

Российский физиологический журнал им. И.М. Сеченова, 2022, T. 108, № 11, стр. 1453-1467
Влияние доксорубицина на реактивность брыжеечных артерий крыс Вистар
Г. Т. Иванова *
Институт физиологии им. И.П. Павлова РАН
Санкт-Петербург, Россия
* E-mail: ivanovagt@infran.ru
Поступила в редакцию 11.07.2022
После доработки 11.09.2022
Принята к публикации 27.09.2022
- EDN: KDHIKP
- DOI: 10.31857/S0869813922110036
Аннотация
Доксорубицин применяется в качестве противоопухолевого препарата, однако он оказывает негативное влияние на сердечно-сосудистую систему. Основное внимание уделяется кардиотоксическому эффекту, а механизмы его действия на сосуды изучены недостаточно. В исследовании оценивалось влияние доксорубицина на реактивность брыжеечных артерий крыс. Экспериментальной группе крыс Вистар вводили однократно внутрибрюшинно доксорубицин (4 мг/кг), контрольным животным – физиологический раствор в равном объеме. Через 6 нед. исследовали реактивность брыжеечных артерий на ацетилхолин (АХ), нитропруссид и фенилэфрин. Для уточнения механизмов эндотелийзависимой вазодилатации использовали блокаторы: NO-синтазы – L-NAME, циклооксигеназы – индометацин, К+-каналов – тетраэтиламмоний. Для оценки реактивности артерий in vivo проводили микрофото- и видеорегистрацию диаметра брыжеечных артерий крыс (микроскоп Биомед МС-1Т-ZOOM и камера Basler BASLER acA4600-10uc), полученные данные обрабатывали в программе MultiMedia Catаlog. Показано, что доксорубицин модифицировал реактивность брыжеечных артерий крыс: повышал констрикторные реакции фенилэфрина и угнетал эндотелийзависимую вазорелаксацию при действии АХ. При этом введение крысам доксорубицина приводило к снижению эффективности NO-зависимых механизмов регуляции тонуса сосудов (оцененных по разнице в амплитуде АХ-индуцированной вазодилатации до и после применения L-NAME), а также к уменьшению вызванной нитропруссидом релаксации артерий по сравнению с контрольными животными. Доксорубицин не оказывал влияния на эффективность опосредованных простагландинами и Ca2+-активируемыми К+-каналами промежуточной и малой проводимости механизмов эндотелийзависимой вазодилатации.
В настоящее время во всем мире наблюдается значительный рост онкологических заболеваний, которые занимают первое место среди причин смертности [1]. Применение препаратов антрациклинового ряда, в частности доксорубицина (DOX), оказывает выраженный противоопухолевый эффект, однако сопровождается значительным токсическим влиянием на различные органы и ткани, в частности на миокард и сосуды. Проблема определения механизмов токсического влияния противоопухолевых препаратов на сердце и сосуды, а также поиск путей предотвращения опасных патологических эффектов – одна из острых проблем современной медицины [2–4].
Основное внимание уделяется изучению токсического действия DOX на структуру и функцию миокарда, однако до настоящего времени механизмы этого влияния точно не определены [5–9]. Выраженная кардиотоксичность позволила считать введение DOX методом моделирования хронической сердечной недостаточности у экспериментальных животных [8, 10, 11]. Bведение различных доз DOX сопровождается ухудшением насосной функции сердца, активизацией процессов ремоделирования миокарда, повреждением гладкомышечных клеток (ГМК) и эндотелия коронарных артерий [10, 12].
Химиотерапевтическое действие DOX связано с его способностью к интеркаляции с ДНК, приводящей к нарушению репарации, и ингибированию расщепляющей нити ДНК топомеразы II типа, в результате чего происходит апоптоз клеток опухоли [13, 14]. Однако подобное влияние DOX оказывает и на другие ткани организма, в частности на кардиомиоциты, что во многом обуславливает кардиотоксичность этого препарата. Кроме того, введение DOX приводит к образованию активных форм кислорода, индуцируя окислительный стресс, к которому особенно чувствительны клетки миокарда и эндотелий сосудов [15, 16], а также к нарушению функции митохондрий, вызывая снижение уровня АТФ [17].
Действие DOX на сосуды изучается, как правило, в экспериментах, проводимых in vitro, с использованием изолированных сосудистых фрагментов, которые либо инкубировались в растворе с данным препаратом [18–21], либо выделены от животных, которым его предварительно вводили [22]. Исследования влияния DOX на сосуды в условиях сохранения естественного кровотока и целостности сосудистой системы единичны. Разнообразие результатов о действии DOX на сосуды, представленных в литературе, является следствием различий в условиях эксперимента: использование крыс, мышей и других животных, различия в сроках наблюдения после введения DOX, многообразие протоколов введения, дозировок препарата, типа сосудов. В исследованиях в той или иной мере отмечается токсическое влияние DOX на функциональное состояние сосудов, сопровождающееся повреждением эндотелия, снижением эндотелийзависимой вазорелаксации, увеличением жесткости сосудистой стенки [4, 23]. Механизмы этих влияний остаются малоизученными. Обсуждается также непосредственное влияние DOX на гладкомышечные клетки сосудов [24].
Целью нашего исследования было оценить реактивность брыжеечных артерий, а также участие отдельных механизмов эндотелийзависимой вазодилатации у крыс, получивших однократное введение 4 мг/кг DOX, в условиях сохранения целостности сосудистой системы и естественного кровотока (in vivo).
МЕТОДЫ ИССЛЕДОВАНИЯ
Эксперименты проведены на половозрелых самцах крыс стока Вистар (Центр коллективного пользования “Биоколлекция” Института физиологии им. И.П. Павлова РАН), исходной массой 280–320 г. Все манипуляции с животными проводились в соответствии с принципами Базельской декларации при одобрении этической комиссии Института физиологии им. И.П. Павлова РАН. Крысы были случайным образом распределены на 2 группы по 30 особей в каждой. Животные первой группы (DOX-группа) получали однократную инъекцию растворенного в физиологическом растворе DOX, внутрибрюшинно, из расчета 4 мг/кг массы тела крысы. Крысам контрольной группы был однократно внутрибрюшинно введен физиологический раствор в количестве, эквивалентном объему вводимого раствора DOX. Все животные получали стандартный пищевой рацион и имели доступ к пище и воде ad libitum. Животные содержались в равных условиях, по 5 особей в клетке, при температуре в помещении 20–22°C и световом режиме 12 ч свет/12 ч темнота. Длительность наблюдения составила 6 нед. после инъекции.
Перед началом и окончанием эксперимента у бодрствующих крыс измеряли артериальное давление (АД) манжеточным методом на хвосте, используя электроманометр фирмы (ELEMA, Швеция). Среднее трех последовательных измерений считали величиной АД.
Через 6 нед. после введения DOX или физиологического раствора у крыс, наркотизированных тилетамин/золазепамом (10 мг/кг массы крысы, Zoletil 100, Vibrac, Франция), при необходимости дополнительно вводили дробно не более 50% от первоначально рассчитанной дозы тилетамин/золазепама, исследовали реактивность брыжеечных артерий in vivo. Эвтаназию животных осуществляли введением избыточной дозы наркоза до остановки дыхания.
Для оценки ацетилхолин-индуцированной релаксации сосудов у наркотизированной крысы через разрез стенки брюшной полости (длиной 1.5 см) выводили петлю тонкого кишечника, расправляли и помещали в специальный желоб, который фиксировали в термостабилизируемой при темпратуре 37°С камере с протекающим физиологическим солевым раствором следующего состава (ммоль/л): NaCl – 120.4; KCl – 5.9; CaCl2 – 2.5; MgCl2 – 1.2; NaH2PO4 – 1.2; NaHCO3 – 15.5; глюкоза – 11.5. Физиологический раствор сатурировали газовой смесью, состоящей из 95% О2 и 5% СО2. Перед тестированием сосуд стабилизировали в ванночке в течение 30 мин. Затем проводили исследования согласно выбранному протоколу. В работу брали сосуды диаметром 200–350 мкм, имеющие выраженный вазоконстрикторный эффект на фенилэфрин (ФЭ).
Для оценки эндотелийзависимой и эндотелийнезависимой вазодилатации и-спользовали введение в раствор ацетилхолин хлорида (АХ, Sigma-Aldrich, США) 1 × 10–5 моль/л или нитропруссида натрия (ICN Biomedicals, США) 1 × × 10–6 моль/л на фоне предварительного сокращения сосуда ФЭ (Sigma-Aldrich, США) 1 × 10–5 моль/л. Амплитуду дилатации выражали в процентах от амплитуды констрикции, вызванной ФЭ.
С целью оценки участия отдельных механизмов в сосудистых реакциях на АХ использовали соответствующие блокаторы: блокатор NO-синтазы L-NAME (Nω-Nitro-L-arginine methyl ester hydrochloride, ICN Biomedicals) 1 × 10–4 моль/л; блокатор циклооксигеназы индометацин (Indomethacin, Sigma-Aldrich), 1 × 10–5 моль/л; неспецифический блокатор К+-каналов – тетраэтиламмоний (ТЭА), (Tetraethylammonium chloride, Sigma-Aldrich) 1 × 10–3 моль/л. Через 30 мин инкубации сосудов в растворе с блокатором вводили ФЭ с последующим добавлением АХ. Вклад различных механизмов в АХ-индуцированную вазорелаксацию рассчитывали как разницу в амплитуде дилатации сосуда при действии АХ до применения соответствующего ингибитора (блокатора) и после инкубации с ним. Маточные растворы АХ, нитропруссида натрия, L-NAME, ТЭА приготовлялись на основе дистиллированной воды. Индометацин растворяли в диметилсульфоксиде (Вектон, Россия). Полученный раствор добавляли в физиологический раствор до достижения необходимой концентрации индометацина. Диметилсульфоксид, при разведении его физиологическим раствором в соотношении 1 : 1000, не вызывал значимых изменений реактивности брыжеечных артерий.
Для уточнения реактивности сосудов на AX исследовали его зависимое от концентрации действие на предварительно сокращенные ФЭ брыжеечные артерии, для этого оценивали кумулятивный эффект постепенного повышения концентрации вводимого в ванночку АХ (1 × 10–10–1 × 10–5 моль/л) на амплитуду вазодилатации.
Для оценки реактивности артерий проводили фото- и видеорегистрацию диаметра брыжеечных артерий крыс in vivo с использованием микроскопа Биомед МС-1Т-ZOOM (Россия) и камеры Basler BASLER acA4600-10uc (Германия), обработка результатов осуществлялась с помощью программы MultiMedia Catаlog (MMC).
При статистической обработке результатов использовали программу Statistica v.12. Полученные данные представляли в виде среднего с его стандартной ошибкой. Для анализа полученных данных использовали однофакторный дисперсионный анализ (ANOVA). Различия считали статистически значимыми при р < 0.05.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Однократное введение 4 мг/кг DOX через 6 нед. не приводило к изменению массы тела у крыс, в среднем она составляла 333 ± 18 г у DOX-группы и 330 ± 20 г у контрольной. DOX также не оказывал влияния на величину АД: 121 ± 7 мм рт. ст. – у DOX-группы и 123 ± 5 мм рт. ст. – у контрольной.
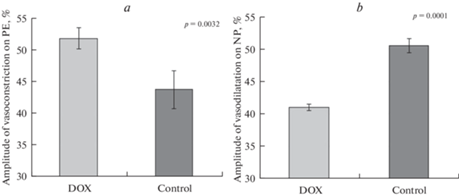
Анализ результатов измерения диаметра брыжеечных артерий in vivo при действии вазоактивных агентов показал, что однократное введение 4 мг/кг DOX через 6 нед. приводит к изменению реактивности сосудов. Так, у крыс DOX-группы было отмечено увеличение амплитуды вазоконстрикции на ФЭ в среднем на 18.5% (р = 0.003) по сравнению с контрольными крысами (рис. 1а). Кроме того, у крыс получавших DOX, наблюдалось снижение реактивности сосудов на экзогенный NO (рис. 1b): амплитуда дилатации предварительно сокращенных ФЭ сосудов крыс DOX-группы при добавлении в камеру нитропруссида натрия была в среднем на 18.8% меньше, чем у контрольных животных (р = 0.0001).
Рис. 1.
Влияние доксорубицина на реактивность брыжеечных артерий крыс. (a) – влияние фенилэфрина (PE, 1 × 10–5 моль/л) на диаметр брыжеечных артерий крыс. По оси ординат – амплитуда вазоконстрикции, выраженная в процентах от исходного диаметра сосуда. (b) – влияние нитропруссида натрия (NP, 1 × 10–6 моль/л) на диаметр предварительно сокращенных фенилэфрином брыжеечных артерий крыс. По оси ординат – амплитуда вазодилатации, выраженная в процентах от амплитулы вызванной фенилэфрином констрикции сосуда. Экспериментальные группы: DOX – крысы, получившие однократное введение 4 мг/кг доксорубицина; Сontrol – контрольная группа. Данные представлены в виде среднего значения ± стандартная ошибка. р – значимость различий между группами.
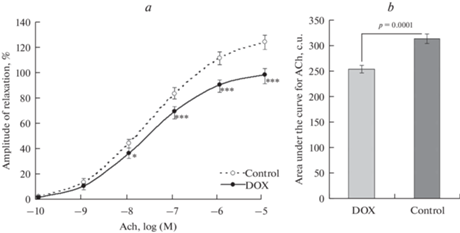
При исследовании эндотелийзависимой дилатации брыжеечных артерий крыс оценивали кумулятивный эффект введения в ванночку с физиологическим раствором АХ в ступенчато повышающейся концентрации от 1 × 10–10 до 1 × 10–5 моль/л. Результаты показали, что у крыс DOX-группы, по сравнению с контрольной, наблюдается ослабление вазодилатации: амплитуда релаксации значимо уменьшалась уже при концентрации АХ 1 × 10–8 моль/л, а при повышении концентрации АХ различия между группами усиливались (рис. 2а). Кроме того, расчет площади под кривой амплитуды вазодилатации при различной концентрации АХ показал, что эффективность дилатации брыжеечных артерий крыс, получивших DOX, была ниже на 19.1% (р = 0.0001) по сравнению с контрольными животными (рис. 2b).
Рис. 2.
Влияние доксорубицина на ацетилхолин-индуцированную дилатацию брыжеечных артерий крыс. (а) – амплитуда дилатации предварительно сокращенных фенилэфрином артерий при действии ацетилхолина (ACh): по оси абсцисс – логарифм концентрации ацетилхолина, по оси ординат – амплитуда релаксации, выраженная в процентах от амплитуды вызванной фенилэфрином констрикции (1 × 10–5 моль/л). (b) – площадь под кривой амплитуды вазодилатации при различной концентрации ацетилхолина: по оси ординат – площадь под кривой амплитуды дилатации артерий при действии ацетилхолина, усл. ед. Экспериментальные группы: DOX – крысы, получившие однократное введение 4 мг/кг доксорубицина; Control – контрольная группа. Данные представлены в виде среднего значения ± стандартная ошибка. * – различия между группами значимы при р < 0.05, *** – различия между группами значимы при р < 0.001.
Для выяснения роли эндотелийзависимой вазодилатации оценивали изменение диаметра брыжеечных артерий при действии АХ в концентрации 1 × 10–5 моль/л. Амплитуда АХ-индуцированной релаксации артерий крыс, получивших DOX, была в среднем на 15.8% меньше, чем у контрольных животных, р = 0.0011 (рис. 3). Была проведена оценка реакции сосудов на АХ в условиях применения блокаторa эндотелиальной NO-синтазы L-NAME. Следует отметить, что диаметр сосудов, измеренный до введения в омывающий раствор L-NAME, значимо не изменялся после инкубации с данным блокатором. Амплитуда дилатации при действии АХ на фоне применения L-NAME у крыс DOX-группы оказалась в среднем на 26.4% больше, чем у контрольных животных. Однако сравнение величины вызванной АХ релаксации в отсутствие блокаторов и при блокаде NO-синтазы показало, что у животных DOX-группы инкубация брыжеечных артерий с L-NAME приводила к снижению амплитуды дилатации на 45.5 ± 4.3% по сравнению с релаксацией без блокаторов, а у контрольных – на 66.8 ± 5.7%, р < 0.001.
Рис. 3.
Влияние доксорубицина на ацетилхолин-индуцированную дилатацию предварительно сокращенных фенилэфрином брыжеечных артерий крыс при отсутствии блокаторов и при блокаде NO-синтазы инкубацией сосудов с L-NAME (1 × 10–4 моль/л). По оси ординат – амплитуда релаксации при действии ацетилхолина (ACh, 1 × 10–5 моль/л), выраженная в процентах от амплитуды вызванной фенилэфрином (1 × 10–6 моль/л) констрикции. Экспериментальные группы: DOX – крысы, получившие однократное введение 4 мг/кг доксорубицина; Сontrol – контрольная группа. Данные представлены в виде среднего значения ± стандартная ошибка. p – значимость различий.

Для оценки роли простагландинов в вызванной АХ вазодилатации у крыс, получавших DOX, проводили предварительную инкубацию сосудов в физиологическом растворе, содержащем блокатор циклооксигеназы индометацин и блокатор NO-синтазы L-NAME. Исследования показали, что DOX не влиял на опосредованную циклооксигеназой АХ-индуцированную релаксацию артерий, так как инкубация с индометацином и L-NAME приводила к равному снижению вазодилатации у исследованных животных: уменьшение амплитуды составляло 12.8 ± 1.8% у контрольной и 11.9 ± 1.7% – у DOX-группы (p > 0.05) по сравнению с амплитудой релаксации в условиях блокады только NО-синтазы (рис. 4).
Рис. 4.
Влияние доксорубицина на ацетилхолин-индуцированную дилатацию предварительно сокращенных фенилэфрином брыжеечных артерий крыс при блокаде циклооксигеназы инкубацией сосудов с индометацином (Indo), блокаде К+-каналов – инкубацией с тетраэтиламмонием (ТЕА), в комплексе с блокатором NО-синтазы L-NAME (1 × 10–4 моль/л). По оси ординат – амплитуда релаксации при действии ацетилхолина (ACh, 1 × 10–5 моль/л), выраженная в процентах от амплитуды вызванной фенилэфрином (1 × 10–6 моль/л) констрикции. Экспериментальные группы: DOX – крысы, получившие однократное введение 4 мг/кг доксорубицина; Сontrol – контрольная группа. Данные представлены в виде среднего значения ± стандартная ошибка. p – значимость различий.

Для оценки роли К+-каналов в индуцированной АХ вазодилатации использовали неспецифический блокатор К+-каналов ТЭА при одновременном ингибировании синтеза NО и простациклина. Показано, что у животных, получавших DOX, амплитуда вызванной АХ релаксации после инкубации сосудов с комплексом трех блокаторов (ТЭА, L-NAME, индометацин) снижалась на 52.9 ± 4.8%, тогда как у контрольных – только на 23.1 ± 4.1%, p < 0.001 по сравнению с дилатацией в условиях блокады NО-синтазы и циклооксигеназы (рис. 4). Одновременное применение трех блокаторов не приводило к полному прекращению АХ-индуцированной дилатации сосудов, при этом амплитуда релаксации брыжеечных артерий у крыс DOX-группы и контрольной группы значимо не отличалась.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Клинические исследования показывают токсический эффект DOX на состояние сердечно-сосудистой системы у больных, получающих химиотерапию данным препаратом [25]. В экспериментальных исследованиях основное внимание уделялось кардиотоксическому действию DOX [5–9], а влияние DOX на состояние сосудов изучено недостаточно. В публикациях существует большой разброс результатов, являющихся следствием как разнообразия видов экспериментальных животных, так и различий в количестве введенного DOX, протоколе его введения и длительности сроков наблюдения, а также типа сосуда. Кроме того, основное количество работ проведено на изолированных сосудах, взятых от подвергнутых введению DOX животных, либо на изолированных сосудистых сегментах, которые инкубировали с DOX. Наше исследование имело цель изучить влияние DOX на реактивность сосудов в условиях in vivo при сохранении естественного кровотока через брыжеечные артерии. Для этого мы использовали видео- и фотофиксацию динамики диаметра брыжеечных артерий, снабжающих кровью тонкий кишечник.
В проведенном исследовании оценивались как эндотелийзависимые реакции сосудов, так и не опосредованные эндотелием. До настоящего времени неясным остается вопрос о влиянии DOX на реактивность ГМК, в частности о его влиянии на величину ФЭ-индуцированной вазоконстрикции. Имеются сообщения как о снижении вазоконстрикции на ФЭ у животных, получавших однократное введение DOX [22, 26], так и o увеличении [27], в частности сообщается об усилении констрикторной реакции на ФЭ при инкубации сосудистых колец с DOX [18].
Полученные нами результаты показали, что однократное введение 4 мг/кг DOX через 6 недель вызывало небольшое, но статистически значимое увеличение констрикторной реакции брыжеечных артерий на ФЭ. Такая повышенная реактивность может приводить к изменению баланса между вазоконстрикторными и вазодилатирующими влияниями и способствовать нарушению адекватных реакций сосудистого тонуса в ответ на различные воздействия, например, на физическую нагрузку. В отличие от исследований, проводимых на изолированных фрагментах сосудов, в которых не отмечалось повышения констрикторной реакции артерий на ФЭ у крыс, получавших DOX [22, 26], в нашем исследовании небольшое повышение реактивности на ФЭ у животных DOX-группы может быть следствием более физиологических условий эксперимента, когда сохранен естественный кровоток по исследуемому сосуду, что давало возможность участия всех вазоактивных агентов, циркулирующих в крови, в регуляции сосудистого тонуса. Стоит учитывать, что при исследовании на фрагментах сосудов in vitro меняется исходный тонус сосуда, сосудистое кольцо растягивается до стандартного напряжения, и его реактивность на вазоактивные агенты зависит в основном от физических характеристик вырезанного из артерии кольца (длина сегмента, толщина стенок, жесткость) и функционального состояния ГМК и эндотелия. В наших экспериментах введение ФЭ в камеру, где располагалась кишечная петля с исследуемым сосудом, производилось на фоне сохраненного исходного тонуса брыжеечных артерий, когда по всем сосудам продолжала циркулировать кровь, в которой изначально присутствовал определенный баланс различных вазоконстрикторных и вазодилатирующих факторов. Можно предположить, что усиление реактивности на ФЭ у животных DOX-группы, по сравнению с контрольной, опосредовано, в частности, различием исходного уровня NO в ГМК, которое в условиях in vivo может различаться между сосудами крыс разных групп. Это предположение подтверждается исследованиями, которые показали, что у крыс, получавших DOX, повышение реактивности сосудов на ФЭ коррелирует со снижением так называемого базального уровня NO (процентного увеличения вызванного ФЭ сокращения при добавлении блокатора NO-синтазы L-NAME) [28].
В наших экспериментах оценивалось влияние DOX на эндотелийзависимую реактивность артерий. Было показано, что введение DOX приводит к развитию эндотелиальной дисфункции, выраженной в снижении АХ-индуцированной релаксации брыжеечных артерий DOX-крыс по сравнению с контрольной группой. Подавляющий эндотелийзависимую вазорелаксацию эффект DOX отмечался и в исследованиях [16, 23], проведенных на сосудах в условиях in vitro.
Для уточнения механизмов эндотелиальной дисфункции перед проведением пробы на АХ мы использовали предварительную инкубацию сосудов с некоторыми блокаторами, в частности L-NAME, индометацином и ТЭА. Поскольку основные механизмы эндотелийзависимой дилатации связывают с синтезом NO, простациклина и эндотелиальной гиперполяризацией [29, 30], в нашем исследовании мы оценивали влияние DOX на эти механизмы вызванной АХ релаксации брыжеечных артерий.
Одним из основных путей эндотелийзависимой вазодилатации считают NO-зависимый механизм [31]. Его влияние определяется как количеством продуцируемого эндотелием NO, так и чувствительностью ГМК к NO. Чтобы выявить причину снижения эндотелийзависимой релаксации брыжеечных артерий, нами была оценена реактивность сосудов на экзогенный источник NO – нитропруссид натрия. Проведенные исследования показали, что введение крысам DOX приводило к снижению амплитуды вазорелаксации на нитропруссид натрия. Подобный эффект отмечали и другие исследователи, например, после введения мышам однократно 4 мг/кг DOX [32]. Полученное нами у крыс DOX-группы уменьшение реактивности на нитропруссид натрия свидетельствует о снижении чувствительности ГМК к NO, что может приводить к угнетению эндотелийзависимой релаксации сосуда при действии различных вазоактивных агентов даже в случае достаточной продукции NO эндотелием.
С другой стороны, мы также оценили влияние DOX на эндотелийзависимую релаксацию сосудов, связанную с активностью NO-синтаз, продуцирующих NO. При использовании L-NAME, ингибирующего NO-синтазу, и, следовательно, прекращающего продукцию NO эндотелием, мы обнаружили значительное снижение величины АХ-индуцированного ответа брыжеечных артерий у животных обеих групп. Однако DOX вызывал повреждение NO-зависимого механизма дилатации брыжеечных артерий, которое отражалось в уменьшении разницы между амплитудой вызванной АХ релаксации до и после блокады NO-синтазы у крыс DOX-группы по сравнению с сосудами контрольных животных. Следует отметить, что вопрос о влиянии DOX на NO-зависимые механизмы вазодилатации неоднозначен. Так, на сосудах in vitro отмечено отсутствие эффекта DOX на опосредованный активностью эндотелиальной NO-синтазы механизм АХ-индуцируемой вазодилатации [19, 32]. Но при этом сообщается о способности DOX увеличивать транскрипцию эндотелиальной NO-синтазы, что было показано при инкубации с DOX изолированных эндотелиальных клеток аорты [33]. Кроме того, при моделировании доксорубициновой кардиотоксичности отмечается повышение уровня активности как нейрональной, так и индуцибельной NO-синтазы в миокарде [34], а введение DOX животным приводило к повышению уровня NO в миокарде, но не в крови [35]. Таким образом, существует потенциальная возможность DOX посредством различных связанных с NO механизмов модулировать реактивность сосудов. Результаты наших экспериментов на сосудах в условиях in vivo показали, что DOX нарушал NO-зависимые механизмы дилатации брыжеечных артерий, снижая участие данного механизма в АХ-индуцированной вазорелаксации. При этом развитие эндотелиальной дисфункции было опосредовано как снижением чувствительности сосуда к NO, так и нарушением продукции NO эндотелием.
Учитывая, что NO оказывает модулирующее влияние на продукцию эндотелием других вазодилататоров [36, 37], то оценку роли отличных от NO путей АХ-индуцированной дилатации артерий мы проводили на фоне ингибирования NO-синтазы.
Оценивая влияние DOX на связанные с простагландинами механизмы эндотелийзависимой вазодилатации, перед введением АХ проводили инкубацию сосудов в растворе с индометацином, селективным блокатором циклооксигеназы, в условиях ингибирования NO-синтазы. Циклооксигеназа ответственна за синтез простациклина и некоторых других простаноидов из арахидоновой кислоты [38], блокада циклооксигеназы введением в омывающий раствор индометацина приводит к прекращению продукции простагландинов. Показано, что основным простагландином, синтезируемым эндотелием, и участвующим в эндотелийзависимой вазодилатации, является простациклин [39]. Результаты наших экспериментов показали, что DOX не оказывал влияния на опосредованные простациклином пути АХ-индуцированной релаксации брыжеечных артерий, так как блокада циклооксигеназы в сосудах крыс обеих групп приводила к равному по величине снижению амплитуды дилатации по сравнению с амплитудой в условиях применения только L-NAME. Таким образом, снижение реактивности брыжеечных артерий на АХ у животных, получавших DOX, в наших исследованиях не было связано с изменением простагландин-зависимых механизмов вазодилатации.
Кроме NO- и простагландин-зависимых путей, одним из важнейших механизмов вазодилатации является эндотелиальная гиперполяризация. Независимо от природы гиперполяризующего фактора, дилатация вследствие эндотелиальной гиперполяризации реализуется через активацию различных К+-каналов [40, 41], локализованных как в ГМК, так и в эндотелии [42]. Для оценки влияния DOX на опосредованную активностью Са2+-активируемых К+-каналов АХ-индуцированную дилатацию было проведено исследование реакции брыжеечных артерий на АХ после инкубации сосудов в физиологическом растворе, содержащем неспецифический блокатор К+-каналов ТЭА, который использовали в комплексе с L-NAME и индометацином для предупреждения влияния NO и простагландинов на эффективность других механизмов вазодилатации. Результаты показали, что после введения DOX роль блокируемых ТЭА К+-каналов в АХ-индуцированной релаксации усиливается, так как у крыс DOX-группы снижение амплитуды вазодилатации после применения комплекса трех блокаторов, включая ТЭА, по сравнению с реакцией сосуда в условиях ингибирования только NO- и простагландин-зависимого пути, было выражено значительно сильнее, чем в контрольной группе. Известно, что ТЭА является неспецифическим блокатором как Са2+-активируемых К+-каналов большой проводимости (ВКСа), так и различных потенциал-зависимых К+-каналов (Кv) [43]. Считается, что роль ВКСа в дилатации, опосредованной эндотелиальной гиперполяризацией, не столь значительна, как у Са2+-активируемых К+-каналов промежуточной (IКСа) и малой проводимости (SКСа) [44, 45]. Однако экспериментальные исследования с применением селективного блокатора ВКСа ибериотоксина показали, что ВКСа могут вовлекаться в опосредованную эндотелиальной гиперполяризацией дилатацию брыжеечных артерий, но степень их участия снижается по мере уменьшения размера сосуда [46]. При этом не было обнаружено различий в уровне экспрессии мРНК ВКСа в тканях брыжеечных артерий 1-го и 4-го порядков ветвления, то есть существует потенциальная возможность экспрессии ВКСа в брыжеечных сосудах разного диаметра. Наши исследования проводились преимущественно на сосудах 2-го порядка, поэтому можно предположить, что у крыс, получавших DOX, ослабление NO-зависимых механизмов АХ‑индуцированной вазодилатации могло частично компенсироваться усилением активности ВКСа. Условия проведения наших экспериментов не дают возможности судить о влиянии DOX на активность блокируемых ТЭА Кv, изоформы которых обнаружены в брыжеечных артериях крыс [47], при этом Кv могут модулировать величину вазодилатации [48, 49]. Считают, что при концентрации менее 10–3 ммоль/л ТЭА достаточно селективен для ВКСа, [43] и используется для изучения участия ВКСа в реактивности брыжеечных артерий [50, 51]. Несмотря на используемую в нашем исследовании концентрацию ТЭА (1 × 10–3 моль/л), нельзя отрицать вероятность одновременной блокады ВКСа и некоторых Kv, поэтому мы не можем исключать возможность модулирующего влияния Kv на АХ-индуцированную дилатацию сосудов при снижении связанных с NO механизмов.
О влиянии DOX на опосредованную IКСаи SКСа вазодилатацию судили по величине АХ-индуцированной релаксации, развиваемой сосудами после применения комплекса трех блокаторов – L-NAME, индометацина и ТЭА. Результаты показали, что амплитуда релаксации сосудов после инкубации с комплексом блокаторов не отличалась у крыс разных групп. Это свидетельствует об отсутствии влияния DOX на активность IКСаи SКСа в обусловленной эндотелиальной гиперполяризацией релаксации сосудов, исследованных in vivo. Следует отметить, что в литературе имеются сведения, полученные в экспериментах на изолированных сосудах, в которых показано уменьшение вазорелаксации, опосредованной эндотелиальной гиперполяризацией, при снижении экспрессии IКСаи SКСа у животных, получавших DOX [43]. Кроме того, показано, что DOX вызывает нарушение структуры эндотелия [43], что приводит не только к нарушению продукции эндотелием вазоактивных агентов, но и препятствует генерации и распространению эндотелиальной гиперполяризации. Отсутствие влияния DOX на опосредованную активностью IКСаи SКСа дилатацию в наших экспериментах можно объяснить существенно меньшим количеством введенного крысам DOX (4 мг/кг однократно), поскольку в работе Idris-Khodja и соавт. [43] крысам вводили DOX в значительно большем количестве (суммарно 15 мг/кг).
Таким образом, наши исследования показали, что DOX влияет на реактивность брыжеечных артерий крыс, вызывая развитие эндотелиальной дисфункции: DOX повышает констрикторные реакции на ФЭ и снижает эндотелийзависимую вазорелаксацию. При этом ослабление АХ-индуцированной дилатации у крыс, получавших DOX, опосредовано нарушением NO-зависимых механизмов. DOX не оказывал влияния на эффективность опосредованных простагландинами механизмов вызванной АХ вазодилатации, а также не изменял связанных с активностью IКСа и SКСа путей АХ-индуцированной дилатации. Предполагается, что снижение эффективности NO-зависимого пути регуляции сосудистого тонуса у крыс после введения DOX может, в некоторой степени, компенсироваться усилением роли ВКСа в стимулированной АХ релаксации брыжеечных артерий.
Список литературы
Bray F, Laversanne M, Weiderpass E, Soerjomataram I (2021) The ever-increasing importance of cancer as a leading cause of premature death worldwide. Cancer 127(16): 3029–3030. https://doi.org/10.1002/cncr.33587
Cardinale D, Colombo A, Bacchiani G, Tedeschi I, Meroni CA, Veglia F, Civelli M, Lamantia G, Colombo N, Curigliano G, Fiorentini C, Cipolla CM (2015) Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation 131(22): 1981–1988. https://doi.org/10.1161/CIRCULATIONAHA.114.013777
Octavia Y, Tocchetti C, Gabrielson K, Janssens S, Crijns H, Moens A (2012) Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J Mol Cell Cardiol 52: 1213–1225. https://doi.org/10.1016/j.yjmcc.2012.03.006
Luu AZ, Chowdhury B, Al-Omran M, Teoh H, Hess DA, Verma S (2018) Role of Endothelium in Doxorubicin-Induced Cardiomyopathy. JACC Basic Transl Sci 3(6): 861–870. https://doi.org/10.1016/j.jacbts.2018.06.005
Christidi E, Brunham LR (2021) Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell Death Dis 12(4): 339. https://doi.org/10.1038/s41419-021-03614-x
Li D, Yang Y, Wang S, He X, Liu M, Bai B, Tian C, Sun R, Yu T, Chu X (2021) Role of acetylation in doxorubicin-induced cardiotoxicity. Redox Biol 46: 102089. https://doi.org/10.1016/j.redox.2021.102089
Sun X, Chen G, Xie Y, Jiang D, Han J, Chen F, Song Y (2020) Qiliqiangxin improves cardiac function and attenuates cardiac remodelling in doxorubicin-induced heart failure rats. Pharmaceutic Biol 58(1): 417–426. https://doi.org/10.1080/13880209.2020.1761403
Tan C, Zeng J, Wu G, Zheng L, Huang M, Huang X (2021) Xinshuitong Capsule extract attenuates doxorubicin-induced myocardial edema via regulation of cardiac aquaporins in the chronic heart failure rats. Biomed Pharmacother 144: 112261. https://doi.org/10.1016/j.biopha.2021.112261
Wu BB, Leung KT, Poon EN-Y (2022) Mitochondrial-Targeted Therapy for Doxorubicin-Induced Cardiotoxicity. Int J Mol Sci 23(3): 1912. https://doi.org/10.3390/ ijms23031912
Wen J, Zhang L, Liu H, Wang J, Li J, Yang Y, Wang Y, Cai H, Li R, Zhao Y (2019) Salsolinol Attenuates Doxorubicin-Induced Chronic Heart Failure in Rats and Improves Mitochondrial Function in H9c2 Cardiomyocytes. Front Pharmacol 10: 1135. https://doi.org/10.3389/fphar.2019.01135
Huang C, Qiu S, Fan X, Jiao G, Zhou X, Sun M, Weng N, Gao S, Tao X, Zhang F, Chen W (2021) Evaluation of the effect of Shengxian Decoction on doxorubicin-induced chronic heart failure model rats and a multicomponent comparative pharmacokinetic study after oral administration in normal and model rats. Biomed Pharmacother 144: 112354. https://doi.org/10.1016/j.biopha.2021.112354
Pajović V, Kovácsházi C, Kosić M, Vasić M, Đukić L, Brenner GB, Giricz Z, Bajić D, Ferdinandy P, Japundžić-Žigon N (2021) Phenomapping for classification of doxorubicin-induced cardiomyopathy in rats. Toxicol Appl Pharmacol 423: 115579. https://doi.org/10.1016/j.taap.2021.115579
Simůnek T, Stérba M, Popelová O, Adamcová M, Hrdina R, Gersl V (2009) Anthracycline-induced cardiotoxicity: overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol Rep 61(1): 154–171. https://doi.org/10.1016/s1734-1140(09)70018-0
Takemura G, Fujiwara H (2007) Doxorubicin-induced cardiomyopathy from the cardiotoxic mechanisms to management. Prog Cardiovasc Dis 49(5): 330–352. https://doi.org/10.1016/j.pcad.2006.10.002
Zhang Y-W, Shi J, Li Y-J, Wei L (2009) Cardiomyocyte death in doxorubicin-induced cardiotoxicity. Archiv Immunol Therap Exp (Warsz) 57(6): 435–445. https://doi.org/10.1007/s00005-009-0051-8
He H, Wang L, Qiao Y, Zhou Q, Li H, Chen S, Yin D, Huang Q, He M (2020) Doxorubicin Induces Endotheliotoxicity and Mitochondrial Dysfunction via ROS/eNOS/NO Pathway. Front Pharmacol 10: 1531. https://doi.org/10.3389/fphar.2019.01531
Tokarska-Schlattner M, Zaugg M, Zuppinger C, Wallimann T, Schlattner U (2006) New Insights into Doxorubicin-Induced Cardiotoxicity: The Critical Role of Cellular Energetics. J Mol Cell Cardiol 41: 389–405. https://doi.org/10.1016/j.yjmcc.2006.06.009
Henidi HA, Al-Abbasi FA, El-Moselhy MA, El-Bassossy HM, Al-Abd AM (2020) Despite Blocking Doxorubicin-Induced Vascular Damage, Quercetin Ameliorates Its Antibreast Cancer Activity. Oxid Med Cell Longev 2020: 8157640. https://doi.org/10.1155/2020/8157640
den Hartog GJ, Boots AW, Haenen GR, van der Vijgh WJ, Bast A (2003) Lack of inhibition of endothelial nitric oxide synthase in the isolated rat aorta by doxorubicin. Toxicol In Vitro 17(2): 165–167. https://doi.org/10.1016/s0887-2333(03)00007-9
Murata T, Yamawaki H, Yoshimoto R, Hori M, Sato K, Ozaki H, Karaki H (2001) Chronic effect of doxorubicin on vascular endothelium assessed by organ culture study. Life Sci 69(22): 2685–2695. https://doi.org/10.1016/s0024-3205(01)01352-2
Sonowal H, Pal P, Shukla K, Saxena A, Srivastava SK, Ramana KV (2018) Aldose reductase inhibitor, fidarestat prevents doxorubicin-induced endothelial cell death and dysfunction. Biochem Pharmacol 150: 181–190. https://doi.org/10.1016/j.bcp.2018.02.018
Bosman M, Krüger DN, Favere K, Wesley CD, Neutel CHG, Van Asbroeck B, Diebels OR, Faes B, Schenk TJ, Martinet W, De Meyer GRY, Van Craenenbroeck EM, Guns P-JDF (2021) Doxorubicin Impairs Smooth Muscle Cell Contraction: Novel Insights in Vascular Toxicity. Int J Mol Sci 22(23): 12812. https://doi.org/10.3390/ijms222312812
Clayton ZS, Brunt VE, Hutton DA, VanDongen NS, D’Alessandro A, Reisz JA, Ziemba BP, Seals DR (2020) Doxorubicin-Induced Oxidative Stress and Endothelial Dysfunction in Conduit Arteries Is Prevented by Mitochondrial-Specific Antioxidant Treatment. JACC Cardio Oncol 2(3): 475–488. https://doi.org/10.1016/j.jaccao.2020.06.010
Shen B, Ye CL, Ye KH, Zhuang L, Jiang JH (2009) Doxorubicin-induced vasomotion and [Ca(2+)](i) elevation in vascular smooth muscle cells from C57BL/6 mice. Acta Pharmacol Sin 30: 1488–1495. https://doi.org/10.1038/aps.2009.145
Tantawy M, Pamittan FG, Singh S, Gong Y (2021) Epigenetic Changes Associated With Anthracycline-Induced Cardiotoxicity. Clin Translat Sci 14(1): 36–46. https://doi.org/10.1111/cts.12857
Gibson NM, Greufe SE, Hydock DS, Hayward R (2013) Doxorubicin-induced vascular dysfunction and its attenuation by exercise preconditioning. J Cardiovasc Pharmacol 62(4): 355–360. https://doi.org/10.1097/FJC.0b013e31829c9993
Bosman M, Favere K, Neutel CHG, Jacobs G, De Meyer GRY, Martinet W, Van Craenenbroeck EM, Guns PDF (2021) Doxorubicin induces arterial stiffness: A comprehensive in vivo and ex vivo evaluation of vascular toxicity in mice. Toxicol Lett 346: 23–33. https://doi.org/10.1016/j.toxlet.2021.04.015
Vanhoutte PM, Shimokawa H, Feletou M, Tang EH (2017) Endothelial dysfunction and vascular disease – a 30th anniversary update. Acta Physiol (Oxf) 219(1): 22–96. https://doi.org/10.1111/apha.12646
Rubanyi GM (1991) Endothelium-derived relaxing and contracting factors. J Cell Biochem 46(1): 27–36. https://doi.org/10.1002/jcb.240460106
Freed JK, Gutterman DD (2017) Communication Is Key: Mechanisms of Intercellular Signaling in Vasodilation. J Cardiovasc Pharmacol 69(5): 264–272. https://doi.org/10.1097/FJC.0000000000000463
Olukman M, Can C, Erol A, Oktem G, Oral O, Cinar MG (2009) Reversal of doxorubicin-induced vascular dysfunction by resveratrol in rat thoracic aorta: Is there a possible role of nitric oxide synthase inhibition? Anadolu Kardiyol Derg 9(4): 260–266.
Idris-Khodja N, Di Marco P, Farhat M, Geny B, Schini-Kerth VB (2013) Grape-Derived Polyphenols Prevent Doxorubicin-Induced Blunted EDH-Mediated Relaxations in the Rat Mesenteric Artery: Role of ROS and Angiotensin II. Evid Based Complement Alternat Med 2013: 516017. https://doi.org/10.1155/2013/516017
Kalivendi SV, Kotamraju S, Zhao H, Joseph J, Kalyanaraman B (2001) Doxorubicin-induced apoptosis is associated with increased transcription of endothelial nitric-oxide synthase. Effect of antiapoptotic antioxidants and calcium. J Biol Chem 276(50): 47266–47276. https://doi.org/10.1074/jbc.M106829200
Deng S, Kruger A, Schmidt A, Metzger A, Yan T, Gödtel-Armbrust U, Hasenfuss G, Brunner F, Wojnowski L (2009) Differential roles of nitric oxide synthase isozymes in cardiotoxicity and mortality following chronic doxorubicin treatment in mice. Naunyn Schmiedebergs Arch Pharmacol 380(1): 25–34. https://doi.org/10.1007/s00210-009-0407-y
Sayed-Ahmed MM, Khattab MM, Gad MZ, Osman AM (2001) Increased plasma endothelin-1 and cardiac nitric oxide during doxorubicin-induced cardiomyopathy. Pharmacol Toxicol 89(3): 140–144. https://doi.org/10.1034/j.1600-0773.2001.d01-148.x
Looft -Wilson RC, Ashley BS, Billig JE, Wolfert MR, Ambrecht LA, Bearden SE (2008) Chronic diet-induced hyperhomocysteinemia impairs eNOS regulation in mouse mesenteric arteries. Am J Physiol Regul Integr Comp Physiol 295(1): R59–R66. https://doi.org/10.1152/ajpregu.00833.2007
Giles TD, Sander GE, Nossaman BD, Kadowitz PJ (2012) Impaired vasodilation in the pathogenesis of hypertension: focus on nitric oxide, endothelial-derived hyperpolarizing factors, and prostaglandins. J Clin Hypertens (Greenwich) 14(4): 198–205. https://doi.org/10.1111/j.1751-7176.2012.00606.x
Larsen BT, Gutterman DD, Hatoum OA (2006) Emerging role of epoxyeicosatrienoic acids in coronary vascular function. Eur J Clin Invest 36(5): 293–300. https://doi.org/10.1111/j.1365-2362.2006.01634.x16634832
Parkington HC, Coleman HA, Tare M (2004) Prostacyclin and endothelium-dependent hyperpolarization. Pharmacol Res 49(6): 509–514. https://doi.org/10.1016/j.phrs.2003.11.012
Jin X, Satoh-Otonashi Y, Zamami Y, Takatori S, Hashikawa-Hobara N, Kitamura Y, Kawasaki H (2011) New molecular mechanisms for cardiovascular disease: contribution of endothelium-derived hyperpolarizing factor in the regulation of vasoconstriction in peripheral resistance arteries. J Pharmacol Sci 116(4): 332–336. https://doi.org/10.1254/jphs.10r30fm
Mandalà M, Gokina N, Barron C, Osol G (2012) Endothelial-derived hyperpolarization factor (EDHF) contributes to PLGF-induced dilation of mesenteric resistance arteries from pregnant rats. J Vasc Res 49: 43–49. https://doi.org/10.1159/000329821
Busse R, Edwards G, Félétou M, Fleming I, Vanhoutte PM, Weston AH (2002) EDHF: bringing the concepts together. Trends Pharmacol Sci 23(8): 374–380. https://doi.org/10.1016/s0165-6147(02)02050-3
Tykocki NR, Boerman EM, Jackson WF (2017) Smooth Muscle Ion Channels and Regulation of Vascular Tone in Resistance Arteries and Arterioles. Compr Physiol 16;7(2): 485–581. https://doi.org/10.1002/cphy.c160011
Grgic I, Kaistha BP, Hoyer J, Kohler R (2009) Endothelial Ca2+ -activated K + channels in normal and impaired EDHF-dilator responses-relevance to cardiovascular pathologies and drug discovery. Br J Pharmacol 157: 509–526. https://doi.org/10.1111/j.1476-5381.2009.00132.x
Crane GJ, Gallagher N, Dora KA, Garland CJ. (2003) Small- and intermediate-conductance calcium-activated K+ channels provide different facets of endothelium-dependent hyperpolarization in rat mesenteric artery. J Physiol 553(Pt1): 183–189. https://doi.org/10.1113/jphysiol.2003.051896
Hilgers RH, Todd J Jr, Webb RC (2006) Regional heterogeneity in acetylcholine-induced relaxation in rat vascular bed: role of calcium-activated K+ channels. Am J Physiol Heart Circ Physiol 291(1): H216–H222. https://doi.org/10.1152/ajpheart.01383.2005
Lu Y, Hanna ST, Tang G, Wang R (2002) Contributions of Kv1.2, Kv1.5 and Kv2.1 subunits to the native delayed rectifier K(+) current in rat mesenteric artery smooth muscle cells. Life Sci 71: 1465–1473. https://doi.org/10.1016/s0024-3205(02)01922-7
González C, Baez-Nieto D, Valencia I, Oyarzún I, Rojas P, Naranjo D, Latorre R (2012) K(+) channels: function-structural overview. Compr Physiol 2(3): 2087–2149. https://doi.org/10.1002/cphy.c110047
Jackson WF (2018) KV channels and the regulation of vascular smooth muscle tone. Microcirculation (1): https://doi.org/10.1111/micc.12421. 10.1111/micc.12421
Lucchesi PA, Belmadani S, Matrougui K (2005) Hydrogen peroxide acts as both vasodilator and vasoconstrictor in the control of perfused mouse mesenteric resistance arteries. J Hypertens 23: 571–579. https://doi.org/10.1097/01.hjh.0000160214.40855.79
Jackson-Weaver O, Paredes DA, Gonzalez Bosc LV, Walker BR, Kanagy NL (2011) Intermittent hypoxia in rats increases myogenic tone through loss of hydrogen sulfide activation of large-conductance Ca2+-activated potassium channels. Circ Res 108: 1439–1447. https://doi.org/10.1161/CIRCRESAHA.110.228999
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова