

Российский физиологический журнал им. И.М. Сеченова, 2022, T. 108, № 3, стр. 304-314
Нейропротекторное действие пальмитоилэтаноламида в литий-пилокарпиновой модели височной эпилепсии
Т. Б. Мелик-Касумов 1, *, М. А. Корнеева 1, А. В. Чуприна 1, А. А. Жабинская 1, А. А. Рожко 1
1 Институт физиологии Национальной академии наук Беларуси
Минск, Беларусь
* E-mail: tigranbmk@gmail.com
Поступила в редакцию 15.12.2021
После доработки 26.01.2022
Принята к публикации 27.01.2022
- EDN: MNPAHU
- DOI: 10.31857/S0869813922030050
Аннотация
Эпилепсия – одно из самых распространенных хронических неврологических заболеваний, при этом около 30% случаев эпилепсии являются фармакорезистентными. В связи с этим остается актуальным более глубокое изучение патогенетических механизмов эпилепсии, а также поиск новых соединений, обладающих противосудорожной или нейропротекторной активностью. Оценка эффектов эндогенных соединений в экспериментальных моделях эпилепсии может быть полезна для решения обеих этих задач. Целью данной работы являлось изучение нейропротекторного действия пальмитоилэтаноламида (PEA) в литий-пилокарпиновой модели височной эпилепсии у крыс Вистар в возрасте 10–11 нед. PEA применяли подкожно в дозе 10 мг/кг через час после купирования эпилептического статуса и далее ежедневно в течение 7 дней. Нейропротекторные свойства PEA оценивали по изменению поведения в открытом поле через 20 дней после эпистатуса, а также по изменению плотности нейронов в области СА1 и хилуса дорсального и вентрального гиппокампа и степени обратного прорастания мшистых волокон в молекулярный слой зубчатой извилины через 21 день после эпилептического статуса. Установлено, что применение PEA в течение первой недели после эпистатуса приводит к сохранению некоторых особенностей поведения, характерных для здоровых животных, сохранению плотности нейронов в области СА1 дорсального гиппокампа, а также уменьшает степень обратного прорастания мшистых волокон как в дорсальном, так и в вентральном гиппокампе. Таким образом, результаты исследования указывают на умеренное нейропротекторное действие PEA в модели височной эпилепсии.
Эпилепсия – одно из самых распространенных хронических неврологических заболеваний, проявляющееся в периодической пароксизмальной электрической активности головного мозга. Около 30% пациентов сталкиваются с фармакорезистентными формами эпилепсии [1]. В таких случаях купировать эпилептические приступы не удается при использовании двух и более противосудорожных препаратов различных классов и(или) поколений [2]. При отсутствии возможности хирургического лечения фармакорезистентной эпилепсии активно применяются нефармакологические методы – кетогенная диета, стимуляция блуждающего нерва [3]. Однако и эти методы достаточно часто не позволяют уменьшить частоту приступов до приемлемого уровня, ввиду чего некоторые авторы относят их к паллиативным [4, 5]. Сохранение высокой частоты эпилептических приступов ожидаемо приводит к усилению оксидативного стресса и нейровоспаления и, как следствие, к дальнейшему повреждению и гибели нейронов с нарушением когнитивных функций и развитием коморбидных психических расстройств [6, 7].
Вышесказанное указывает на актуальность более глубокого исследования патогенетических механизмов эпилепсии, равно как и на необходимость дальнейшего поиска соединений, обладающих противосудорожной или нейропротекторной активностью. Два этих направления могут объединяться в исследованиях противоэпилептического действия эндогенных соединений, в частности, амидов жирных кислот. Пальмитоилэтаноламид (PEA) является одним из представителей класса эндогенных N-этаноламидов. Применение его в экспериментальных моделях эпилепсии показало умеренное противосудорожное действие [8–10]. Среди механизмов противосудорожного действия PEA выделяют активацию каннабиноидных рецепторов CB1 и CB2 [8, 11], а также ванилоидных рецепторов первого типа TRPv1 [12] преимущественно за счет “эффекта окружения”, который заключается в конкурентном уменьшении деградации эндогенных каннабиноидов гидролазой амидов жирных кислот [13]. Замедление деградации эндогенных каннабиноидов в свою очередь может оказывать непосредственный противосудорожный эффект [14, 15]. Тем не менее существенность вклада “эффекта окружения” в реализацию эффектов каннабимиметиков в последнее время подвергается сомнению [16]. Вместе с тем известно, что наряду с классическими эндоканнабиноидами PEA является лигандом рецепторов, активируемых пролифератором пероксисом α (PPARα) [10, 12, 17]. По-видимому, за счет активации PPARα реализуются такие эффекты эндогенных N-этаноламидов, как усиление чувства насыщения, контроль массы тела, стимуляция липолиза [18]. Противосудорожное действие PEA, вероятно, также объясняется активацией PPARα, так как показано, что другие агонисты этих рецепторов проявляют противосудорожные свойства в пентилентеразоловой и литий-пилокарпиновой моделях эпилепсии [19, 20].
Учитывая то, что PPARα является фактором транскрипции, активация которого приводит к увеличению экспрессии генов, регулирующих интенсивность воспалительных процессов, представляется актуальным исследование нейропротекторного действия PEA в модели височной эпилепсии, так как по современным представлениям нейровоспаление играет существенную роль в прогрессировании нейрональной гибели в ходе эпилептогенеза [7, 21]. Характерное для медиальной височной эпилепсии обратное прорастание мшистых волокон, приводящее к формированию патологически возбудимых нейронных сетей, по некоторым представлениям может быть также связано со степенью нейровоспаления и снижения плотности нейронов [22]. В связи с этим в настоящей работе нейропротекторные свойства PEA в модели височной эпилепсии у крыс оценивали по изменению свободного поведения, плотности нейронов в областях гиппокампа, а также степени обратного прорастания мшистых волокон в молекулярный слой зубчатой извилины гиппокампа.
МЕТОДЫ ИССЛЕДОВАНИЯ
Литий-пилокарпиновая модель височной эпилепсии
Исследование выполнено в соответствии с принципами Базельской декларации и рекомендациями комиссии по биоэтике Института физиологии НАН Беларуси. Эпилептический статус (SE) индуцировали у самцов крыс линии Вистар в возрасте 10–11 нед. За сутки до эпистатуса животным внутрибрюшинно вводили хлорид лития (Sigma-Aldrich, 127 мг/кг). На следующий день проводили подкожную инъекцию метскополамина бромида (Sigma-Aldrich, 1 мг/кг) для блокирования нежелательных периферических холинергических эффектов пилокарпина и через 30 мин – внутрибрюшинную инъекцию пилокарпина (Sigma-Aldrich, 25 мг/кг). Крыс помещали в индивидуальные клетки и проводили видеорегистрацию судорог. Тяжесть судорог оценивали на основании модифицированной шкалы Расина [23]: 0 – отсутствие судорог, 1 – лицевые клонусы, 2 – клонические судороги шеи и миоклонические вздрагивания, 3 – клонические судороги передних конечностей без подъема на задние лапы, 4 – клонические судороги передних конечностей с подъемом на задние лапы, 5 – генерализованные клонические судороги с падением на бок, 6 – последующие тонические судороги передних и/или задних конечностей. Развитие эпилептического статуса считали состоявшимся, если у животного после первоначального приступа не ниже 4 баллов по шкале Расина судорожная активность сохранялась далее на уровне не ниже 3 баллов. Через 90 мин после манифестации эпистатуса для его купирования животным внутрибрюшинно проводили инъекцию диазепама (Белмедпрепараты, 20 мг/кг). Через час после этого подкожно вводили PEA (Sigma-Aldrich, SE + PEA, 10 мг/кг, n = 12), растворенный в чистом диметилсульфоксиде (Sigma-Aldrich, 2 мл/кг). Группе отрицательного контроля вводили чистый диметилсульфоксид в дозе 2 мл/кг (SE + Vehicle, n = 12). Далее животным ежедневно в течение 7 суток повторяли подкожные инъекции PEA или чистого диметилсульфоксида. Группа интактного контроля (Control, n = 12) не получала никаких экспериментальных воздействий.
Тест “Открытое поле”
Первый тест проводили за сутки до развития эпилептического статуса непосредственно перед введением хлорида лития. Открытое поле представляло собой арену 1.2 м в диаметре с высотой стенок 0.5 м. Поле было расчерчено на 16 секторов вдоль стенок, 8 секторов в центре и один центральный круг. Все процедуры, связанные с тестированием, проводились с 9.00 до 12.00. Крыс помещали в центр поля и засекали время выхода из центрального круга (замирание), подсчитывали число пересеченных периферических и центральных секторов, число стоек на задних лапах, актов груминга, суммарное число актов уринации и дефекации. Наблюдение продолжали в течение 3 мин. Повторный тест проводили на 20-е сутки после эпилептического статуса.
Морфологические исследования
Морфологические исследования включали в себя оценку плотности нейронов в областях СА1 и хилуса дорсального и вентрального гиппокампа (окрашивание по Нисслю), а также оценку распространения мшистых волокон и степени их прорастания в молекулярный слой зубчатой извилины (окрашивание по Тимму). На 21-е сутки после эпилептического статуса животных внутрибрюшинно наркотизировали тиопенталом натрия (Синтез, 100 мг/кг), вскрывали грудную и брюшную полости и проводили трансаортальную перфузию. Для этого делали надрез на верхушке сердца, через левый желудочек в аорту вводили металлический катетер, который фиксировали зажимом. Для оттока крови и перфузионных растворов делали надрез на правом предсердии. Далее животное перфузировали физиологическим раствором с фосфатным буфером (рН 7.4) в течение 10 мин при поддержании давления на уровне 80 мм рт. ст. Критерием удовлетворительной перфузии считали побледнение кожных покровов и печени. После этого перфузионную систему переключали на раствор сульфида натрия (Sigma-Aldrich, 150 мМ) c фосфатным буфером (pH 7.4) и продолжали перфузию еще 5 мин. Критерием удовлетворительной перфузии считали почернение печени и подергивание конечностей. По завершении перфузии животных декапитировали, мозг доставали из черепной коробки, разделяли на полушария и помещали в раствор сульфида натрия еще на 45–60 мин. Затем полушария фиксировали в 10%-ном формалине в течение суток. Далее на 1.5 ч перекладывали в 2.5%-ный глутаровый альдегид и снова фиксировали в формалине 24 ч. Далее готовили парафиновые блоки, после чего из них изготавливали серийные фронтальные (правое полушарие, дорсальный гиппокамп) и поперечные (левое полушарие, вентральный гиппокамп, в промежутке –2.5... –4.5 мм от брегмы) срезы толщиной 8 мкм, которые окрашивали раствором по Тимму. Для этого готовили 200 мл раствора состава: 5.1 г лимонной кислоты, 4.7 г цитрата натрия, 3.4 гидрохинона, 170 мг нитрата серебра, 40 г гуммиарабика. Срезы содержали в растворе в темноте при температуре 26°С в течение 45 мин, далее при 60°С еще 20 мин. После промывки срезов применяли контрокрашивание тионином по Нисслю. В гиппокампе с использованием программы ImageJ определяли плотность нейронов в области CA1 и хилуса, а также степень прорастания мшистых волокон в молекулярный слой зубчатой извилины. Последний показатель оценивали по величине относительной оптической плотности (OD) по формуле:
Статистическая обработка данных
Статистическую обработку производили в программе Statistica 10. Проверку нормальности распределения проводили с помощью критерия Шапиро–Уилка. Для межгруппового сравнения применяли однофакторный дисперсионный анализ, для анализа изменений при повторном проведении теста в группе – однофакторный дисперсионный анализ с повторными измерениями. Для апостериорного попарного сравнения использовали критерий Тьюки. Различия считали достоверными при р < 0.05. Данные на графиках представлены в виде среднего значения и стандартной ошибки.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Анализ результатов тестов в открытом поле показал, что большинство показателей, а именно длительность периода замирания, количество актов груминга, количество пройденных секторов в центре поля и количество актов уринации/дефекации ни в одной из экспериментальных групп не претерпевало существенных изменений после 20 дней эксперимента. Среди проанализированных данных наиболее информативными оказались показатели количества пройденных секторов по краю поля и количество стоек на задних лапах (рис. 1).
Рис. 1.
Изменение количества пройденных периферических секторов (а) и количества стоек на задних лапах (b) в открытом поле на начальном этапе (Before SE) и через 20 дней (20 days after SE) в группе здоровых крыс (Control), в группе отрицательного контроля (SE + Vehicle) и в группе крыс, получавших PEA (SE + PEA); * – достоверные отличия от соответствующего значения контроля, ^ – достоверные изменения по сравнению с первоначальными данными в группе.
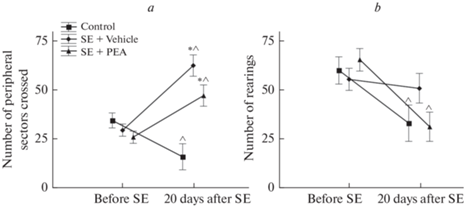
Установлено, что у контрольных животных при повторном помещении в открытое поле через 20 суток происходит уменьшение пройденной дистанции по краю поля в среднем в 2.2 раза (p = 0.04). У здоровых животных уменьшается также и количество стоек на задних лапах – в среднем в 1.8 раза (p = 0.028). Вместе с тем, у животных в группе SE + Vehicle напротив, наблюдалось увеличение двигательной активности: количество пройденных периферических секторов возросло в 2.1 раза (p < 0.001). При этом двигательная активность этих животных после повторного предъявления теста была в 3.9 раза больше аналогичных значений контрольной группы (p < 0.001). Кроме того, для группы отрицательного контроля не обнаружено характерное для здоровых животных уменьшение количества стоек. Применение PEA в аналогичных условиях также приводило к увеличению количества пройденных секторов по краю поля. В данном случае показатель увеличился в среднем в 1.8 раза (p < 0.001), достоверно отличался от аналогичных значений контроля в 3 раза (p < 0.001) и не отличался от значений группы SE + Vehicle (p = 0.15). Кроме того, у крыс, получавших после эпистатуса PEA, так же как и у здоровых животных, отмечалось уменьшение в 2.1 раза количества стоек на задних лапах (p < 0.01).
Морфология дорсальной и вентральной части гиппокампа крыс в группе SE + Vehicle имела ряд особенностей, характерных для литий-пилокарпиновой модели височной эпилепсии [25]. Как в дорсальном (рис. 2), так и в вентральном (рис. 3) гиппокампе в области Аммонова рога (в большей степени в СА1) и хилуса гиппокампа на фоне глиоза часто встречались нейроны с признаками некробиоза (перицеллюлярный отек, гиперхромные клетки). Сходная картина отмечалась в случае группы SE + PEA.
Рис. 2.
Результаты исследования морфологии дорсального гиппокампа. (a) –репрезентативные микрофотографии дорсального гиппокампа (Hipp, увеличение ×40), СА1 области (CA1, увеличение ×400) и хилуса (Hilus (H), увеличение ×200); 1 – внешний молекулярный слой, 2 – внутренний молекулярный слой; (b) – показатель относительной оптической плотности (OD) в молекулярном слое зубчатой извилины; (с) – плотность нейронов в области СА1; (d) – плотность нейронов в хилусе. * – достоверно отличается от значений группы контроля; # – достоверно отличается от значений группы SE + Vehicle.
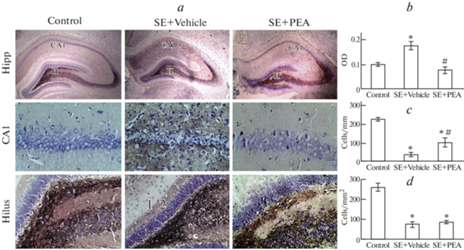
Рис. 3.
Результаты исследования морфологии вентрального гиппокампа. (a) –репрезентативные микрофотографии дорсального гиппокампа (Hipp, увеличение ×40), СА1 области (CA1, увеличение ×400) и хилуса (Hilus (H), увеличение ×200); (b) – показатель относительной оптической плотности (OD) в молекулярном слое зубчатой извилины; (с) – плотность нейронов в области СА1; (d) – плотность нейронов в хилусе. * – достоверно отличается от значений группы контроля; # – достоверно отличается от значений группы SE + Vehicle.
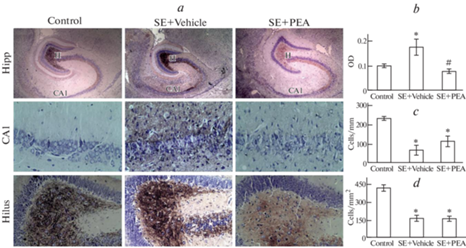
Морфометрический анализ показал, что в группе SE + Vehicle во всех исследованных областях плотность нейронов ожидаемо была значительно меньше показателей контрольной группы (рис. 2c–d и 3c–d) – в среднем в 2.5–6.9 раз (для всех зон p < 0.001). Аналогичные изменения были обнаружены практически во всех зонах у крыс, получавших PEA после развития эпилептического статуса. Вместе с тем, в группе SE + PEA отмечено некоторое сохранение плотности нейронов в дорсальной области CA1 (рис. 2с). Здесь плотность нейронов была в 2.3 раза меньше контрольных значений (p < 0.001), однако в 3.1 раза больше значений группы SE + Vehicle (p = 0.036).
Анализ относительной оптической плотности в молекулярном слое зубчатой извилины показал, что у животных в группе SE + Vehicle через 21 сутки после развития эпистатуса степень обратного прорастания мшистых волокон была значительно выше, чем в контроле (рис. 2b и 3b). Так, в дорсальном гиппокампе OD для группы SE + Vehicle был в среднем в 1.7 раза выше (рис. 2b, p < 0.01), чем у контрольных животных, в вентральном – в 1.9 раза (рис. 3b, p = 0.016). В аналогичных условиях у крыс, получавших PEA, не было обнаружено достоверных отличий от контрольных показателей, тогда как отличия от группы SE + Vehicle обнаружены как в дорсальном (рис. 2b, p < 0.001), так и в вентральном (рис. 3b, p < 0.01) гиппокампе.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Результаты исследования указывают на умеренно выраженный нейропротекторный эффект PEA в модели височной эпилепсии с учетом выбранного дизайна. В условиях свободного поведения в открытом поле у здоровых животных отмечалось уменьшение двигательной активности и количества стоек. Угасание обоих показателей при повторном тесте может говорить о снижении у здоровых животных интереса к открытому полю как к знакомому пространству. У крыс в группе отрицательного контроля, напротив, количество стоек не изменялось, что говорит о сохранении интереса к полю как к новому пространству. Применение PEA нивелирует этот эффект модели, сохраняя когнитивные функции животных. Развитие модели приводит также к тому, что двигательная активность животных в открытом поле растет. Такой эффект отмечен ранее также при однократном использовании теста [26]. В данном случае PEA не оказывает существенного влияния на показатель. Важно отметить, что увеличение двигательной активности выше первоначальной указывает в большей степени на развитие характерной для модели гиперреактивности и тревожности [27], нежели на повышенный интерес к открытому полю.
Результаты гистологических исследований показывают, что наиболее существенно применение PEA влияет на плотность нейронов в дорсальной области СА1, а также на степень прорастания мшистых волокон в молекулярный слой зубчатой извилины, причем как в дорсальном, так и в вентральном гиппокампе. Согласно современным представлениям о механизмах развития пилокарпиновой модели височной эпилепсии пилокарпин после достижения индивидуальной пороговой дозы активирует глутаматергические нейронные сети, вызывая развитие эпилептического статуса. При этом гибель нейронов, отмечаемая впоследствии, определяется эксайтотоксическим действием глутамата [26]. Обратное прорастание мшистых волокон – характерный патоморфологический признак височной эпилепсии. Предполагается, что при этом происходит формирование нейронных сетей, которые компенсируют функции утраченных нейронов других областей гиппокампа и вместе с тем обладают патологической сверхвозбудимостью [28, 29]. При этом ключевым периодом являются первые дни после начала эпилептогенеза.
Нейропротекторное действие эндогенных амидов пальмитиновой кислоты было показано ранее в моделях других патологий. В частности, показан антиневропатический эффект PEA при нейротоксическом действии оксалиплатина в эксперименте [30]. В другом исследовании доказано, что наряду с противовоспалительным действием препарат на основе PEA оказывал выраженный нейропротекторный эффект в мышиной модели сосудистой деменции [31]. В настоящем исследовании был использован дизайн, позволяющий в некоторой степени разграничить противосудорожное и предполагаемое нейропротекторное действие PEA в условиях эпилептогенеза. Так, PEA вводился через час после купирования эпилептического статуса диазепамом, после чего инъекции повторялись ежесуточно еще в течение 7 дней, то есть в период наиболее существенных изменений в медиальной височной доле. В этот период отмечается формирование так называемого “гиппокампального склероза” – гибель нейронов преимущественно в области Аммонова рога с одновременным развитием астро- и микроглиоза, в особенности в хилусе и секторе Соммера – области перехода СА1 в подножие гиппокампа [32]. Вместе с тем, в латентный период по определению не отмечаются спонтанные судороги, в частности, у крыс Вистар они обычно не отмечаются ранее 11 суток после эпилептического статуса [33]. Таким образом, представляется маловероятным, что обнаруженный нейропротекторный эффект PEA определяется его противосудорожным действием, описанным ранее. Вероятно, сохранение плотности нейронов в дорсальной области СА1 определяется непосредственным влиянием PEA на процессы нейровоспаления и обмена веществ через активацию PPARα. В этой связи представляется интересным то, что в вентральной области СА1, несмотря на общую тенденцию, достоверного эффекта PEA не произвел. Последние исследования показывают, что септо-темпоральная гетерогенность гиппокампа, в том числе различная степень подверженности эпилептогенезу, может определяться отличиями в особенностях метаболизма в дорсальном и вентральном гиппокампе. Так, установлено, что в дорсальном гиппокампе катаболические процессы в системе “гликолиз–окислительное фосфорилирование” несколько больше смещены к последнему, чем в вентральном гиппокампе [34]. Эта особенность может играть существенную роль в большей подверженности дорсального гиппокампа эпилептогенезу [35, 36], так как гликолиз может иметь некоторое преимущество при повышенном энергопотреблении в условиях активного нейровоспаления. В том числе поэтому активация PPARα как основной механизм описанного нейропротекторного действия PEA представляется наиболее вероятной. PPARα является транскрипционным фактором и одним из главных регуляторов липидного обмена, его активация приводит к ускорению β-окисления жирных кислот в митохондриях и к кетогенезу, увеличивая таким образом долю окислительного фосфорилирования в общем катаболизме [37]. Поэтому активация его рецепторов в дорсальном гиппокампе может иметь больший эффект, чем в вентральном. Не стоит также забывать и об упомянутом ранее непосредственном противовоспалительном действии агонистов PPARα. Агонисты других рецепторов PPAR также в эксперименте показали противосудорожный и нейропротекторный эффекты. В частности, агонист PPARγ росиглитазон предотвращает чрезмерную автофагию в височной доле в течение 24 ч после эпистатуса [38]. При этом сохранение нейронов происходит на фоне повышения активности антиоксидантного фермента супероксиддисмутазы. По-видимому, ключевую роль в таком нейропротекторном эффекте росиглитазона играет снижение реактивности микроглии в ответ на повреждающее действие длительных судорог [39]. Среди других агонистов PPARα рецепторов свою эффективность в пентилентетразоловой и литий-пилокарпиновой модели показал фенофибрат [20]. Вместе с тем, эффекты PEA представляют особый интерес, так как это соединение вырабатывается в организме человека и животных, и может играть существенную роль в естественных процессах предотвращения нейродеструкции.
Список литературы
Santana-Gomez CE, Engel J Jr, Staba R (2021) Drug Resistant Epilepsy and the hypothesis of intrinsic severity: What about the High-Frequency Oscillations? Epilepsia Open. https://doi.org/10.1002/epi4.12565
Hakami T (2021) Efficacy and tolerability of antiseizure drugs. Ther Adv Neurol Disord 14:17562864211037430. https://doi.org/10.1177/17562864211037430
Guery D, Rheims S (2021) Clinical Management of Drug Resistant Epilepsy: A Review on Current Strategies. Neuropsychiatr Dis Treat 17: 2229–2242. https://doi.org/10.2147/NDT.S256699
Gough SM, Casella A, Ortega KJ, Hackam AS (2021) Neuroprotection by the Ketogenic Diet: Evidence and Controversies. Front Nutr 8: 782657. https://doi.org/10.3389/fnut.2021.782657
Marras CE, Colicchio G, De Palma L, De Benedictis A, Di Gennaro G, Cavaliere M, Cesaroni E, Consales A, Asioli S, Caulo M, Villani F, Zamponi N (2020) Health Technology Assessment Report on Vagus Nerve Stimulation in Drug-Resistant Epilepsy. Int J Environ Res Public Health 17(17): 6150. https://doi.org/10.3390/ijerph17176150
Novais F, Loureiro S, Andrea M, Figueira ML, Pimentel J, Pestana LC (2020) May the right-side epileptogenic zone be a predictor of psychiatric comorbidity in people with refractory epilepsy? Laterality 25(3): 275–284. https://doi.org/10.1080/1357650X.2019.1662431
Bazhanova ED, Kozlov AA, Litovchenko AV (2021) Mechanisms of Drug Resistance in the Pathogenesis of Epilepsy: Role of Neuroinflammation. A Literature Review. Brain Sci 11(5): 663. https://doi.org/10.3390/brainsci11050663
Sheerin AH, Zhang X, Saucier DM, Corcoran ME (2004) Selective Antiepileptic Effects of N-Palmitoylethanolamide, a Putative Endocannabinoid. Epilepsia 45(10): 1184–1188. https://doi.org/10.1111/j.0013-9580.2004.16604.x
Lambert DM, Vandevoorde S, Diependaele G, Govaerts SJ, Robert AR (2001) Anticonvulsant Activity of N-Palmitoylethanolamide, a Putative Endocannabinoid, in Mice. Epilepsia 42(3): 321–327. https://doi.org/10.1046/j.1528-1157.2001.41499.x
Citraro R, Russo E, Scicchitano F, van Rijn CM, Cosco D, Avagliano C, Russo R, D’Agostino G, Petrosino S, Guida F, Gatta L, van Luijtelaar G, Maione S, Di Marzo V, Calignano A, De Sarro G (2013) Antiepileptic action of N-palmitoylethanolamine through CB1 and PPAR-a receptor activation in a genetic model of absence epilepsy. Neuropharmacology 69: 115–126. https://doi.org/10.1016/j.neuropharm.2012.11.017
Aghaei I, Rostampour M, Shabani M, Naderi N, Motamedi F, Babaei P, Khakpour-Taleghani B (2015) Palmitoylethanolamide attenuates PTZ-induced seizures through CB1 and CB2 receptors. Epilepsy Res 117: 23–28. https://doi.org/10.1016/j.eplepsyres.2015.08.010
Ambrosino P, Soldovieri MV, Russo C, Taglialatela M (2013) Activation and desensitization of TRPV1 channels in sensory neurons by the PPARα agonist palmitoylethanolamide. Br J Pharmacol 168(6): 1430–1444. https://doi.org/10.1111/bph.12029
Okine BN, Madasu MK, McGowan F, Prendergast C, Gaspar JC, Harhen B, Roche M, Finn DP (2016) N-palmitoylethanolamide in the anterior cingulate cortex attenuates inflammatory pain behaviour indirectly via a CB1 receptor-mediated mechanism. Pain 157(12): 2687–2696. https://doi.org/10.1097/j.pain.0000000000000687
de Castro Medeiros D, Cota VR, Oliveira ACP, Moreira FA, Moraes MFD (2020) The Endocannabinoid System Activation as a Neural Network Desynchronizing Mediator for Seizure Suppression. Front Behav Neurosci 14: 603245. https://doi.org/10.3389/fnbeh.2020.603245
Shubina L, Aliev R, Kitchigina V (2015) Attenuation of kainic acid-induced status epilepticus by inhibition of endocannabinoid transport and degradation in guinea pigs. Epilepsy Res 111: 33–44. https://doi.org/10.1016/j.eplepsyres.2015.01.003
Cogan PS (2020) The 'entourage effect' or 'hodge-podge hashish': the questionable rebranding, marketing, and expectations of cannabis polypharmacy. Expert Rev Clin Pharmacol 13(8): 835–845. https://doi.org/10.1080/17512433.2020.1721281
Melis M, Pillolla G, Luchicchi A, Muntoni AL, Yasar S, Goldberg SR, Pistis M (2008) Endogenous fatty acid ethanolamides suppress nicotineinduced activation of mesolimbic dopamine neurons through nuclear receptors. J Neurosci 28(51): 13985–13994. https://doi.org/10.1523/JNEUROSCI.3221-08.2008
O'Sullivan SE, Kendall DA (2010) Cannabinoid activation of peroxisome proliferator-activated receptors: potential for modulation of inflammatory disease. Immunobiology 215(8): 611–616. https://doi.org/10.1016/j.imbio.2009.09.007
Saha L, Bhandari S, Bhatia A, Banerjee D, Chakrabarti A (2014) Anti-kindling Effect of Bezafibrate, a Peroxisome Proliferator-activated Receptors Alpha Agonist, in Pentylenetetrazole Induced Kindling Seizure Model. J Epilepsy Res 4(2). https://doi.org/10.14581/jer.14011
Porta N, Vallée L, Lecointe C, Bouchaert E, Staels B, Bordet R, Auvin S (2009) Fenofibrate, a peroxisome proliferator-activated receptor-α agonist, exerts anticonvulsive properties. Epilepsia 50(4): 943–948. https://doi.org/10.1111/j.1528-1167.2008.01901.x
Sun L, Shan W, Yang H, Liu R, Wu J, Wang Q (2021) The Role of Neuroinflammation in Post-traumatic Epilepsy. Front Neurol 12: 646152. https://doi.org/10.3389/fneur.2021.646152
Twible C, Abdo R, Zhang Q (2021) Astrocyte Role in Temporal Lobe Epilepsy and Development of Mossy Fiber Sprouting. Front Cell Neurosci 15: 725693. https://doi.org/10.3389/fncel.2021.725693
Racine RJ (1972) Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 323: 281–294. https://doi.org/10.1016/0013-4694(72)90177-0
Liu C, Lin Y, Tang N, Liu H, Hsieh C (2012) Neuroprotective Effect of Uncaria rhynchophylla in Kainic Acid-Induced Epileptic Seizures by Modulating Hippocampal Mossy Fiber Sprouting, Neuron Survival, Astrocyte Proliferation, and S100B Expression. Evid Based Complement Alternat Med 2012: 194790. https://doi.org/10.1155/2012/194790
Linard B, Ferrandon A, Koning E, Nehlig A, Raffo E (2010) Ketogenic diet exhibits neuroprotective effects in hippocampus but fails to prevent epileptogenesis in the lithium-pilocarpine model of mesial temporal lobe epilepsy in adult rats. Epilepsia 51(9): 1829–1836. https://doi.org/10.1111/j.1528-1167.2010.02667.x
Smolensky IV, Zubareva OE, Kalemenev SV, Lavrentyeva VV, Dyomina AV, Karepanov AA, Zaitsev AV (2019) Impairments in cognitive functions and emotional and social behaviors in a rat lithium-pilocarpine model of temporal lobe epilepsy. Behav Brain Res 372: 112044. https://doi.org/10.1016/j.bbr.2019.112044
Minjareza B, Camarenab HO, Haramatia J, Rodríguez-Yañezc Y, Mena-Munguíaa S, Buriticáb J, García-Lealb O (2017) Behavioral changes in models of chemoconvulsant-induced epilepsy: A review. Neurosci Biobehav Rev 83: 373–380. https://doi.org/10.1016/j.neubiorev.2017.10.016
Kulikov AA, Naumova AA, Aleksandrova EP, Glazova MV, Chernigovskaya EV (2021) Audiogenic kindling stimulates aberrant neurogenesis, synaptopodin expression, and mossy fiber sprouting in the hippocampus of rats genetically prone to audiogenic seizures. Epilepsy Behav 125: 108445. https://doi.org/10.1016/j.yebeh.2021.108445
Buckmaster PS (2014) Does mossy fiber sprouting give rise to the epileptic state? Adv Exp Med Biol 813: 161–168. https://doi.org/10.1007/978-94-017-8914-1_13
Di Cesare Mannelli L, Pacini A, Corti F, Boccella S, Luongo L, Esposito E, Cuzzocrea S, Maione S, Calignano A, Ghelardini C (2015) Antineuropathic Profile of N-Palmitoylethanolamine in a Rat Model of Oxaliplatin-Induced Neurotoxicity. PLoS One 10(6): e0128080. https://doi.org/10.1371/journal.pone.0128080
Siracusa R, Impellizzeri D, Cordaro M, Crupi R, Esposito E, Petrosino S, Cuzzocrea S (2017) Anti-Inflammatory and Neuroprotective Effects of Co-UltraPEALut in a Mouse Model of Vascular Dementia. Front Neurol 8: 233. https://doi.org/10.3389/fneur.2017.00233
Walker MC (2015) Hippocampal Sclerosis: Causes and Prevention. Semin Neurol 35(3): 193–200. https://doi.org/10.1055/s-0035-1552618
Chauvière L, Doublet T, Ghestem A, Siyoucef SS, Wendling F, Huys R, Jirsa V, Bartolomei F, Bernard C (2012) Changes in interictal spike features precede the onset of temporal lobe epilepsy. Ann Neurol 71(6): 805–814. https://doi.org/10.1002/ana.23549
Brancati GE, Rawas C, Ghestem A, Bernard C, Ivanova AI (2021) Spatio-temporal heterogeneity in hippocampal metabolism in control and epilepsy conditions. Proc Natl Acad Sci U S A 118(11): e2013972118. https://doi.org/10.1073/pnas.2013972118
Häussler U, Bielefeld L, Froriep UP, Wolfart J, Haas CA (2012) Septotemporal position in the hippocampal formation determines epileptic and neurogenic activity in temporal lobe epilepsy. Cereb Cortex 22(1): 26–36. https://doi.org/10.1093/cercor/bhr054
Lévesque M, Biagini G, de Curtis M, Gnatkovsky V, Pitsch J, Wang S, Avoli M (2021) The pilocarpine model of mesial temporal lobe epilepsy: Over one decade later, with more rodent species and new investigative approaches. Neurosci Biobehav Rev 130: 274–291. https://doi.org/10.1016/j.neubiorev.2021.08.020
Grabacka M, Pierzchalska M, Dean M, Reiss K (2016) Regulation of Ketone Body Metabolism and the Role of PPARα. Int J Mol Sci 17(12): 2093. https://doi.org/10.3390/ijms17122093
Peng Y, Chen L, Qu Y, Wang D, Zhu Y, Zhu Y (2021) Rosiglitazone Prevents Autophagy by Regulating Nrf2-Antioxidant Response Element in a Rat Model of Lithium-pilocarpine-induced Status Epilepticus. Neuroscience 455: 212–222. https://doi.org/10.1016/j.neuroscience.2020.10.026
Peng J, Wang K, Xiang W, Li Y, Hao Y, Guan Y (2019) Rosiglitazone polarizes microglia and protects against pilocarpine-induced status epilepticus. CNS Neurosci Ther 25(12): 1363–1372. https://doi.org/10.1111/cns.13265
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова