na,K-АТФазы, ряд данных свидетельствует о способности уабаина в сверхнизких концентрациях активировать Na,K-АТФазу. В течение уже весьма длительного времени обсуждаются самые различные причины этого феномена [105–108], однако его механизм до сих пор остается неясным. В частности, предполагается, что эффекты уабаина, включая активацию α1-изоформы Na,K-АТФазы, могут быть обусловлены изменением соотношения внутриклеточных концентраций Na+ и K+ [108, 109]. Насколько значим такой механизм для мышечных клеток, в которых уабаин в наномолярных концентрациях специфически активирует α2-изоформу Na,K-АТФазы [31, 105], неизвестно. Однако, если предположить, что локальное повышение внутриклеточной концентрации Na+ в микродомене около молекулы Na,K-АТФазы, оккупированной молекулой КТС, способно активировать расположенные поблизости свободные молекулы Na,K-АТФазы, это могло бы быть механизмом активирующего действия КТС. Для реализации этого эффекта как раз и необходимы условия специфической субклеточной локализации α2-изоформы Na,K-АТФазы, которые обсуждались выше.
Чувствительность различных изоформ α-субъединицы Na,K-АТФазы к КТС может различаться, но у грызунов это различие выражено в наибольшей степени. У этих животных α1-изоформа мало чувствительна к уабаину (константа блокирования составляет десятки и сотни мкМ), напротив, изоформы α2- и α3- высокочувствительны к уабаину (константа блокирования составляет десятки и сотни нМ). У других млекопитающих, включая человека, чувствительность α1-изоформы к уабаину лежит в наномолярном диапазоне концентраций [8, 9, 14, 24, 110].
Специфический рецептор к КТС формируют несколько экстраклеточных участков α-субъединицы Na,K-АТФазы [9, 10, 14]. Участок между трансмембранными доменами М1–М2 определяет чувствительность к уабаину. Замены аминокислот в позициях 111 и 122 на этом участке позволяют получать генетически модифицированных мышей с измененной чувствительностью к уабаину у α1- и α2-изоформ Na,K-АТФазы. Таких животных широко используют для изучения функциональной специализации этих изоформ [14], в частности, при изучении их роли в адаптации скелетной мышцы к физической нагрузке [44].
Поскольку α-субъединица Na,K-АТФазы является специфическим рецептором для КТС, было высказано предположение о существовании эндогенных лигандов к этому рецептору. К настоящему времени выделен ряд эндогенных аналогов КТС, также имеющих стероидную структуру, из которых наиболее изучены эндогенные аналоги уабаина и маринобуфагенина. Эти циркулирующие лиганды предположительно синтезируются в коре надпочечников, гипоталамусе и гипофизе [8, 9, 13, 15, 18, 101], и их концентрация в крови и цереброспинальной жидкости в норме лежит в субнаномолярном диапазоне. Однако при различных физиологических и патофизиологических процессах [14, 15, 18, 110, 111], в частности, при интенсивной физической активности [44, 112], концентрация этих лигандов может существенно возрастать.
Хотя роль эндогенных КТС в сердечно-сосудистой системе, ЦНС, почках и других органах в норме и патологии интенсивно исследуется [10, 18, 41, 47, 113–116], сравнительно мало известно об их возможной функции в скелетных мышцах, содержащих один из самых больших пулов Na,K-АТФазы. К настоящему времени продемонстрирована способность уабаина в наномолярных концентрациях стимулировать синтез гликогена, что предполагает возможность такого механизма регуляции в скелетной мышце [11, 43]. В опытах на культуре скелетных мышечных клеток человека была показана регуляция наномолярными концентрациями уабаина секреции IL-6, а также модуляция IL-6/STAT3-сигнальных путей, осуществляемая через уабаин-чувствительную α1-изоформу Na,K-АТФазы [45]. В опытах с применением антител, связывающих циркулирующие КТС, было показано, что физиологическая роль эндогенного уабаина и α2-изоформы Na,K-АТФазы может заключаться в динамической адаптации скелетной мышцы к физической нагрузке [44]. Опыты с хроническим введением крысам уабаина, вдвое повышающим уровень уабаина в крови, показали, что циркулирующий уабаин способен модулировать электрогенез диафрагмальной мышцы через активацию α2-изоформы Na,K-АТФазы, и эффект может развиваться без каких-либо экспрессионных изменений данного белка. Предположительно, такая модуляция может иметь протективное значение в условиях нарушения мышечного электрогенеза при двигательной дисфункции [31, 96].
В настоящее время интенсивно изучается участие эндогенных КТС в механизмах регуляции, реализуемых через так называемую сигнальную функцию Na,K-АТФазы. Предполагается существование двух пулов Na,K-АТФазы, один из которых функционирует как ионный насос. Другой пул, преимущественно локализованный в кавеолах, образует молекулярный комплекс с EGFR и Src-киназой [17, 20] и участвует в мембранной и внутриклеточной сигнальной трансдукции. Выявлен участок α-субъединицы (так называемая NaKtide последовательность), отвечающий за реализацию сигнальной функции Na,K-АТФазы и предположительно специфичный для α1-изоформы Na,K-АТФазы [20]. Предполагается, что связывание молекулы КТС с комплексом α1Na,K-АТФаза/Src вызывает конформационные изменения, которые запускают ряд сигнальных внутриклеточных каскадов (ERK1/2 и др.), влияющих на процессы апоптоза, пролиферацию, синтез белка, сократительные свойства, оказывающих нейропротекторное действие и др. [9, 17, 18, 20, 114]. Роль Src-киназы в скелетной мышце изучена недостаточно. В опытах на скелетных мышечных клетках человека показана возможность Src-зависимой регуляции синтеза гликогена наномолярными концентрациями уабаина [11, 43] и обсуждается участие Src-киназы в реализации сигнальной функции уабаин-чувствительной α1-изоформы Na,K-АТФазы [45]. Участие α2- и α3-изоформ в сигнальной трансдукции подвергается сомнению [20, 21].
VI. ПЕРСПЕКТИВЫ
Проведенный нами анализ позволяет высказать некоторые предположения относительно перспектив дальнейших исследований функциональной пластичности α2-изоформы Na,K-АТФазы. В опытах на экспериментальных моделях миодистрофии Дюшенна и дисферлинопатии, а также в условиях моделирования гравитационной разгрузки, различающихся молекулярными механизмами двигательной дисфункции, выявлены специфические нарушения функционирования α2-изоформы Na,K-АТФазы, сопровождающиеся перераспределением мембранного холестерина [26, 36, 56]. Перспективным представляется продолжение этих исследований с использованием других моделей нарушений двигательной активности. В частности, модели бокового амиотрофического склероза (Amyotrophic Lateral Sclerosis, ALS) – заболевания, характеризующегося дегенерацией и гибелью мотонейронов, что приводит к денервации и атрофии скелетных мышц. Изоформ-специфичность функционирования Na,K-АТФазы и состояние липид-упорядоченной фазы плазматической мембраны при ALS не исследованы. Одной из моделей ALS может служить линия трансгенных мышей FUS1-513 с эктопной нейроспецифической экспрессией укороченного гена FUS человека [117].
Известно, что нарушение функционирования Na,K-АТФазы является одним из самых ранних ответов на гипоксию [118]. В скелетной мышце гипоксия также вызывает существенные функциональные нарушения [119]. Хотя и имеются данные о деполяризации мышечных волокон при гипоксии, предположительно обусловленные нарушением функции Na,K-АТФазы [120], изоформ-специфичность этих нарушений до сих пор не исследована. Новейшие данные свидетельствуют о терапевтическом потенциале циркулирующих КТС [31, 115, 116]. Эндогенный уабаин, повышение уровня которого выявлено при экспериментальной гипоксии у крыс [121], а также Na,K-АТФаза рассматриваются в качестве возможных факторов адаптации к высокогорью и гипоксии [13, 122]. В связи с вышесказанным использование модели гипоксии представляется перспективным для дальнейшего анализа функциональной пластичности α2-изоформы Na,K-АТФазы.
Нарушение гомеостаза глюкозы, при котором уровень глюкозы в крови превышает нормальный (гипергликемия), является преддиабетическим состоянием. Это происходит в результате нарушения стимулируемой инсулином утилизации глюкозы. Существует корреляция между количеством/активностью Na,K-АТФазы, экспрессируемой скелетными мышцами, и уровнем глюкозы, механизм которой не вполне ясен [123]. Вовлечение α2-изоформы Na,K-АТФазы в метаболизм глюкозы, связанный с ожирением, остается предметом изучения [124]. Дальнейшее исследование взаимосвязи между функционированием α2-изоформы Na,K-АТФазы скелетных мышц и уровнем глюкозы также представляется весьма перспективным.
Список литературы
Sejersted OM, Sjogaard G (2000) Dynamics and consequences of potassium shifts in skeletal muscle and heart during exercise. Physiol Rev 80: 1411–1481. https://doi.org/10.1152/physrev.2000.80.4.1411
Clausen T (2003) Na+–K+ pump regulation and skeletal muscle contractility. Physiol Rev 83: 1269–1324. https://doi.org/10.1152/physrev.00011.2003
Clausen T (2013) Quantification of Na+,K+ pumps and their transport rate in skeletal muscle: Functional significance. J Gen Physiol 142: 327–345. https://doi.org/10.1085/jgp.201310980
Clausen T (2015) Excitation of skeletal muscle is a self-limiting process, due to run-down of Na+,K+ gradients, recoverable by stimulation of the Na+,K+ pumps. Physiol Rep 3(4): e12373. https://doi.org/10.14814/phy2.12373
DiFranco M, Hakimjavadi H, Lingrel JB, Heiny JA (2015) Na,K-ATPase α2 activity in mammalian skeletal muscle T-tubules is acutely stimulated by extracellular K+. J Gen Physiol 146: 281–294. https://doi.org/10.1085/jgp.201511407
Matyushkin DP, Krivoi II, Drabkina TM (1995) Synaptic feed-backs mediated by potassium ions. Gen Physiol Biophys 14: 369–381.
Skou JC (1957) The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim Biophys Acta 23: 394–401. https://doi.org/10.1016/0006- 3002(57)90343-8
Blanco G, Mercer RW (1998) Isozymes of the Na-K-ATPase: Heterogeneity in structure, diversity in function. Am J Physiol 275: F633–F655. https://doi.org/10.1152/ajprenal.1998.275.5.F633
Mijatovic T, Van Quaquebeke E, Delest B, Debeir O, Darro F, Kiss R (2007) Cardiotonic steroids on the road to anti-cancer therapy. Biochim Biophys Acta 1776: 32–57. https://doi.org/10.1016/j.bbcan.2007.06.002
Bagrov AY, Shapiro JI, Fedorova OV (2009) Endogenous cardiotonic steroids: Physiology, pharmacology, and novel therapeutic targets. Pharmacol Rev 61: 9–38. https://doi.org/10.1124/pr.108.000711
Pirkmajer S, Chibalin AV (2016) Na,K-ATPase regulation in skeletal muscle. Am J Physiol Endocrinol Metab 311(1): E1–E31. https://doi.org/10.1152/ajpendo.00539.2015
Clausen MV, Hilbers F, Poulsen H (2017) The Structure and Function of the Na,K-ATPase Isoforms in Health and Disease. Front Physiol 8: 371. https://doi.org/10.3389/fphys.2017.00371
Blaustein MP (1993) Physiological effects of endogenous ouabain: control of intracellular Ca2+ stores and cell responsiveness. Am J Physiol Cell Physiol 264: C1367–C1387. https://doi.org/10.1152/ajpcell.1993.264.6.C1367
Lingrel JB (2010) The physiological significance of the cardiotonic steroid/ouabain-binding site of the Na,K-ATPase. Annu Rev Physiol 72: 395–412. https://doi.org/10.1146/annurev-physiol-021909-135725
Blaustein MP, Hamlyn JM (2020) Ouabain, Endogenous Ouabain and Ouabain-like Factors: The Na+ Pump/Ouabain Receptor, its linkage to NCX, and its Myriad Functions. Cell Calcium 102159. https://doi.org/10.1016/j.ceca.2020.102159
Matchkov VV, Krivoi II (2016) Specialized functional diversity and interactions of the Na,K‑ATPase. Front Physiol 7: 179. https://doi.org/10.3389/fphys.2016.00179
Xie Z, Askari A (2002) Na+/K+-ATPase as a signal transducer. Eur J Biochem 269: 2434–2439. https://doi.org/10.1046/j.1432-1033.2002.02910.x
Schoner W, Scheiner-Bobis G (2007) Endogenous and exogenous cardiac glycosides and their mechanisms of action. Am J Cardiovasc Drugs 7: 173–189. https://doi.org/10.2165/00129784-200707030-00004
Reinhard L, Tidow H, Clausen MJ, Nissen P (2013) Na+,K+-ATPase as a docking station: protein-protein complexes of the Na+,K+-ATPase. Cell Mol Life Sci 70: 205–222. https://doi.org/10.1007/s00018-012-1039-9
Cui X, Xie Z (2017) Protein Interaction and Na/K-ATPase-Mediated Signal Transduction. Molecules 22: 990. https://doi.org/10.3390/molecules22060990
Yu H, Cui X, Zhang J, Xie JX, Banerjee M, Pierre SV, Xie Z (2018) Heterogeneity of signal transduction by Na-K-ATPase alpha-isoforms: role of Src interaction. Am J Physiol Cell Physiol 314: C202–C210. https://doi.org/10.1152/ajpcell.00124.2017
Orlowski J, Lingrel JB (1988) Tissue-specific and developmental regulation of rat Na,K-ATPase catalytic α isoform and β subunit mRNAs. J Biol Chem 263: 10436–10442. https://doi.org/10.1016/S0021-9258(19)81535-1
He S, Shelly DA, Moseley AE, James PF, James JH, Paul RJ, Lingrel JB (2001) The α1- and α2‑isoforms of Na-K-ATPase play different roles in skeletal muscle contractility. Am J Physiol Regul Integr Comp Physiol 281: R917–R925.https://doi.org/10.1152/ajpregu.2001.281.3.R917
Cherniavsky Lev M, Karlish SJ, Garty H (2015) Cardiac glycosides induced toxicity in human cells expressing α1-, α2-,or α3-isoforms of Na-K-ATPase. Am J Physiol Cell Physiol 309: C126–C135. https://doi.org/10.1152/ajpcell.00089.2015
Radzyukevich TL, Neumann JC, Rindler TN, Oshiro N, Goldhamer DJ, Lingrel JB, Heiny JA (2013) Tissue-specific role of the Na,K-ATPase α2 isozyme in skeletal muscle. J Biol Chem 288: 1226–1237. https://doi.org/10.1074/jbc.M112.424663
Kravtsova VV, Petrov AM, Matchkov VV, Bouzinova EV, Vasiliev AN, Benziane B, Zefirov AL, Chibalin AV, Heiny JA, Krivoi II (2016) Distinct α2 Na,K-ATPase membrane pools are differently involved in early skeletal muscle remodeling during disuse. J Gen Physiol 147: 175–188. https://doi.org/10.1085/jgp.201511494
Kutz LC, Mukherji ST, Wang X, Bryant A, Larre I, Heiny JA, Lingrel JB, Pierre SV, Xie Z (2018) Isoform-specific role of Na/K-ATPase α1 in skeletal muscle. Am J Physiol Endocrinol Metab 314(6): E620–E629. https://doi.org/10.1152/ajpendo.00275.2017
Krivoi I, Vasiliev A, Kravtsova V, Dobretsov M, Mandel F (2003) Porcine kidney extract contains factor(s) that inhibit the ouabain-sensitive isoform of Na,K-ATPase (α2) in rat skeletal muscle: A convenient electrophysiological assay. Ann NY Acad Sci 986: 639–641. https://doi.org/10.1111/j.1749-6632.2003.tb07272.x
Radzyukevich TL, Moseley AE, Shelly DA, Redden GA, Behbehani MM, Lingrel JB, Paul RJ, Heiny JA (2004) The Na,K-ATPase α2 subunit isoform modulates contractility in the perinatal mouse diaphragm. Am J Physiol Cell Physiol 287: C1300–C1310. https://doi.org/10.1152/ajpcell.00231.2004
Heiny JA, Kravtsova VV, Mandel F, Radzyukevich TL, Benziane B, Prokofiev AV, Pedersen SE, Chibalin AV, Krivoi II (2010) The nicotinic acetylcholine receptor and the Na,K-ATPase α2 isoform interact to regulate membrane electrogenesis in skeletal muscle. J Biol Chem 285: 28614–28626. https://doi.org/10.1074/jbc.M110.150961
Kravtsova VV, Bouzinova EV, Matchkov VV, Krivoi II (2020) Skeletal Muscle Na,K-ATPase as a Target for Circulating Ouabain. Int J Mol Sci 21: 2875.https://doi.org/10.3390/ijms21082875
Cornelius F, Habeck M, Kanai R, Toyoshima C, Karlish SJ (2015) General and specific lipid-protein interactions in Na,K-ATPase. Biochim Biophys Acta 1848: 1729–1743. https://doi.org/10.1016/j.bbamem.2015.03.012
Levitan I, Singh DK, Rosenhouse-Dantsker A (2014) Cholesterol binding to ion channels. Front Physiol 5: 65. https://doi.org/10.3389/fphys.2014.00065
Krivoi II, Petrov AM (2019) Cholesterol and the Safety Factor for Neuromuscular Transmission. Int J Mol Sci 20: 1046. https://doi.org/10.3390/ijms20051046
Wyckelsma VL, McKenna MJ (2016) Effects of Age on Na+,K+-ATPase Expression in Human and Rodent Skeletal Muscle. Front Physiol 7: 316. https://doi.org/10.3389/fphys.2016.00316
Kravtsova VV, Bouzinova EV, Chibalin AV, Matchkov VV, Krivoi II (2020) Isoform-Specific Na,K-ATPase and Membrane Cholesterol Remodeling in the Motor Endplates in Distinct Mouse Models of Myodystrophy. Am J Physiol Cell Physiol 318: C1030–C1041. https://doi.org/10.1152/ajpcell.00453.2019
Guo Q, Mi X, Sun X, Li X, Fu W, Xu S, Wang Q, Arfat Y, Wang H, Chang H, Gao Y (2017) Remarkable plasticity of Na+,K+-ATPase, Ca2+-ATPase and SERCA contributes to muscle disuse atrophy resistance in hibernating Daurian ground squirrels. Sci Rep 7: 10509. https://doi.org/10.1038/s41598-017-10829-6
Shenkman BS, Kozlovskaya IB (2019) Cellular Responses of Human Postural Muscle to Dry Immersion. Front Physiol 10: 187. https://doi.org/10.3389/fphys.2019.00187
Shenkman BS (2020) How Postural Muscle Senses Disuse? Early Signs and Signals. Int J Mol Sci 21: 5037. https://doi.org/10.3390/ijms21145037
Vilchinskaya NA, Krivoi II, Shenkman BS (2018) AMP-Activated Protein Kinase as a Key Trigger for the Disuse-Induced Skeletal Muscle Remodeling. Int J Mol Sci 19: 3558. https://doi.org/10.3390/ijms19113558
Blaustein MP, Chen L, Hamlyn JM, Leenen FH, Lingrel JB, Wier WG, Zhang J (2016) Pivotal role of α2 Na+ pumps and their high affinity ouabain binding site in cardiovascular health and disease. J Physiol 594: 6079–6103. https://doi.org/10.1113/JP272419
Krivoi II, Drabkina TM, Kravtsova VV, Vasiliev AN, Vashchinkina EV, Prokofiev AV, Kubasov IV (2006) Role of the Na+,K+-ATPase α2 isoform in the positive inotropic effect of ouabain and marinobufagenin in the rat diaphragm. Biophysics 51: 799–804. https://doi.org/10.1134/S0006350906050228
Kotova O, Al-Khalili L, Talia S, Hooke C, Fedorova OV, Bagrov AY, Chibalin AV (2006) Cardiotonic steroids stimulate glycogen synthesis in human skeletal muscle cells via a Src- and ERK1/2-dependent mechanism. J Biol Chem 281: 20085–20094. https://doi.org/10.1074/jbc.M601577200
Radzyukevich TL, Lingrel JB, Heiny JA (2009) The cardiac glycoside binding site on the Na,K-ATPase α2 isoform plays a role in the dynamic regulation of active transport in skeletal muscle. Proc Natl Acad Sci USA 106: 2565–2570. https://doi.org/10.1073/pnas.0804150106
Pirkmajer S, Bezjak K, Matkovic U, Dolinar K, Jiang LQ, Miš K, Gros K, Milovanova K, Pirkmajer KP, Marš T, Kapilevich L, Chibalin AV (2020) Ouabain Suppresses IL-6/STAT3 Signaling and Promotes Cytokine Secretion in Cultured Skeletal Muscle Cells. Front Physiol 11: 566584. https://doi.org/10.3389/fphys.2020.566584
Hazelwood L, Free RB, Cabrera DM, Skinbjerg M, Sibley DR (2008) Reciprocal modulation of function between the D1 and D2 dopamine receptors and the Na+,K+-ATPase. J Biol Chem 283(52): 36441–36453. https://doi.org/10.1074/jbc.M805520200
Sibarov DA, Bolshakov AE, Abushik PA, Krivoi II, Antonov SM (2012) Na+,K+-ATPase functionally interacts with the plasma membrane Na+,Ca2+ exchanger to prevent Ca2+ overload and neuronal apoptosis in excitotoxic stress. J Pharmacol Exp Ther 343: 596–607. https://doi.org/10.1124/jpet.112.198341
Matos M, Augusto E, Agostinho P, Cunha RA, Chen J-F (2013) Interaction between adenosine A2A receptors and α2 Na,K-ATPase controlling glutamate uptake in astrocytes. J Neuroci 33(47): 18492–18502. https://doi.org/10.1523/JNEUROSCI1828-13.2013
Illarionava NB, Brismar H, Aperia A, Gunnarson E (2014) Role of Na,K-ATPase α1 and α2 isoforms in the support of astrocyte glutamate uptake. PLoS One 9(6): e98469. https://doi.org/10.1371/journal.pone.0098469
Akkuratov EE, Westin L, Vazquez-Juarez E, de Marothy M Melnikova AK, Blom H, Lindskog M, Brismar H, Aperia A (2020) Ouabain Modulates the Functional Interaction Between Na,K-ATPase and NMDA Receptor. Mol Neurobiol 57(10): 4018–4030. https://doi.org/10.1007/s12035-020-01984-5
Krivoi II, Drabkina TM, Kravtsova VV, Vasiliev AN, Eaton MJ, Skatchkov SN, Mandel F (2006) On the functional interaction between nicotinic acetylcholine receptor and Na+,K+-ATPase. Pflugers Arch 452: 756–765. https://doi.org/10.1007/s00424-006-0081-6
Vyskocil F, Nikolsky E, Edwards C (1983) An analysis of the mechanisms underlying the non–quantal release of acetylcholine at the mouse neuromuscular junction. Neuroscience 9(2): 429–435. https://doi.org/10.1016/0306-4522(83)90305-6
Nikolsky EE, Zemkova H, Voronin VA, Vyskocil F (1994) Role of non-quantal acetylcholine release in surplus polarization of mouse diaphragm fibres at the endplate zone. J Physiol 477: 497–502. https://doi.org/10.1113/jphysiol.1994.sp020210
Vyskocil F, Malomouzh AI, Nikolsky EE (2009) Non-quantal acetylcholine release at the neuromuscular junction. Physiol Res 58: 763–784.
Chibalin AV, Heiny JA, Benziane B, Prokofiev AV, Vasiliev AN, Kravtsova VV, Krivoi II (2012) Chronic nicotine exposure modifies skeletal muscle Na,K-ATPase activity through its interaction with the nicotinic acetylcholine receptor and phospholemman. PLoS One 7: e33719. https://doi.org/10.1371/journal.pone.0033719
Petrov AM, Kravtsova VV, Matchkov VV, Vasiliev AN, Zefirov AL, Chibalin AV, Heiny JA, Krivoi II (2017) Membrane lipid rafts are disturbed in the response of rat skeletal muscle to short-term disuse. Am J Physiol Cell Physiol 312: C627–C637. https://doi.org/10.1152/ajpcell.00365.2016
Hezel M, de Groat WC, Galbiati F (2010) Caveolin-3 promotes nicotinic acetylcholine receptor clustering and regulates neuromuscular junction activity. Mol Biol Cell 21(2): 302–310. https://doi.org/10.1091/mbc.E09-05-0381
Prince RJ, Sine SM (1999) Acetylcholine and epibatidine binding to muscle acetylcholine receptors distinguish between concerted and uncoupled models. J Biol Chem 274: 19623–19629. https://doi.org/10.1074/jbc.274.28.19623
Mourot A, Rodrigo J, Kotzyba-Hibert F, Bertrand S, Bertrand D, Goeldner M (2006) Probing the Reorganization of the Nicotinic Acetylcholine Receptor during Desensitization by Time-Resolved Covalent Labeling Using [3H]AC5, a Photoactivatable Agonist. Mol Pharmacol 69: 452–461. https://doi.org/10.1124/mol.105.017566
Lester RA, Dani JA (1995) Acetylcholine receptor desensitization induced by nicotine in rat medial habenula neurons. J Neurophysiol 74: 195–206. https://doi.org/10.1152/jn.1995.74.1.195
Benowitz NL, Zevin S, Jacob P (1997) Sources of variability in nicotine and cotinine levels with use of nicotine nasal spray, transdermal nicotine, and cigarette smoking. Br J Clin Pharmacol 43: 259–267. https://doi.org/10.1111/j.1365-2125.1997.00566.x
Larsson L, Orlander J, Ansved T, Edstrom L (1988) Effects of chronic nicotine exposure on contractile enzyme-histochemical and biochemical properties of fast- and slow-twitch muscles in the rat. Acta Physiol Scand 134: 519–527.https://doi.org/10.1111/j.1748-1716.1998.tb08526.x
Nakatani T, Nakashima T, Kita T, Ishihara A (2003) Effects of exposure to cigarette smoke at different dose levels on extensor digitorum longus muscle fibres in Wistar-Kyoto and spontaneously hypertensive rats. Clin Exp Pharmacol Physiol 30: 671–677. https://doi.org/10.1046/j.1440-1681.2003.03898.x
Degens H, Gayan-Ramirez G, van Hees HWH (2015) Smoking-induced Skeletal Muscle Dysfunction. From Evidence to Mechanisms. Am J Respir Crit Care Med 191(6): 620–625. https://doi.org/10.1164/rccm.201410-1830pp
Wang H, Sun X (2005) Desensitized nicotinic receptors in brain. Brain Res Rev 48: 420–437. https://doi.org/10.1016/j.brainresrev.2004.09.003
Wang L, McComb JG, Weiss MH, McDonough AA, Zlokovic BV (1994) Nicotine downregulates α2 isoform of Na,K-ATPase at the blood-brain barrier and brain in rats. Biochem Biophys Res Commun 199: 1422–1427. https://doi.org/10.1006/bbrc.1994.1389
Bao H, Sun H, Xiao Y, Zhang Y, Wang X, Xu X, Liu Z, Fang J, Li Z (2015) Functional interaction of nicotinic acetylcholine receptors and Na+/K+ ATPase from Locusta migratoria manilensis (Meyen). Sci Rep 5: 8849. https://doi.org/10.1038/sRep08849
Zhu D, Xiong WC, Mei L (2006) Lipid rafts serve as a signaling platform for nicotinic acetylcholine receptor clustering. J Neuroci 26: 4841–4851. https://doi.org/10.1523/JNEUROSCI2807-05.2006
Willmann R, Pun S, Stallmach L, Sadasivam G, Santos AF, Caroni P, Fuhrer C (2006) Cholesterol and lipid microdomains stabilize the postsynapse at the neuromuscular junction. EMBO J 25: 4050–4060. https://doi.org/10.1038/sj.emboj.7601288
Brannigan G, LeBard DN, Henin J, Eckenhoff RG, Klein ML (2010) Multiple binding sites for the general anesthetic isoflurane identified in the nicotinic acetylcholine receptor transmembrane domain. Proc Natl Acad Sci USA 107(32): 14122–14127. https://doi.org/10.1073/pnas.1008534107
Haviv H, Habeck M, Kanai R, Toyoshima C, Karlish SJ (2013) Neutral phospholipids stimulate Na,K-ATPase activity: a specific lipid-protein interaction. J Biol Chem 288: 10073–10081. https://doi.org/10.1074/jbc.M112.446997
Habeck M, Kapri-Pardes E, Sharon M, Karlish SJ (2017) Specific phospholipid binding to Na,K-ATPase at two distinct sites. Proc Natl Acad Sci USA 114(11): 2904–2909. https://doi.org/10.1073/pnas.1620799114
Zhang J, Li X, Yu H, Larre I, Dube PR, Kennedy DJ, Tang WHW Westfall K, Pierre SV, Xie Z, Chen Y (2020) Regulation of Na/K-ATPase expression by cholesterol: isoform specificity and the molecular mechanism. Am J Physiol Cell Physiol 319: C1107–C1119. https://doi.org/10.1152/ajpcell.00083.2020
Chen Y, Li X, Ye Q, Tian J, Jing R, Xie Z (2011) Regulation of α1 Na/K-ATPase expression by cholesterol. J Biol Chem 286: 15517–15524. https://doi.org/10.1074/jbc.M110.204396
Lifshitz Y, Petrovich E, Haviv H, Goldshleger R, Tal DM, Garty H, Karlish SJD (2007) Purification of the human α2 isoform of Na,K-ATPase expressed in Pichia pastoris. Stabilization by lipids and FXYD1. Biochemistry 46: 14937–14950. https://doi.org/10.1021/bi701812c
Kapri-Pardes E, Katz A, Haviv H, Mahmmoud Y, Ilan M, Khalfin-Penigel I, Carmeli S, Yarden O, Karlish SJD (2011) Stabilization of the α2 isoform of Na,K-ATPase by mutations in a phospholipid binding pocket. J Biol Chem 286: 42888–42899. https://doi.org/10.1074/jbc.M111.293852
Kravtsova VV, Petrov AM, Vasiliev AN, Zefirov AL, Krivoi II (2015) Role of cholesterol in the maintenance of endplate electrogenesis in rat diaphragm. Bull Exp Biol Med 158: 298–300. https://doi.org/10.1007/s10517-015-2745-8
Boon H, Kostovski E, Pirkmajer S, Song M, Lubarski IIversen PO, Hjeltnes N, Widegren U, Chibalin AV (2012) Influence of chronic and acute spinal cord injury on skeletal muscle Na+‑K+-ATPase and phospholemman expression in humans. Am J Physiol Endocrinol Metab 302: E864–E871. https://doi.org/10.1152/ajpendo.00625.2011
Perry BD, Levinger P, Morris HG, Petersen AC, Garnham AP, Levinger I, McKenna MJ (2015) The effects of knee injury on skeletal muscle function, Na+,K+-ATPase content, and isoform abundance. Physiol Rep 3: e12294. https://doi.org/10.14814/phy2.12294
Kravtsova VV, Timonina NA, Zakir’yanova GF, Sokolova AV, Mikhailov VM, Zefirov AL, Krivoi II (2018) The Structural and Functional Characteristics of the Motor End Plates of Dysferlin-Deficient Mice. Neurochem J 12: 305–310. https://doi.org/10.1134/S1819712418040049
Kravtsova VV, Bouzinova EV, Machkov VV, Timonina NA, Zakyrjanova GF, Zefirov AL, Krivoi II (2019) Abnormal membrane localization of α2 isoform of Na,K-ATPase in m. soleus of dysferlin-deficient mice. Bull Exp Biol Med 166: 593–597. https://doi.org/10.1007/s10517-019-04398-z
Bodine SC, Baehr LM (2014) Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF-1 and MAFbx/Atrogin-1. Am J Physiol Endocrinol Metab 307: E469–E484. https://doi.org/10.1152/ajpendo.00204.2014
Vilchinskaya NA, Mochalova EP, Nemirovskaya TL, Mirzoev TM, Turtikova OV, Shenkman BS (2017) Rapid decline in MyHC I(β) mRNA expression in rat soleus during hindlimb unloading is associated with AMPK dephosphorylation. J Physiol 595: 7123–7134. https://doi.org/10.1113/JP275184
Gorza L, Sorge M, Seclì L, Brancaccio M (2021) Master Regulators of Muscle Atrophy: Role of Costamere Components. Cells 10: 61. https://doi.org/10.3390/cells10010061
Kravtsova VV, Matchkov VV, Bouzinova EV, Vasiliev AN, Razgovorova IA, Heiny JA, Krivoi II (2015) Isoform-specific Na,K-ATPase alterations precede disuse-induced atrophy of rat soleus muscle. Biomed Res Int 720172. https://doi.org/10.1155/2015/720172
Bryndina IG, Shalagina MN, Protopopov VA, Sekunov AV, Zefirov AL, Zakirjanova GF, Petrov AM (2021) Early Lipid Raft-Related Changes: Interplay between Unilateral Denervation and Hindlimb Suspension. Int J Mol Sci 22: 2239. https://doi.org/10.3390/ijms22052239
Rudolf R, Khan MM, Labeit S Deschenes MR (2014) Degeneration of neuromuscular junction in age and dystrophy. Front Aging Neuroci 6: 99. https://doi.org/10.3389/fnagi.2014.00099
Tintignac LA, Brenner HR, Rüegg MA (2015) Mechanisms regulating neuromuscular junction development and function and causes of muscle wasting. Physiol Rev 95: 809–852. https://doi.org/10.1152/physrev.00033.2014
Slater CR (2020) “Fragmentation” of NMJs: a sign of degeneration or regeneration? A long journey with many junctions. Neuroscience 439: 28–40. https://doi.org/10.1016/j.neuroscience.2019.05.017
Chibalin AV, Benziane B, Zakyrjanova GF, Kravtsova VV, Krivoi II (2018) Early endplate remodeling and skeletal muscle signaling events following rat hindlimb suspension. J Cell Physiol 233: 6329–6336. https://doi.org/10.1002/jcp.26594
Pirkmajer S, Petric M, Chibalin AV (2021) The role of AMPK in regulation of Na+,K+-ATPase in skeletal muscle: does the gauge always plug the sink? J Muscle Res Cell Motil 42(1): 77–97. https://doi.org/10.1007/s10974-020-09594-3
Carnio S, LoVerso F, Baraibar MA, Longa E, Khan MM, Maffei M, Reischl M, Canepari M, Loefler S, Kern H, Blaauw B, Friguet B, Bottinelli R, Rudolf R, Sandri M (2014) Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Rep 8: 1509–1521. https://doi.org/10.1016/j.celRep.2014.07.061
Cervero C, Montull N, Tarabal O, Piedrafita L, Esquerda JE, Calderó J (2016) Chronic treatment with the AMPK agonist AICAR prevents skeletal muscle pathology but fails to improve clinical outcome in a mouse model of severe spinal muscular atrophy. Neurotherapeutics 13: 198–216. https://doi.org/10.1007/s13311-015-0399-x
Dial AG, Ng SY, Manta A, Ljubicic V (2018) The Role of AMPK in Neuromuscular Biology and Disease. Trends Endocrinol Metab 29: 300–312. https://doi.org/10.1016/j.tem.2018.02.010
Ambery AG, Tackett L, Penque BA, Brozinick JT, Elmendorf JS (2017) Exercise training prevents skeletal muscle plasma membrane cholesterol accumulation, cortical actin filament loss, and insulin resistance in C57BL/6J mice fed a western-style high-fat diet. Physiol Rep 5: e13363. https://doi.org/10.14814/phy2.13363
Kravtsova VV, Vilchinskaya NA, Rozlomii VL, Shenkman BS, Krivoi II (2019) Low Ouabain Doses and AMP-Activated Protein Kinase as Factors Supporting Electrogenesis in Skeletal Muscle. Biochemistry (Moscow) 84: 1085–1092.https://doi.org/10.1134/S0006297919090116
Juel C (2016) Nitric oxide and Na,K-ATPase activity in rat skeletal muscle. Acta Physiol (Oxf) 216(4): 447–453. https://doi.org/10.1111/apha.12617
Vitadello M, Sorge M, Percivalle E, Germinario E, Danieli-Betto D, Turco E, Tarone G, Brancaccio M, Gorza L (2020) Loss of melusin is a novel, neuronal NO synthase/FoxO3-independent master switch of unloading-induced muscle atrophy. J Cachexia Sarcopenia Muscle 11: 802–819. https://doi.org/10.1002/jcsm.12546
Sharlo KA, Paramonova II, Lvova ID, Mochalova EP, Kalashnikov VE, Vilchinskaya NA, Tyganov SA, Konstantinova TS, Shevchenko TF, Kalamkarov GR, Shenkman BS (2021) Plantar Mechanical Stimulation Maintains Slow Myosin Expression in Disused Rat Soleus Muscle via NO-Dependent Signaling. Int J Mol Sci 22: 1372. https://doi.org/10.3390/ijms22031372
Zhao C, Yu Y, Zhang Y, Shen J, Jiang L, Sheng G, Zhang W, Xu L, Jiang K, Mao S, Jiang P, Gao F (2019) β-Catenin Controls the Electrophysiologic Properties of Skeletal Muscle Cells by Regulating the α2 Isoform of Na+/K+-ATPase. Front Neuroci 13: 831. https://doi.org/10.3389/fnins.2019.00831
Doris PA, Bagrov AY (1998) Endogenous sodium pump inhibitors and blood pressure regulation: an update on recent progress. Proc Soc Exp Biol Med 218: 156–167. https://doi.org/10.3181/00379727-218-44283
Blaustein MP, Golovina VA (2001) Structural complexity and functional diversity of endoplasmic reticulum Ca2+ stores. Trends Neurosci 24: 602–608. https://doi.org/10.1016/S0166-2236(00)01891-9
Sacchetto R, Margreth A, Pelosi M, Carafoli E (1996) Colocalization of the dihydropyridine receptor, the plasma-membrane calcium ATPase isoform 1 and the sodium/calcium exchanger to the junctional membrane domain of transverse tubules of rabbit skeletal muscle. Eur J Biochem 237: 483–488.
Altamirano F, Eltit JM, Robin G, Linares N, Ding X, Pessah IN, Allen PD, López JR (2014) Ca2+ influx via the Na+/Ca2+ exchanger is enhanced in malignant hyperthermia skeletal muscle. J Biol Chem 289: 19180–19190. https://doi.org/10.1074/jbc.M114.550764
Gao J, Wymore RS, Wang Y, Gaudette GR, Krukenkamp IB, Cohen IS, Mathias RT (2002) Isoform-specific stimulation of cardiac Na/K pumps by nanomolar concentrations of glycosides. J Gen Physiol 119: 297–312. https://doi.org/10.1085/jgp.20028501
Holthouser KA, Mandal A, Merchant ML, Schelling JR, Delamere NA, Valdes RR Jr, T-yagi SC, Lederer ED, Khundmiri SJ (2010) Ouabain stimulates Na-K-ATPase through a sodium/hydrogen exchanger-1 (NHE-1)-dependent mechanism in human kidney proximal tubule cells. Am J Physiol Renal Physiol 299: F77–F90. https://doi.org/10.1152/ajprenal.00581.2009
Ketchem CJ, Conner CD, Murray RD, DuPlessis M, Lederer ED, Wilkey D, Merchant M, Khundmiri SJ (2016) Low dose ouabain stimulates Na-K ATPase α1 subunit association with angiotensin II type 1 receptor in renal proximal tubule cells. Biochim Biophys Acta 1863: 2624–2636. https://doi.org/10.1016/j.bbamcr.2016.07.008
Tverskoi AM, Sidorenko SV, Klimanova EA, Akimova OA, Smolyaninova LV, Lopina OD, Orlov SN (2016) Effects of ouabain on proliferation of human endothelial cells correlate with Na+,K+-ATPase activity and intracellular ratio of Na+ and K+. Biochemistry (Moscow) 81: 876–883. https://doi.org/10.1134/S0006297916080083
Orlov SN, Klimanova EA, Tverskoi AM, Vladychenskaya EA, Smolyaninova LV, Lopina OD (2017) Na+i,K+i-Dependent and -Independent Signaling Triggered by Cardiotonic Steroids: Facts and Artifacts. Molecules 22: 635. https://doi.org/10.3390/molecules22040635
Dobretsov M, Stimers JR (2005) Neuronal function and alpha3 isoform of the Na/K-ATPase. Front Biosci 10: 2373–2396. https://doi.org/10.2741/1704
Khalaf FK, Dube P, Mohamed A, Tian J, Malhotra D, Haller ST, Kennedy DJ (2018) Cardiotonic steroids and the sodium trade balance: new insights into trade-off mechanisms mediated by the Na+/K+-ATPase. Int J Mol Sci 19: 2576. https://doi.org/10.3390/ijms19092576
Bauer N, Müller-Ehmsen J, Krämer U, Hambarchian N, Zobel C, Schwinger RH Neu H, Kirch U, Grünbaum EG, Schoner W (2005) Ouabain-like compound changes rapidly on physical exercise in humans and dogs: Effects of β-blockade and angiotensin-converting enzyme inhibition. Hypertension 45: 1024–1028. https://doi.org/10.1161/01.HYP.0000165024.47728.f7
Hamlyn JM, Manunta P (2015) Endogenous cardiotonic steroids in kidney failure: A review and an hypothesis. Adv Chronic Kidney Dis 22: 232–244. https://doi.org/10.1053/j.ackd.2014.12.005
Lichtstein D, Ilani A, Rosen H, Horesh N, Singh SV, Buzaglo N, Hodes A (2018) Na+,K+-ATPase Signaling and Bipolar Disorder. Int J Mol Sci 19: 2314. https://doi.org/10.3390/ijms19082314
Markov AG, Fedorova AA, Kravtsova VV, Bikmurzina AE, Okorokova LS, Matchkov VV, Cornelius V, Amasheh S, Krivoi1 II (2020) Circulating Ouabain Modulates Expression of Claudins in Rat Intestine and Cerebral Blood Vessels. Int J Mol Sci 21: 5067. https://doi.org/10.3390/ijms21145067
Agalakova NI, Kolodkin NI, Adair CD, Trashkov AP, Bagrov AY (2021) Preeclampsia: Cardiotonic Steroids, Fibrosis, Fli1 and Hint to Carcinogenesis. Int J Mol Sci 22: 1941. https://doi.org/10.3390/ijms22041941
Shelkovnikova TA, Peters OM, Deykin AV, Connor-Robson N, Robinson H, Ustyugov AA, Bachurin SO, Ermolkevich TG, Goldman IL, Sadchikova ER, Kovrazhkina EA, Skvortsova VI, Ling SC, Da Cruz S, Parone PA, Buchman VL, Ninkina NN (2013) Fused in sarcoma (FUS) protein lacking nuclear localization signal (NLS) and major RNA binding motifs triggers proteinopathy and severe motor phenotype in transgenic mice. J Biol Chem 288: 25266–25274. https://doi.org/10.1074/jbc.M113.492017
Bogdanova A, Petrushanko IY, Hernansanz-Agustin P, Martínez-Ruiz A (2016) “Oxygen Sensing” by Na,K-ATPase: These Miraculous Thiols. Front Physiol 7: 314. https://doi.org/10.3389/fphys.2016.00314
Chuang C-C, Zhou T, Olfert IM, Zuo L (2018) Hypoxic Preconditioning Attenuates Reoxygenation-Induced Skeletal Muscle Dysfunction in Aged Pulmonary TNF-α Overexpressing Mice. Front Physiol 9:1 720. https://doi.org/10.3389/fphys.2018.01720
Vyskocil F, Di Gregorio F, Gorio A (1985) The facilitating effect of gangliosides on the electrogenic (Na+/K+) pump and on the resistance of the membrane potential to hypoxia in neuromuscular pReparation. Pflugers Arch 403: 1–6. https://doi.org/10.1007/BF00583273
De Angelis C, Haupert GT Jr (1998) Hypoxia triggers release of an endogenous inhibitor of Na+-K+-ATPase from midbrain and adrenal. Am J Physiol 274: F182–F188. https://doi.org/10.1152/ajprenal.1998.274.1.F182
Lewis P, O’Halloran KD (2016) Diaphragm Muscle Adaptation to Sustained Hypoxia: Lessons from Animal Models with Relevance to High Altitude and Chronic Respiratory Diseases. Front Physiol 7: 623. https://doi.org/10.3389/fphys.2016.00623
Iannello S, Milazzo P, Belfiore F (2007) Animal and human tissue Na,K-ATPase in normal and insulin-resistant states: regulation, behaviour and interpretative hypothesis on NEFA effects. Obes Rev 8: 231–251. https://doi.org/10.1111/j.1467-789X.2006.00276.x
Kawakami K, Onaka T, Iwase M, Homma I, Ikeda K (2005) Hyperphagia and obesity in Na,K-ATPase alpha2 subunit-defective mice. Obes Res 13: 1661–1671. https://doi.org/10.1038/oby.2005.204
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова