

Российский физиологический журнал им. И.М. Сеченова, 2022, T. 108, № 4, стр. 490-504
Действие анакинры на экспрессию генов рецепторов, активируемых пероксисомным пролифератором в мозге крыс в литий-пилокарпиновой модели эпилепсии
А. И. Рогинская 1, А. В. Дёмина 1, А. А. Коваленко 1, М. В. Захарова 1, А. П. Шварц 1, Т. Б. Мелик-Касумов 2, О. Е. Зубарева 1, *
1 Институт эволюционной физиологии и биохимии им. И.М. Сеченова РАН
Санкт-Петербург,, Россия
2 Институт физиологии Национальной академии наук Беларуси
Минск, Беларусь
* E-mail: ZubarevaOE@mail.ru
Поступила в редакцию 24.02.2022
После доработки 03.03.2022
Принята к публикации 04.03.2022
- EDN: KRJRRX
- DOI: 10.31857/S0869813922040070
Аннотация
В последние годы широко обсуждается роль нейровоспаления в механизмах эпилептогенеза. Одним из факторов, влияющих на воспалительные процессы в мозге, может быть изменение экспрессии ядерных транскрипционных факторов, в частности, рецепторов, активируемых пероксисомным пролифератором (PPARs). Агонисты этих рецепторов обладают выраженным нейропротекторным действием в моделях эпилепсии. Исследования, проведенные на клетках различных тканей организма, выявляют тесную функциональную связь, существующую между генами PPARs, провоспалительного цитокина интерлейкина-1β (IL-1β) и противовоспалительного цитокина – антагониста рецепторов интерлейкина 1 (IL-1ra). Целью данной работы явилось изучение особенностей экспрессии генов Ppars в структурах мозга крыс в литий-пилокарпиновой модели эпилепсии и оценка возможного влияния IL-1ra (препарат анакинра) на эти показатели. Пилокарпин вводили крысам Вистар в возрасте 7–8 нед., через сутки после инъекций LiCl. Введение анакинры осуществляли в течение недели после пилокарпина (первая инъекция 100 мкг/кг через час после судорог, далее – 5 дней по 100 мкг/кг и 2 дня по 50 мкг/кг), после чего производился забор образцов мозга для биохимического анализа. Оценку экспрессии генов Ppara, Ppard и Pparg производили методом обратной транскрипции с последующей полимеразной цепной реакцией в реальном времени в дорзальном и вентральном гиппокампе, височной коре и миндалине. Показано, что пилокарпин-индуцированные судороги приводят к усилению экспрессии генов Ppard и Pparg в вентральном гиппокампе и снижению экспрессии гена Ppara во всех обследованных областях мозга. Анакинра усиливает снижение экспрессии гена Ppara, не влияет на продукцию мРНК Ppard и нивелирует усиление экспрессии гена Pparg. Таким образом, экспрессия генов Ppars в мозге меняется в процессе эпилептогенеза, анакинра разнонаправлено регулирует продукцию мРНК Ppara и Pparg, но не влияет на экспрессию гена Ppard.
Височная эпилепсия является одним из наиболее распространенных и трудно поддающихся лечению неврологических заболеваний [1]. Около трети случаев эпилепсии остаются лекарственно-устойчивыми [2]. Во многом сложности лечения эпилепсии связаны с недостаточной изученностью ее патогенетических механизмов. Наряду с традиционными представлениями, рассматривающими в качестве основного патогенетического механизма эпилепсии нарушение баланса активности возбуждающих (глутаматергических) и тормозных (ГАМК-ергических) систем мозга [3], в последние годы широко обсуждается возможная роль нейровоспаления в развитии эпилептических процессов [4, 5]. Нейровоспаление связано, в частности, с повышенным синтезом в клетках мозга провоспалительных цитокинов – интерлейкина 1β (IL-1β), интерлейкина-6 (IL-6) и фактора некроза опухоли α (TNFα) [6]. Роль провоспалительных цитокинов в развитии психоневрологических нарушений в экспериментальных моделях эпилепсии была доказана, в том числе с помощью введений антагониста рецепторов интерлейкина-1 (препарат анакинра, аналог эндогенного белка IL-1ra) [7–9]. Негативное действие провоспалительных цитокинов в мозге ограничивается рядом нейропротекторных механизмов, один из которых связан с активацией рецепторов, активируемых пероксисомными пролифераторами (PPARs) [10].
PPARs – это ядерные транскрипционные факторы, регулирующие экспрессию целого ряда генов, участвующих в обмене углеводов и липидов, и других процессах, включая клеточную дифференцировку и апоптоз [11–13]. Семейство PPARs включает три типа рецепторов: PPARα, PPARβ и PPARγ, экспрессия которых выявлена в различных клетках организма, включая клетки центральной нервной системы [14]. Несмотря на то, что отдельные виды PPARs отличаются друг от друга по распределению в различных тканях, некоторой специфичности лигандов и физиологическим функциям [15], общим для них является участие в регуляции воспалительных процессов [16, 17]. Предполагается, что именно с противовоспалительным действием во многом связаны нейропротекторные свойства агонистов PPARs, показанные в различных моделях эпилепсии, включая литий-пилокарпиновую модель [18–21]. При этом особенности экспрессии генов PPARs при развитии эпилептических процессов исследованы недостаточно. Следует также отметить тесную двустороннюю функциональную связь, существующую между генами, кодирующими PPARs, IL-1β и IL-1ra, в различных клетках организма. Предполагается, что PPARs могут ингибировать воспалительные сигнальные пути, связанные с интерлейкином-1β в клетках мозга [22]. Доказано, что экспрессия гена IL-1ra в клетках печени регулируется PPARα [23]. С другой стороны, IL-1β вызывает подавление продукции мРНК PPARγ в клетках бурой жировой ткани [24]. Однако возможные эффекты активации рецепторов IL-1 на экспрессию генов PPARs в мозге при развитии эпилептических процессов в мозге остаются неисследованными.
Целью данной работы явилось изучение особенностей экспрессии генов PPARs в структурах мозга крыс в литий-пилокарпиновой модели эпилепсии и оценка возможного влияния анакинры на эти показатели.
МЕТОДЫ ИССЛЕДОВАНИЯ
Эксперименты проводились в соответствии с протоколом работы с лабораторными животными, утвержденным этическим комитетом Института эволюционной физиологии и биохимиии РАН и основанным на директиве Европейского сообщества о гуманном обращении с лабораторными животными (Directive #86/609 for the Care of Laboratory Animals) и инструкциях ARRIVE.
Схема экспериментов
Эксперименты выполнены на самцах крыс Вистар в возрасте 7–8 нед. Крысы содержались в стандартных условиях, со свободным допуском к воде и пище, при 12-часовом цикле освещения (с 8:00 до 20:00 – день, с 20:00 до 8:00 – ночь). Отбор крыс в различные группы был случайным. Схема эксперимента представлена на рис. 1. Использована литий-пилокарпиновая (PC) модель эпилепсии. Особенностью этой модели является то, что однократное введение РС индуцирует развитие длительных эпилептических процессов в мозге [25]. За сутки до введения РC всем крысам, включая контрольных, была произведена инъекция хлорида лития (LiCl) в/б в дозе 127 мг/кг (LiCl; Sigma-Aldrich, Сент-Луис, Миссури, США). За час до введения пилокарпина, животным был введен метилбромидскополамин (1 мг/кг, в/б; Sigma-Aldrich) для блокады периферических мускариновых рецепторов. PC (Sigma-Aldrich) вводили в/б, дробно в дозе 20–40 мг/кг (по 10 мг/кг, 2–4 инъекции с интервалом 30 мин) до развития судорог 4 балла по шкале Racine [26]. По истечении 75 мин, после развития 4-й стадии, эпилептический статус (SE) блокировали введением диазепама (10 мг/кг, в/б; Sigma-Aldrich).
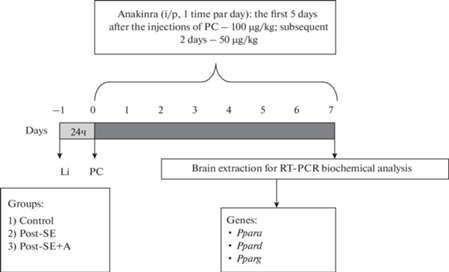
Эксперименты выполнены на 26 животных, которых разделили на три группы: 1) контроль (животным вводился LiCl и физиологический раствор), 2) крысы, перенесшие судороги, нелеченые (LiCl, PC, физиологический раствор), 3) крысы, перенесшие судороги, леченые (LiCl, PC +анакинра). Анакинра (НИИ Особо чистых биопрепаратов, Санкт-Петербург, Россия) вводилась в течение недели, в/б, растворенная в физиологическом растворе, первая инъекция 100 мкг/кг через час после судорог, далее 1 раз в день, 5 дней по 100 мкг/кг в день, 2 дня по 50 мкг/кг в день. Объем вводимого препарата – 0.5 мл/100 г массы тела.
Доза препарата анакинра была подобрана в предшествующей работе, в которой были обнаружено благоприятное воздействие лечения на психоневрологическое состояние животных, которое проверялось с помощью поведенческих и гистологических методов [7].
Крыс декапитировали через 7 дней после введения РС. Мозг быстро извлекали, замораживали и хранили при –80°С. Дорзальную и вентральную области гиппокампа (DH, VH), височную кору (TC) и миндалевидное тело (Am) выделяли по атласу мозга крыс [27] с помощью микротома-криостата OTF5000 (BrightInstruments, Лутон, Великобритания). Данные области мозга были выбраны в связи с их важной ролью в процессах эпилептогенеза. Для оценки экспрессии генов интереса: Ppara (белок – PPARα), Ppard (белок – PPARβ/δ) и Pparg (белок – PPARγ), а также генов домашнего хозяйства, описанных ниже, использовали метод обратной транскрипции с последующей полимеразной цепной реакцией в реальном времени. Для этого сначала экстрагировали тотальную РНК. Выделение проводили с использованием реагента ExtractRNA (Евроген, Москва, Россия) в соответствии с инструкциями производителя. Образцы РНК обрабатывали ДНКазой RQ1 (Promega, Мэдисон, Висконсин, США) в течение 15 мин с последующим осаждением LiCl и промывкой этанолом. Концентрацию и чистоту РНК оценивали с помощью спектрофотометра NanoDrop ™ Lite (ThermoFisherScientific, Уолтем, Массачусетс, США).
Далее проводили обратную транскрипцию. Для синтеза кДНК использовали 1 мкг (VH и Am) или 2 мкг (TC и DH) тотальной РНК, олиго-dT (0.5 мкг на 1 мкг РНК, ООО “ДНК Синтез”, Москва, Россия) и обратную транскриптазу M-MLV (100 ЕД на 1 мкг РНК; Promega, Мэдисон, Висконсин, США) согласно инструкции производителя.
Для проведения полимеразной цепной реакции (ПЦР) в реальном времени применяли технологию TaqMan. ПЦР проводили в триплетах, с использованием полимеразы TaqM (“Алкор Био”, Санкт-Петербург, Россия), зондов и праймеров, синтезированных ООО “ДНК-Синтез” (Москва, Россия). Последовательности использованных праймеров и зондов представлены в табл. 1. Референсные гены для нормализации данных экспрессии были выбраны на основе анализа экспрессии 9 стабильных генов, путем всестороннего ранжирования с использованием онлайн-инструмента RefFinder (https://www.heartcure.com.au/reffinder/) и сравнительных алгоритмов delta CT. В табл. 1 приведены последовательности праймеров и зондов генов домашнего хозяйства, которые были использованы в данной работе. Относительную экспрессию генов рассчитывали с использованием метода 2–ΔΔCt [36], нормирование проводили относительно среднего значения контрольной группы и среднего геометрического для трех наиболее стабильных референсных генов анализируемых областей мозга: DH и VH – Gapdh, Pgk1, Ywhaz; TC – Gapdh, Hprt1, Pgk1; Am – Pgk1, Ppia, Rpl13a.
Таблица 1.
Последовательности праймеров и зондов
| Символ гена и его номер в базе данных | Кодируемый белок | Последовательности праймеров и зондов (прямой, обратный, TaqMan зонд) |
Ссылки |
|---|---|---|---|
| Ppara NM_013196.2 |
Рецептор, активируемый пероксисомным пролифератором – альфа PPARα | AATCCACGAAGCCTACCTGA GTCTTCTCAGCCATGCACAA FAM-AGGCCCGGGTCATACTCGCAGGAA-BHQ1 |
[28] (праймеры) Зонд подобран авторами |
| Ppard NM_013141.2 |
Рецептор, активируемый пероксисомным пролифератором – бета/дельта PPARβ/δ | CAAACCCACGGTAAAGGCGG TGGCTGTTCCATGACTGACC HEX-CCAGGCCTGCAGGCGCCACGCCA-BHQ2 |
Праймеры и зонд подобраны авторами |
| Pparg NM_013124.3 |
Рецептор, активируемый пероксисомным пролифератором – гамма PPARγ | CCTGAAGCTCCAAGAATACC GATGCTTTATCCCCACAGAC HEX-CCCTCATGGCCATCGAGTGCC-BHQ2 |
[29] (праймеры) Зонд подобран авторами |
| Gapdh NM_017008 |
Глицеральдегид-3-фосфатдегидро-геназа GAPDH |
TGCACCACCAACTGCTTAG GGATGCAGGGATGATGTTC R6G-ATCACGCCACAGCTTTCCAGAGGG-BHQ2 |
[30] |
| Rpl13a NM_173340 |
60S рибосомный белок L13a RPL13A | GGATCCCTCCACCCTATGACA CTGGTACTTCCACCCGACCTC FAM-CTGCCCTCAAGGTTGTGCGGCT-BHQ1 |
[31] (праймеры) [32] (зонд) |
| Ppia NM_017101 |
Пептидил-пролилцис-транс-изомераза A/Циклофилин A PPIA/CyPA | AGGATTCATGTGCCAGGGTG CTCAGTCTTGGCAGTGCAGA ROX-CACGCCATAATGGCACTGGTGGCA-BHQ1 |
[33] |
| Hprt1 NM_012583 |
Гипоксантин-гуанинфосфорибозилтрансфераза HGPRT |
TCCTCAGACCGCTTTTCCCGC TCATCATCACTAATCACGACGCTGG FAM-CCGACCGGTTCTGTCATGTCGACCCT-BHQ1 |
[34] (праймеры) [32] (зонд) |
| Pgk1 NM_053291 |
Фосфоглицерат-киназа 1 PGK 1 |
ATGCAAAGACTGGCCAAGCTAC AGCCACAGCCTCAGCATATTTC R6G-TGCTGGCTGGATGGGCTTGGA-BHQ2 |
[35] (праймеры) [32] (зонд) |
| Ywhaz NM_013011 |
14-3-3 белокзета/дельта/
Белок-ингибитор киназы C 1 KCIP-1 |
GATGAAGCCATTGCTGAACTTG GTCTCCTTGGGTATCCGATGTC ROX-TGAAGAGTCGTACAAAGACAGCACGC-BHQ1 |
[35] (праймеры) [32] (зонд) |
Статистический анализ
Статистический анализ был выполнен с использованием пакета программ SPSS Statistics 23 (IBM, Армонк, США). Для проверки нормальности распределения использовался критерии Колмогорова–Смирнова. Равенство дисперсий оценивалось по критерию Ливеня. В случае равенства дисперсий использовался однофакторный дисперсионный анализ с апостериорными сравнениями групп с помощью критерия Тьюки. В случае, когда предположение об однородности дисперсии было нарушено, применялся критерий Уэлча и апостериорный критерий Геймса–Ховелла. Для всех тестов групповые различия считались статистически значимыми при уровне р ≤ 0.05. Данные на рисунках представлены в виде среднего и стандартной ошибки.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
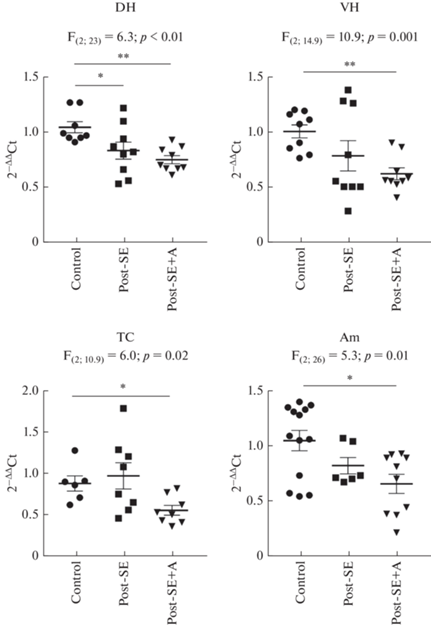
Показано, что у животных, перенесших судороги, экспрессия гена Pparа (рис. 2) снижается в дорзальном гиппокампе (F(2;23) = 6.3; p < 0.01), вентральном гиппокампе (F(2;14.9) = 10.9; p < 0.01), височной коре (F(2;10.9) = 6.0; p = 0.02) и амигдале (F(2;26) = 5.3; p = 0.01). Анакинра не только не уменьшила эти изменения, но даже усилила их. Апостериорные сравнения выявляют достоверное снижение у нелеченых крыс только в дорзальном гиппокампе, у леченых – во всех обследованных структурах мозга.
Рис. 2.
Экспрессия гена Ppara в структурах мозга контрольных крыс (Control) и через 7 дней после пилокарпин-индуцированного эпилептического статуса, в течение которых вводили физиологический раствор (Post-SE) либо анакинру (Post-SE+A). DH – дорзальный гиппокамп, VH – вентральный гиппокамп, TC – височная кора, Am – миндалина. F – критерий Фишера, однофакторный дисперсионный анализ, либо критерий Уэлча. * – p < 0.05; ** – p < 0.01 соответственно, апостериорные сравнения с помощью критерия Тьюки или Геймса–Ховелла.
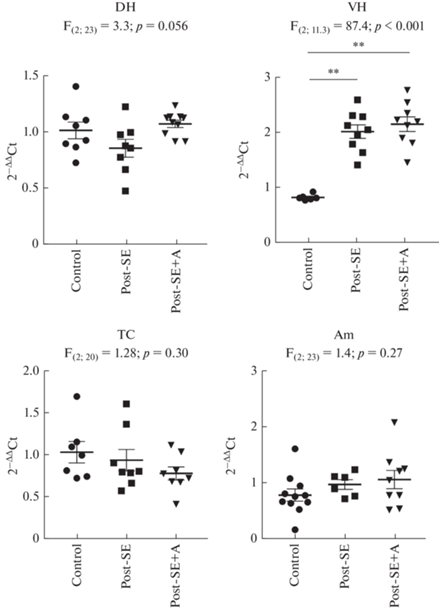
В отношении гена Ppard (рис. 3) достоверные различия между группами были найдены только в вентральном гиппокампе. У нелеченых экспериментальных животных выявлено усиление экспрессии данного гена (F(2;11.3) = 87.4; p < 0.01). Анакинра не влияла на эти изменения.
Рис. 3.
Экспрессия гена Ppard в структурах мозга контрольных крыс (Control) и через 7 дней после пилокарпин-индуцированного эпилептического статуса, в течение которых вводили физиологический раствор (Post-SE) либо анакинру (Post-SE + A). DH – дорзальный гиппокамп, VH – вентральный гиппокамп, TC – височная кора, Am – миндалина. F –критерий Уэлча. ** – p < 0.01, апостериорные сравнения с помощью критерия Геймса–Ховелла.
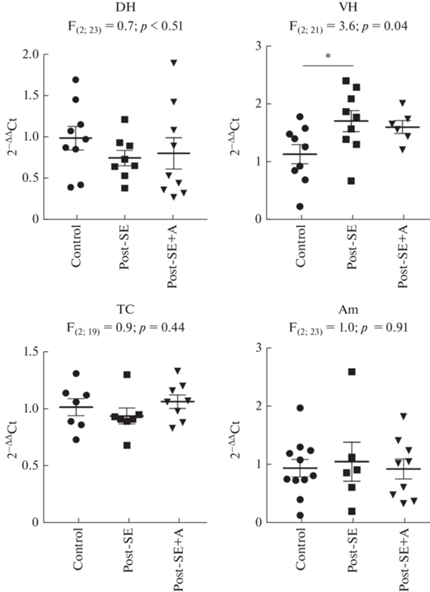
Изменения экспрессии гена Pparg (рис. 4) также были наиболее выражены в вентральном гиппокампе (F(2;21) = 3.6; p = 0.04), однако в этом случае анакинра нивелировала действие пилокарпина: апостериорные сравнения с группой контроля выявляют увеличение уровня мРНК Pparg у нелеченых животных, перенесших пилокарпин-индуцированные судороги, но не у экспериментальных крыс, которым вводили анакинру.
Рис. 4.
Экспрессия гена Pparg в структурах мозга контрольных крыс (Control) и через 7 дней после пилокарпин-индуцированного эпилептического статуса, в течение которых вводили физиологический раствор (Post-SE) либо анакинру (Post-SE + A). DH – дорзальный гиппокамп, VH – вентральный гиппокамп, TC – височная кора, Am – миндалина. F – критерий Фишера, однофакторный дисперсионный анализ. * – p < 0.05, апостериорные сравнения с помощью критерия Тьюки.
Таким образом, пилокарпин-индуцированный эпилептический статус приводил к снижению экспрессии гена Ppara в обследованных областях мозга, а также к усилению экспрессии генов Ppard и Pparg в вентральном гиппокампе. Анакинра усиливала эффекты эпилептического статуса в отношении продукции мРНК Ppara, нивелировала увеличение экспрессии гена Pparg в вентральном гиппокампе и не влияла на продукцию мРНК Ppard.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Первой задачей проведенного исследования было изучение особенностей экспрессии генов семейства Ppar в литий-пилокарпиновой модели эпилепсии. Нами показано, что через 7 дней после пилокарпин-индуцированного эпилептического статуса происходит изменение экспрессии генов Ppara, Ppard и Pparg, причем характер и выраженность этих изменений зависит от типа рецептора и структуры мозга. Ранее особенности экспрессии генов этих рецепторов в экспериментальных моделях эпилепсии исследованы не были. Однако результаты фармакологических исследований, доказавших нейропротекторные свойства агонистов PPARα и PPARγ в различных моделях эпилепсии [18–21, 36, 37] показывают, что обнаруженные нами изменения могут иметь функциональное значение.
Наиболее выраженные изменения обнаружены для гена Ppara, причем направленность этих изменений носила иной характер, чем у генов Ppard и Pparg: продукция мРНК PPARβ/δ и PPARγ увеличивалась, в то время как PPARα – снижалась. Уменьшение экспрессии Ppara может иметь негативные последствия, приводить к снижению антиоксидантных и противовоспалительных процессов [38], способствуя развитию связанных с судорогами неврологических нарушений. С другой стороны, оно, возможно, носит компенсаторный характер, связанный с вовлеченностью PPARα (но не PPARβ и PPARγ) в регуляцию аппетита, массы тела и пищевого поведения [39–41], поскольку было показано, что активация PPARα вызывает сытость и замедляет увеличение массы тела [39]. Известно, что введение пилокарпина и индуцируемый им эпилептический статус приводит к длительному снижению массы тела экспериментальных животных [7]. Возможно, выявленное нами уменьшение экспрессии гена Ppara отражает запуск защитных механизмов, направленных на предотвращение дальнейшей потери массы.
Защитным механизмом, вероятно, является и показанное в данной работе усиление экспрессии гена Pparg в вентральном гиппокампе. Гиппокамп является одной из структур мозга наиболее уязвимых для действия судорог: повреждение и гибель нейронов обнаруживается уже в первые часы и дни после введения пилокарпина [42]. Активация PPARγ опосредует нейропротекцию, помогает уменьшить гибель нейронов при различных видах нервной патологии. В частности, агонисты PPARγ показали большой нейропротекторный потенциал в экспериментальных моделях болезней Паркинсона [43] и Альцгеймера [44], они стимулируют нейрогенез в модели нейродегенеративной патологии, вызванной введением бактериального эндотоксина [45]. Агонист PPARγ росиглитазон предотвращает гибель нейронов в височной коре и гиппокампе [46], а также уменьшает активацию астроцитов и ослабляет когнитивный дефицит [47], развивающиеся после пилокарпин-индуцированного эпилептического статуса. Нейропротекторные свойства агонистов PPARγ реализуются за счет регуляции разных сигнальных путей [48]: благодаря активации Wnt-пути, связанного с клеточной дифференцировкой и регенерацией [49]; активации Nrf2-пути, ответственного за защиту клеток от оксидативного стресса [50], подавления каскадов, опосредованных NF-κB [51], также связанных с оксидативным стрессом и воспалением. Вовлеченность PPARγ в регуляцию воспалительных процессов показана во многих работах (обзор [52]). В частности, выявлено, что агонист этих рецепторов пиоглитазон уменьшает активацию эндотоксин-стимулированных микроглиальных клеток, подавляя продукцию индуцибельной NO-синтазы и провоспалительных цитокинов IL-6, TNF-α и IL-1β [53].
В отличие от PPARα и PPARγ возможная роль PPARβ/δ в эпилептогенезе остается малоизученной. При этом их нейропротекторные свойства показаны на мышах, нокаутных по гену Ppard, при ишемии, вызванной окклюзией средней мозговой артерии [54]. Применение агониста PPARβ/δ GW0742 снижало продукцию IL-1β и TNF-α в мозге мышей в модели индуцированного коллагеназой внутримозгового кровоизлияния [55]. Эти данные свидетельствуют, что активация PPARβ/δ, так же как и активация PPARγ и PPARα, способствует подавлению нейровоспаления. Поэтому активацию экспрессии Ppard, выявленную нами в вентральном гиппокампе, можно рассматривать как защитную реакцию, направленную на уменьшение воспалительных процессов.
При достаточно большом количестве данных о влиянии агонистов PPARs на воспалительные процессы, в частности на продукцию IL-1β в клетках различных тканей, обратный процесс – эффекты активации рецепторов IL-1 на экспрессию генов Ppara, Ppard и Pparg исследованы недостаточно. Нам известна лишь работа Mracek с соавт. [24], показавших, что стимуляция клеток бурой жировой ткани интерлейкином-1β подавляет в них экспрессию гена Pparg. В данной работе нами показано, что активация рецепторов ИЛ-1 может играть определенную роль в усилении экспрессии гена Pparg, вызванной пилокарпин-индуцированным эпилептическим статусом. В отличие от Pparg, экспрессия Ppard не менялась после введения антагониста рецепторов ИЛ-1. Возможно, данный факт свидетельствует о разной роли, которую могут играть эти рецепторы в патогенезе эпилепсии.
Уменьшение экспрессии гена Ppara, выявленное у нелеченых, перенесших судороги крыс, не только не блокировалось введением анакинры, но даже было более выраженным. Можно предположить, что это связано с различной экспрессией PPARα в нейронах и микроглии исследованных областей мозга. Известно, что изменение нейроглиального соотношения является одним из ключевых признаков нейродеструкции в литий-пилокарпиновой модели: количество нейронов в ходе этого процесса уменьшается, в то время как пролиферация и функциональная активность микроглии увеличивается [56]. Исходя из этого, обнаруженный нами эффект уменьшения экспрессии гена Ppara после перенесенных судорог можно объяснить в том числе гибелью нейронов. Это уменьшение может частично компенсироваться повышенной экспрессией PPARα в активированной микроглии. Вместе с тем, в случае применения анакинры, которая может подавлять нейровоспаление за счет антагонизма к рецептору IL-1, экспрессия рецептора на клетках микроглии выражена в меньшей степени, что сказывается на еще большем снижении экспрессии гена Ppara. Это предположение, тем не менее, требует дальнейшего экспериментального подтверждения.
Таким образом, результаты проведенного исследования показывают, что в ходе первой недели после эпилептического статуса в литий-пилокарпиновой модели происходит разнонаправленное изменение экспрессии генов Ppars в гиппокампе: экспрессия Ppara уменьшается в дорсальной области гиппокампа, тогда как экспрессия Ppard и Pparg увеличивается в его вентральной области. Применение анакинры усиливает отмеченный эффект в случае Ppara, при этом он проявляется также в других отделах мозга. В случае Pparg анакинра, напротив, нивелирует увеличение экспрессии гена в вентральном гиппокампе. Наконец, в случае Ppard применение анакинры не влияет на продукцию его мРНК. Отличия во влиянии анакинры на экспрессию различных подтипов PPARs в использованной модели височной эпилепсии позволяет предполагать разную роль, которую они могут играть в регуляции процессов эпилептогенеза.
Список литературы
Fiest KM, Sauro KM, Wiebe S, Patten SB, Kwon C-S, Dykeman J, Pringsheim T, Lorenzetti DL, Jetté N (2017) Prevalence and incidence of epilepsy. Neurology 88: 296–303. https://doi.org/10.1212/WNL.0000000000003509
Fattorusso A, Matricardi S, Mencaroni E, Dell’Isola GB, Di Cara G, Striano P, Verrotti A (2021) The Pharmacoresistant Epilepsy: An Overview on Existant and New Emerging Therapies. Front Neurol 12: 674483. https://doi.org/10.3389/fneur.2021.674483
Sears SM, Hewett SJ (2021) Influence of glutamate and GABA transport on brain excitatory/inhibitory balance. Exp Biol Med 246: 1069–1083. https://doi.org/10.1177/1535370221989263
Pracucci E, Pillai V, Lamers D, Parra R, Landi S (2021) Neuroinflammation: A Signature or a Cause of Epilepsy? Int J Mol Sci 22: 6981. https://doi.org/10.3390/ijms22136981
Devinsky O, Vezzani A, Najjar S, De Lanerolle NC, Rogawski MA (2013) Glia and epilepsy: excitability and inflammation. Trends Neurosci 36: 174–184 . https://doi.org/10.1016/j.tins.2012.11.008
Vezzani A, Balosso S, Ravizza T (2008) The role of cytokines in the pathophysiology of epilepsy. Brain Behav Immun 22: 797–803. https://doi.org/10.1016/j.bbi.2008.03.009
Dyomina A V, Zubareva OE, Smolensky I V, Vasilev DS, Zakharova M V, Kovalenko AA, Schwarz AP, Ischenko AM, Zaitsev A V (2020) Anakinra Reduces Epileptogenesis, Provides Neuroprotection, and Attenuates Behavioral Impairments in Rats in the Lithium–Pilocarpine Model of Epilepsy. Pharmaceuticals 13: 340. https://doi.org/10.3390/ph13110340
Mazarati AM, Pineda E, Shin D, Tio D, Taylor AN, Sankar R (2010) Comorbidity between epilepsy and depression: Role of hippocampal interleukin-1β. Neurobiol Dis 37: 61–467. https://doi.org/10.1016/j.nbd.2009.11.001
Marchi N, Fan Q, Ghosh C, Fazio V, Bertolini F, Betto G, Batra A, Carlton E, Najm I, Granata T, Janigro D (2009) Antagonism of peripheral inflammation reduces the severity of status epilepticus. Neurobiol Dis 33: 171–181. https://doi.org/10.1016/j.nbd.2008.10.002
Korbecki J, Bobiński R, Dutka M (2019) Self-regulation of the inflammatory response by peroxisome proliferator-activated receptors. Inflamm Res 68: 443–458. https://doi.org/10.1007/s00011-019-01231-1
Zolezzi JM, Santos MJ, Bastías-Candia S, Pinto C, Godoy JA, Inestrosa NC (2017) PPARs in the central nervous system: roles in neurodegeneration and neuroinflammation. Biol Rev 92: 2046–2069. https://doi.org/10.1111/brv.12320
Hong F, Pan S, Guo Y, Xu P, Zhai Y (2019) PPARs as Nuclear Receptors for Nutrient and Energy Metabolism. Molecules 24: 2545. https://doi.org/10.3390/molecules24142545
Fidaleo M, Fanelli F, Ceru M, Moreno S (2014) Neuroprotective Properties of Peroxisome Proliferator-Activated Receptor Alpha (PPARα) and its Lipid Ligands. Curr Med Chem 21: 2803–2821. https://doi.org/10.2174/0929867321666140303143455
Heneka M, Landreth G (2007) PPARs in the brain. Biochim Biophys Acta – Mol Cell Biol Lipids 1771: 1031–1045. https://doi.org/10.1016/j.bbalip.2007.04.016
Grygiel-Górniak B (2014) Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications – a review. Nutr J 13: 17. https://doi.org/10.1186/1475-2891-13-17
Blanquart C, Barbier O, Fruchart JC, Staels B, Glineur C (2003) Peroxisome proliferator-activated receptors: regulation of transcriptional activities and roles in inflammation. J Steroid Biochem Mol Biol 85: 267–273. https://doi.org/10.1016/S0960-0760(03)00214-0
Strosznajder AK, Wójtowicz S, Jeżyna MJ, Sun GY, Strosznajder JB (2021) Recent Insights on the Role of PPAR-β/δ in Neuroinflammation and Neurodegeneration, and Its Potential Target for Therapy. NeuroMolecular Med 23: 86–98. https://doi.org/10.1007/s12017-020-08629-9
Hong S, Xin Y, HaiQin W, GuiLian Z, Ru Z, ShuQin Z, HuQing W, Li Y, Ning B, YongNan L (2013) The PPARγ agonist rosiglitazone prevents neuronal loss and attenuates development of spontaneous recurrent seizures through BDNF/TrkB signaling following pilocarpine-induced status epilepticus. Neurochem Int 63: 405–412. https://doi.org/10.1016/j.neuint.2013.07.010
Sun H, Huang Y, Yu X, Li Y, Yang J, Li R, Deng Y, Zhao G (2008) Peroxisome proliferator-activated receptor gamma agonist, rosiglitazone, suppresses CD40 expression and attenuates inflammatory responses after lithium pilocarpine-induced status epilepticus in rats. Int J Dev Neurosci 26: 505–515. https://doi.org/10.1016/j.ijdevneu.2008.01.009
Porta N, Vallée L, Lecointe C, Bouchaert E, Staels B, Bordet R, Auvin S (2009) Fenofibrate, a peroxisome proliferator-activated receptor-α agonist, exerts anticonvulsive properties. Epilepsia 50(4): 943–948. https://doi.org/10.1111/j.1528-1167.2008.01901.x
Wong S-B, Cheng S-J, Hung W-C, Lee W-T, Min M-Y (2015) Rosiglitazone Suppresses In Vitro Seizures in Hippocampal Slice by Inhibiting Presynaptic Glutamate Release in a Model of Temporal Lobe Epilepsy. PLoS One 10: e0144806. https://doi.org/10.1371/journal.pone.0144806
O’Léime CS, Cryan JF, Nolan YM (2017) Nuclear deterrents: Intrinsic regulators of IL-1β-induced effects on hippocampal neurogenesis. Brain Behav Immun 66: 394–412. https://doi.org/10.1016/j.bbi.2017.07.153
Stienstra R, Mandard S, Tan NS, Wahli W, Trautwein C, Richardson TA, Lichtenauer-Kaligis E, Kersten S, Müller M (2007) The Interleukin-1 receptor antagonist is a direct target gene of PPARα in liver. J Hepatol 46: 869–877. https://doi.org/10.1016/j.jhep.2006.11.019
Mráček T, Cannon B, Houštěk J (2004) IL-1 and LPS but not IL-6 inhibit differentiation and downregulate PPAR gamma in brown adipocytes. Cytokine 26: 9–15. https://doi.org/10.1016/j.cyto.2003.12.001
Ahmed Juvale II, Che Has AT (2020) The evolution of the pilocarpine animal model of status epilepticus. Heliyon 6: e04557. https://doi.org/10.1016/j.heliyon.2020.e04557
Phelan KD, Shwe UT, Williams DK, Greenfield LJ, Zheng F (2015) Pilocarpine-induced status epilepticus in mice: A comparison of spectral analysis of electroencephalogram and behavioral grading using the Racine scale. Epilepsy Res 117: 90–96. https://doi.org/10.1016/j.eplepsyres.2015.09.008
Paxinos G, Watson C (2007) The rat brain in stereotaxic coordinates. Elsevier.
Cernecka H, Doka G, Srankova J, Pivackova L, Malikova E, Galkova K, Kyselovic J, Krenek P, Klimas J (2016) Ramipril restores PPARβ/δ and PPARγ expressions and reduces cardiac NAD-PH oxidase but fails to restore cardiac function and accompanied myosin heavy chain ratio shift in severe anthracycline-induced cardiomyopathy in rat. Eur J Pharmacol 791: 244–253. https://doi.org/10.1016/j.ejphar.2016.08.040
Chistyakov DV, Aleshin SE, Astakhova AA, Sergeeva MG, Reiser G (2015) Regulation of peroxisome proliferator-activated receptors (PPAR) α and -γ of rat brain astrocytes in the course of activation by toll-like receptor agonists. J Neurochem 134: 113–124. https://doi.org/10.1111/JNC.13101
Lin W, Burks CA, Hansen DR, Kinnamon SC, Gilbertson TA (2004) Lin. J Neurophysiol 92: 2909–2919. https://doi.org/10.1152/jn.01198.2003
Swijsen A, Nelissen K, Janssen D, Rigo J-M, Hoogland G (2012) Validation of reference genes for quantitative real-time PCR studies in the dentate gyrus after experimental febrile seizures. BMC Res Notes 5: 685. https://doi.org/10.1186/1756-0500-5-685
Schwarz AP, Malygina DA, Kovalenko AA, Trofimov AN, Zaitsev AV (2020) Multiplex qPCR assay for assessment of reference gene expression stability in rat tissues/samples. Mol Cell Probes 53: 101611. https://doi.org/10.1016/j.mcp.2020.101611
Malkin SL, Amakhin D V, Veniaminova EA, Kim KK, Zubareva OE, Magazanik LG, Zaitsev A V (2016) Changes of AMPA receptor properties in the neocortex and hippocampus following pilocarpine-induced status epilepticus in rats. Neuroscience 327: 146–155. https://doi.org/10.1016/j.neuroscience.2016.04.024
Cook NL, Vink R, Donkin JJ, van den Heuvel C (2009) Validation of reference genes for normalization of real-time quantitative RT-PCR data in traumatic brain injury. J Neurosci Res 87: 34–41. https://doi.org/10.1002/jnr.21846
Langnaese K, John R, Schweizer H, Ebmeyer U, Keilhoff G (2008) Selection of reference genes for quantitative real-time PCR in a rat asphyxial cardiac arrest model. BMC Mol Biol 9: 53. https://doi.org/10.1186/1471-2199-9-53
Adabi Mohazab R, Javadi-Paydar M, Delfan B, Dehpour AR (2012) Possible involvement of PPAR-gamma receptor and nitric oxide pathway in the anticonvulsant effect of acute pioglitazone on pentylenetetrazole-induced seizures in mice. Epilepsy Res 101: 28–35. https://doi.org/10.1016/j.eplepsyres.2012.02.015
Saha L, Bhandari S, Bhatia A, Banerjee D, Chakrabarti A (2014) Anti-kindling Effect of Bezafibrate, a Peroxisome Proliferator-activated Receptors Alpha Agonist, in Pentylenetetrazole Induced Kindling Seizure Model. J Epilepsy Res 4: 45–54. https://doi.org/10.14581/jer.14011
Wójtowicz S, Strosznajder AK, Jeżyna M, Strosznajder JB (2020) The Novel Role of PPAR Alpha in the Brain: Promising Target in Therapy of Alzheimer’s Disease and Other Neurodegenerative Disorders. Neurochem Res 45: 972–988. https://doi.org/10.1007/s11064-020-02993-5
Fu J, Gaetani S, Oveisi F, Lo Verme J, Serrano A, Rodríguez de Fonseca F, Rosengarth A, Luecke H, Di Giacomo B, Tarzia G, Piomelli D (2003) Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-α. Nature 425: 90–93. https://doi.org/10.1038/nature01921
Sihag J, Jones PJH (2018) Oleoylethanolamide: The role of a bioactive lipid amide in modulating eating behaviour. Obes Rev 19: 178–197. https://doi.org/10.1111/obr.12630
Pan W, Liu C, Zhang J, Gao X, Yu S, Tan H, Yu J, Qian D, Li J, Bian S, Yang J, Zhang C, Huang L, Jin J (2019) Association Between Single Nucleotide Polymorphisms in PPARA and EPAS1 Genes and High-Altitude Appetite Loss in Chinese Young Men. Front Physiol 10: 59. https://doi.org/10.3389/fphys.2019.00059
Curia G, Longo D, Biagini G, Jones RSG, Avoli M (2008) The pilocarpine model of temporal lobe epilepsy. J Neurosci Methods 172: 143–157. https://doi.org/10.1016/j.jneumeth.2008.04.019
Carta AR (2013) PPARγ: Therapeutic Prospects in Parkinson’s Disease. Curr Drug Targets 14: 743–751. https://doi.org/10.2174/1389450111314070004
Chang KL, Wong LR, Pee HN, Yang S, Ho PC-L (2019) Reverting Metabolic Dysfunction in Cortex and Cerebellum of APP/PS1 Mice, a Model for Alzheimer’s Disease by Pioglitazone, a Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) Agonist. Mol Neurobiol 56: 7267–7283. https://doi.org/10.1007/s12035-019-1586-2
Ormerod BK, Hanft SJ, Asokan A, Haditsch U, Lee SW, Palmer TD (2013) PPARγ activation prevents impairments in spatial memory and neurogenesis following transient illness. Brain Behav Immun 29: 28–38. https://doi.org/10.1016/j.bbi.2012.10.017
Peng J, Wang K, Xiang W, Li Y, Hao Y, Guan Y (2019) Rosiglitazone polarizes microglia and protects against pilocarpine-induced status epilepticus. CNS Neurosci Ther 25: 1363–1372. https://doi.org/10.1111/cns.13265
Hong S, Xin Y, HaiQin W, GuiLian Z, Ru Z, ShuQin Z, HuQing W, Li Y, Yun D (2012) The PPARγ agonist rosiglitazone prevents cognitive impairment by inhibiting astrocyte activation and oxidative stress following pilocarpine-induced status epilepticus. Neurol Sci 33: 559–566. https://doi.org/10.1007/s10072-011-0774-2
Prashantha Kumar BR, Kumar AP, Jose JA, Prabitha P, Yuvaraj S, Chipurupalli S, Jeyarani V, Manisha C, Banerjee S, Jeyabalan JB, Mohankumar SK, Dhanabal SP, Justin A (2020) Minutes of PPAR-γ agonism and neuroprotection. Neurochem Int 140: 104814. https://doi.org/10.1016/j.neuint.2020.104814
Inestrosa N, Godoy J, Quintanilla R, Koenig C, Bronfman M (2005) Peroxisome proliferator-activated receptor γ is expressed in hippocampal neurons and its activation prevents β-amyloid neurodegeneration: role of Wnt signaling. Exp Cell Res 304: 91–104. https://doi.org/10.1016/j.yexcr.2004.09.032
Cai W, Yang T, Liu H, Han L, Zhang K, Hu X, Zhang X, Yin K-J, Gao Y, Bennett MVL, Leak RK, Chen J (2018) Peroxisome proliferator-activated receptor γ (PPARγ): A master gatekeeper in CNS injury and repair. Prog Neurobiol 163–164: 27–58. https://doi.org/10.1016/j.pneurobio.2017.10.002
Wu J-S, Tsai H-D, Cheung W-M, Hsu CY, Lin T-N (2016) PPAR-γ Ameliorates Neuronal Apoptosis and Ischemic Brain Injury via Suppressing NF-κB-Driven p22phox Transcription. Mol Neurobiol 53: 3626–3645. https://doi.org/10.1007/s12035-015-9294-z
Bernardo A, Minghetti L (2006) PPAR-gamma agonists as Regulators of Microglial Activation and Brain Inflammation. Curr Pharm Des 12: 93–109. https://doi.org/10.2174/138161206780574579
Ji H, Wang H, Zhang F, Li X, Xiang L, Aiguo S (2010) PPARγ agonist pioglitazone inhibits microglia inflammation by blocking p38 mitogen-activated protein kinase signaling pathways. Inflamm Res 59: 921–929. https://doi.org/10.1007/s00011-010-0203-7
Arsenijevic D, de Bilbao F, Plamondon J, Paradis E, Vallet P, Richard D, Langhans W, Giannakopoulos P (2006) Increased Infarct Size and Lack of Hyperphagic Response after Focal Cerebral Ischemia in Peroxisome Proliferator-Activated Receptor β-Deficient Mice. J Cereb Blood Flow Metab 26: 433–445. https://doi.org/10.1038/sj.jcbfm.9600200
Tang X, Yan K, Wang Y, Wang Y, Chen H, Xu J, Lu Y, Wang X, Liang J, Zhang X (2020) Activation of PPAR-β/δ Attenuates Brain Injury by Suppressing Inflammation and Apoptosis in a Collagenase-Induced Intracerebral Hemorrhage Mouse Model. Neurochem Res 45: 837–850. https://doi.org/10.1007/s11064-020-02956-w
Borges K (2003) Neuronal and glial pathological changes during epileptogenesis in the mouse pilocarpine model. Exp Neurol 182: 21–34. https://doi.org/10.1016/S0014-4886(03)00086-4
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова