

Российский физиологический журнал им. И.М. Сеченова, 2022, T. 108, № 5, стр. 649-666
Особенности регуляции ангиогенеза в атеросклеротических бляшках каротидного синуса на поздних стадия развития атеросклероза
А. Н. Евдокименко 1, *, К. Н. Куличенкова 1, Т. С. Гулевская 1, М. М. Танашян 1
1 Научный центр неврологии
Москва, Россия
* E-mail: evdokimenko@neurology.ru
Поступила в редакцию 31.01.2022
После доработки 07.04.2022
Принята к публикации 07.04.2022
- EDN: ONJSTA
- DOI: 10.31857/S0869813922050041
Аннотация
Повышение неоваскуляризации атеросклеротической бляшки способствует дестабилизации ее структуры, прогрессированию атеросклероза и повышению риска осложнений, включая инфаркт миокарда и ишемический инсульт. Целью исследования стала комплексная оценка экспрессии разнонаправленных регуляторных факторов ангиогенеза в атеросклеротической бляшке каротидного синуса. В исследование включено 33 пациента с атеросклеротическим стенозом каротидного синуса ≥ 60%, которым была выполнена каротидная эндартерэктомия с последующим патоморфологическим исследованием удаленных атеросклеротических бляшек на всем их протяжении. Оценена структура бляшки, плотность расположения микрососудов в 1 см2 бляшки, а также экспрессия сосудистых эндотелиальных факторов роста (VEGFA, VEGFB, VEGFC и VEGFD) и их рецепторов (VEGFR1, VEGFR2, VEGFR3), FGF2, PDGF-B и TSP-1 с последующим корреляционным анализом полученных результатов. По результатам гистологического исследования 13 бляшек отнесены к атероматозным (тип Va), 12 – к осложненным (с изъязвлением покрышки и/или массивным кровоизлиянием в бляшку, тип VI), 6 – к кальцинозным (тип Vb) и 2 – к фиброзным (тип Vc). Новообразованные сосуды выявлены во всех бляшках (267.5 сосудов/1 см2 [Q1 – 140.9; Q3 – 534.8]). Большинство новообразованных сосудов имели высокопроницаемый фенотип – нарушенная целостность эндотелия; отсутствие слоя перицитов; периваскулярные кровоизлияния. Обнаружена гиперэкспрессия VEGFD, значимо преобладавшая над прочими оцененными факторами (р < 0.001), и выраженная экспрессия VEGFA, VEGFR2, VEGFR3, FGF2 и PDGF-B. Иммунореактивность к VEGFB, VEGFC и TSP-1 была выражена в наименьшей степени, VEGFR1 выявлялся в следовых количествах. Количество микрососудов в бляшке значимо коррелировало с экспрессией VEGFA, VEGFD, FGF2, PDGF-B и VEGFR2 (p < 0.01). Связь прочих структурных компонентов бляшки с содержанием в ней ангиогенных факторов отсутствовала. Таким образом, в атеросклеротических бляшках каротидного синуса на поздних стадиях развития атеросклероза продемонстрирован резко выраженный проангиогенный профиль экспрессии различных регуляторных факторов ангиогенеза с несостоятельностью механизма стабилизации новообразованной сосудистой сети.
Атеросклероз – хроническое, медленно прогрессирующее заболевание, ассоциированное с дисфункцией эндотелия; адгезией, активацией и миграцией моноцитов/макрофагов; окислительным стрессом; накоплением липидов в интиме; синтезом внеклеточного матрикса; миграцией и пролиферацией гладкомышечных клеток (ГМК) с формированием их секреторного фенотипа и пр. [1]. В процессе атерогенеза в бляшке создаются локальные условия (гипоксия, воспаление, окислительный стресс и др.), которые активируют ангиогенные факторы, запускающие образование сосудов преимущественно из предсуществующих vasa vasorum адвентиции [2]. Впервые новообразованные сосуды в атеросклеротической бляшке были обнаружены в 1936 г. [3], и в настоящее время повышение неоваскуляризации ассоциируется с ускорением прогрессирования атеросклероза, дестабилизацией структуры бляшки и повышением риска кардиальных и цереброваскулярных осложнений, включая инфаркт миокарда и ишемический инсульт [4–6].
Ангиогенез в бляшке исходно развивается как компенсаторная реакция, направленная на восстановление оксигенации и регенерацию интимы [3] и наблюдается уже на самых ранних его стадиях (стадия жировых пятен и полосок и ранее), что было продемонстрировано в ряде морфологических исследований [7]. Тем не менее, различные стимулы, в том числе гипоксия, воспаление, липопротеины низкой плотности и окислительный стресс, могут поддерживать длительный ангиогенный ответ, который приобретает патологический характер в связи с отсутствием фазы разрешения и стабилизации новообразованных сосудов и приводит к разрастанию “незрелой”, высокопроницаемой сосудистой сети [1].
Высокопроницаемый фенотип новообразованных сосудов в бляшках связан с многочисленными очагами нарушения целостности эндотелия, неполноценными плотными контактами между эндотелиальными клетками и отсутствием слоя перицитов [4]. В недавно проведенных морфологических исследованиях продемонстрировано, что тонкостенные сосуды являются не только причиной кровоизлияний в бляшку, дополнительным источником поступления в бляшку липопротеидов, белков плазмы и воспалительных клеток, но также и источником эндотелиальных клеток-предшественников, принимающих активное участие в увеличении площади новообразованной “незрелой” сосудистой сети [8].
Ангиогенез – это динамический процесс, регулируемый неустойчивым равновесием между ангиогенными и ангиостатическими факторами, который в конечном итоге приводит к увеличению площади, стабилизации или регрессу сосудистой сети [1]. В многочисленных исследованиях определен широкий спектр регуляторов ангиогенеза, включая его стимуляторы, ингибиторы и стабилизаторы сосудистой сети [3], среди которых наиболее изученными являются сосудистый эндотелиальный фактор роста А (VEGFA), основной фактор роста фибробластов (FGF2), фактор роста тромбоцитов бета (PDGF-B) и тромбоспондин-1 (TSP-1). Большинство работ по изучению ангиогенеза посвящено ключевому, по общепринятому мнению, регуляторному пути VEGFA/VEGFR2, тогда как прочим членам семейства сосудистых эндотелиальных факторов роста (в частности, VEGFВ, VEGFС и VEGFD) и их рецепторам (VEGFR1 и VEGFR3) до недавнего времени отводилась незначительная роль в регуляции ангиогенеза. Тем не менее, в последние годы стало появляться все больше работ, свидетельствующих об их активном участии в ангиогенезе, а также о сложной, во многом слабо изученной системе взаимодействий как между сосудистыми эндотелиальными факторами роста и их рецепторами, так и между ними и прочими регуляторами ангиогенеза, а именно FGF2, PDGF-B и TSP-1 [6, 9–11]. Кроме того, описан ряд компенсаторных механизмов, направленных на восстановление ангиогенеза в случае блокады одного из регуляторных путей [12], вследствие чего многие авторы указывают на необходимость комплексного изучения разнонаправленных регуляторных путей.
Несмотря на то, что пусковые факторы и механизмы регуляции ангиогенеза в целом являются универсальными вне зависимости от запустившего его патологического процесса, значительная роль в координации разнонаправленных путей принадлежит комплексу локальных, специфичных для каждого заболевания/состояния, факторов [1]. Большинство проведенных исследований касалось роли и регуляции ангиогенеза при онкологических заболеваниях и включало оценку одного, значительно реже двух из наиболее широко изученных регуляторных факторов. В то же время проблеме регуляции ангиогенеза в атеросклеротической бляшке посвящено значительно меньше работ. Исследования проводились преимущественно на клеточных и животных моделях атеросклероза, не способных в полной мере воспроизвести сложный и многокомпонентный процесс. В особенности это касается поздних стадий развития атеросклероза, а также изъязвления покрышки и неоваскуляризации бляшки, которые до недавнего времени были слабо воспроизводимы. У животных неоваскуляризация бляшки оценивалась преимущественно косвенно, в адвентиции, в связи с редкой встречаемостью сосудов в самой бляшке в большинстве классических животных моделей атеросклероза (ApoE–/– и LDLR–/–) [13]. Кроме того, комплексные исследования с одновременной оценкой разнонаправленных звеньев регуляции ангиогенеза в атеросклеротической бляшке человека отсутствуют, что в совокупности и определило цель данного исследования.
МЕТОДЫ ИССЛЕДОВАНИЯ
В исследование включено 33 пациента (22 мужчины и 11 женщин; средний возраст 65 ± 8.6 лет) с атеростенозом каротидного синуса (КС) ≥ 60% (84% ± 9%) по данным дооперационного дуплексного сканирования ветвей дуги аорты, которым была выполнена операция каротидной эндартерэктомии с последующим патоморфологическим исследованием удаленных атеросклеротических бляшек на всем их протяжении. Все процедуры, выполненные в исследованиях с участием людей, соответствовали этическим стандартам Национального комитета по исследовательской этике и Хельсинкской декларации 1964 г. и ее последующим изменениям или сопоставимым нормам этики. От каждого из включенных в исследование участников было получено информированное добровольное согласие.
Бляшки в зависимости от длины разрезали на 4–9 поперечных блоков толщиной 0.3 см, которые фиксировали в 10%-ном забуференном растворе формалина по стандартной гистологической методике и заливали в парафин. С каждого парафинового блока делали серийные срезы толщиной 5 мкм, которые окрашивали гематоксилином и эозином, по методам ван Гизона и Вейгерта для оценки структуры бляшки и выявления коллагеновых и эластических волокон соответственно, а также иммуногистохимически с использованием антител к CD34 (выявление кровеносных сосудов), CD68 (оценка общей макрофагальной реакции), сосудистым эндотелиальным факторам роста (VEGFA, VEGFB, VEGFC и VEGFD) и их рецепторам (VEGFR1, VEGFR2, VEGFR3), FGF2, PDGF-B и TSP-1.
Для визуализации иммунопероксидазной реакции использовали систему детекции Ultravision Quanto Detection System HRP DAB (Thermo Fisher Scientific, США) в соответствии с инструкцией следующим образом. Срезы депарафинировали, дегидратировали в ряду спиртов нисходящей концентрации, промывали в дистиллированной воде, обрабатывали раствором UltraVision Peroxide Block (Thermo Fisher Scientific, США) для инактивации тканевых пероксидаз, после чего демаскировали антиген в цитратном буфере с рН 6 (Abcam, США) на водяной бане при температуре 80°С в течение 20 мин. После остывания раствора (около 30 мин) препараты выдерживали в трис-буферном солевом растворе (TBS) c добавлением 0.1%-ного раствора Triton x100 (Sigma-Aldrich, США) в течение 20 мин, блокировали неспецифическое связывание раствором UltraVision Protein Block (Thermo Fisher Scientific, США) и инкубировали срезы с раствором антител во влажной камере. Разведения антител, время и условия инкубации представлены в табл. 1.
Таблица 1.
Подробная информация об использованных первичных антителах и условиях иммуногистохимической реакции
| Антиген | Первичное антитело | ||||
|---|---|---|---|---|---|
| Клон | Вид-хозяин | Производитель/ каталожный номер | Разведение | Время и условия инкубации | |
| VEGF-A | VG-1 | Мышь | Abcam, США/ab1316 | 1 : 200 | 4°С, в течение ночи |
| VEGF-B | J14-I | Мышь | Santa Cruz Biotechnology, США/sc-80442 | 1 : 50 | 4°С, в течение ночи |
| VEGF-C | MM0006-2 E65 | Мышь | Santa Cruz Biotechnology, США/sc-101583 | 1 : 50 | 4°С, в течение ночи |
| VEGF-D | EPR8457 | Кролик | Abcam, США/ab 155288 | 1 : 300 | 25°С, 20 мин |
| VEGFR1 | D-2 | Мышь | Santa Cruz Biotechnology, США/sc-271789 | 1 : 200 | 4°С, в течение ночи |
| VEGFR2 | A-3 | Мышь | Santa Cruz Biotechnology, США/sc-6251 | 1 : 50 | 4°С, в течение ночи |
| VEGFR3 | D-6 | Мышь | Santa Cruz Biotechnology, США/sc-28297 | 1 : 200 | 4°С, в течение ночи |
| FGF2 | 3D9 | Мышь | GeneTex, США/GTX84502 | 1 : 50 | 25°С, на шейкере, 60 мин |
| TSP-1 | 3F357 | Мышь | Santa Cruz Biotechnology, США/sc-73158 | 1 : 100 | 4°С, в течение ночи |
| PDGF-B | – | Кролик | GeneTex, США/GTX54575 | 1 : 200 | 25°С, на шейкере, 60 мин |
| CD34 | QBEnd/10 | Мышь | ThermoScientific, США/MS-363-P | 1 : 200 | 4°С, в течение ночи |
| CD68 | Kp-1 | Мышь | CellMarque, США/188M-96 | 1 : 200 | 25°С, 30 мин |
После инкубации с антителами препараты промывали раствором TBST (трис-буфер с NaCl и Твин 20), помещали в раствор усилителя первичных антител Primary Antibody Amplifier Quanto (Thermo Fisher Scientific, США) на 10 мин, повторно промывали TBST, выдерживали 10 мин в конъюгированной с полимером пероксидазе хрена HRP polymer Quanto (Thermo Fisher Scientific, США), промывали в дистиллированной воде, окрашивали в течение 5 мин 3-3-диаминобензидином (DAB) в составе приготовленной согласно инструкции смеси DAB Quanto Substrate и DAB Quanto Chromogen (Thermo Fisher Scientific, США), после чего докрашивали гематоксилином. В качестве отрицательного контроля проводилась иммуногистохимическая реакция без первичных антител.
Окрашенные срезы переводили в цифровую форму с помощью сканера гистологических препаратов Pannoramic MIDI II (3DHISTECH Ltd., Венгрия) при 400-кратном увеличении изображения и анализировали в программе CaseViewer (3DHISTECH Ltd., Венгрия). Оценивали объемную долю атероматоза (%), объемную долю кальцификатов (%), степень выраженности в бляшке пылевидного обызвествления (включая мелкие кальцификаты), свежих и организованных кровоизлияний, а также макрофагальной реакции.
Степень выраженности пылевидного обызвествления и кровоизлияний оценивали полуколичественно по 5-бальной шкале: 0 – компонент отсутствует, 1 – компонент присутствует в следовых количествах, 2 – компонент занимает до 25% площади среза, 3 – от 25 до 50% площади среза, 4 – более 50% площади среза.
Макрофагальную реакцию, а также экспрессию ангиогенных факторов и их рецепторов в бляшке также оценивали полуколичественно по 5-бальной шкале: 0 – отсутствие экспрессии или отдельные иммунореактивные клетки; 1 – единичные небольшие скопления иммунореактивных клеток; 2 – скопления иммунореактивных клеток занимают в совокупности до 15% площади среза; 3 – скопления иммунореактивных клеток занимают в совокупности от 15 до 30% площади среза; 4 – скопления иммунореактивных клеток занимают в совокупности более 30% площади среза. Интенсивность экспрессии маркеров оценивали полуколичественно: + слабая экспрессия; ++ умеренная экспрессия; +++ выраженная экспрессия.
С учетом крайне неравномерного расположения сосудов в бляшке, количество новообразованных сосудов (CD34+) анализировали во всех поперечных срезах бляшки и рассчитывали усредненную величину “количество сосудов в 1 см2 бляшки” (сумма всех сосудов во всех срезах, разделенная на сумму площадей всех срезов в см2).
Статистическую обработку проводили в программе Statistica (StatSoft Inc., США) версии 13.3. Для выявления различий между независимыми группами применяли непараметрический критерий Манна–Уитни, анализа парных измерений – критерий Вилкоксона, зависимости – коэффициент корреляции Спирмена. Результаты статистического анализа считали значимыми при уровне р < 0.05. Результаты представлены в виде медианы [квартиль 1; квартиль 3] (Ме [Q1; Q3]).
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Проанализирована структура 33 атеросклеротических бляшек. Результаты гистологического исследования бляшек представлены в табл. 2. В соответствии с международной классификацией [14], 13 бляшек отнесено к типу V (атероматозные бляшки), 12 – к типу VI (осложненные атеросклеротические поражения с изъязвлением покрышки и/или массивным кровоизлиянием в бляшку), 6 – к типу Vb или VII (кальцинозные бляшки) и 2 – к типу Vc или VIII (фиброзные бляшки).
Таблица 2.
Характеристика структурных компонентов атеросклеротических бляшек каротидного синуса (n = 33)
| № | Наименование компонента | n бляшек, в которых компонент выявлен | % бляшек, в которых компонент выявлен | Ме [Q1; Q3] |
|---|---|---|---|---|
| 1 | Атероматоз, % площади бляшки | 31 | 94 | 27.6 [8.3; 38.5] |
| 2 | Очаги пылевидного обызвествления | 30 | 91 | НП |
| 3 | Кальцификаты различного размера, % площади бляшки | 28 | 85 | 5.6 [2.5; 13.1] |
| 4 | Свежее/организующееся кровоизлияние | 17 | 52 | НП |
| 5 | Организованные кровоизлияния (скопления гемосидерина, сидерофагов) | 19 | 58 | НП |
| 6 | Изъязвление покрышки | 7 | 21 | НП |
| 7 | Неоваскуляризация, количество CD34+ сосудов в 1 см2 бляшки | 33 | 100 | 267.5 [140.9; 534.8] |
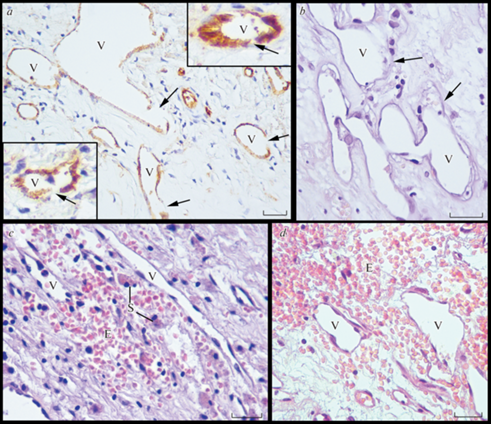
Новообразованные сосуды обнаружены во всех бляшках. Количество сосудов варьировало в широких пределах, составив от 15 до 1320 в 1см2. Большинство из них имели незрелую структуру – тонкую стенку, представленную только слоем эндотелиальных клеток (ЭК), при этом слой перицитов, как правило, отсутствовал; целостность эндотелиального слоя во многих случаях была нарушена, между эндотелиоцитами наблюдались щели; отмечались признаки нарушения проницаемости – в 73% бляшек выявлены периваскулярные скопления эритроцитов, сидерофагов и/или глыбок гемосидерина (рис. 1).
Рис. 1.
Новообразованные сосуды (V) бляшки каротидного синуса человека с участками нарушения целостности эндотелиального слоя (указаны стрелками) и периваскулярными кровоизлияниями (скопления эритроцитов (E) и сидерофагов (S)). a – иммуногистохимическая реакция с антителами к CD34; b‒d – окраска гематоксилином и эозином. Масштабный отрезок соответствует 25 мкм.
Оценена экспрессия 4-х классических стимуляторов ангиогенеза – сосудистых эндотелиальных факторов роста (VEGFA, VEGFB, VEGFC, VEGFD) и рецепторов к ним (VEGFR1, VEGFR2, VEGFR3), альтернативного активатора ангиогенеза (FGF2), ингибитора ангиогенеза (TSP-1) и стабилизатора новообразованной сосудистой сети (PDGF-B). Сводные данные по интенсивности и распространенности экспрессии ангиогенных факторов представлены в табл. 3 и 4.
Таблица 3.
Экспрессия регуляторов ангиогенеза и их рецепторов в различных структурных компонентах атеросклеротической бляшки каротидного синуса человека
| Фактор роста/ рецептор | Эндотелиоциты | Миофибробласты/ гладкомышечные клетки | Макрофаги | Внеклеточный матрикс |
|---|---|---|---|---|
| VEGFA (n = 32) | ++ | ++ | +++ | + |
| VEGFB (n = 32) | + | + | ++ | – |
| VEGFC (n = 27) | ++ | ++ | ++ | – |
| VEGFD (n = 33) | +++ | +++ | +++ | – |
| VEGFR1 (n = 5) | – | – | + | – |
| VEGFR2 (n = 32) | ++ | +++ | ++ | ++ |
| VEGFR3 (n = 33) | + | + | +++ | – |
| FGF2 (n = 32) | ++ | ++ | +++ | +++ |
| TSP-1 (n = 17) | + | + | ++ | ++ |
| PDGF-B (n = 33) | ++ | ++ | +++ | +++ |
Таблица 4.
Распространенность экспрессии регуляторов ангиогенеза и их рецепторов в клетках атеросклеротических бляшек каротидного синуса человека
| Фактор роста/рецептор | Степень выраженности экспрессии* | ||||
|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | |
| VEGFA (n = 32) | 0 | 1 (3%) | 14 (44%) | 13 (41%) | 4 (12%) |
| VEGFB (n = 32) | 6 (18%) | 13 (41%) | 13 (41%) | 0 | 0 |
| VEGFC (n = 27) | 1 (4%) | 11 (40%) | 14 (52%) | 1 (4%) | 0 |
| VEGFD (n = 33) | 0 | 0 | 3 (9%) | 12 (36%) | 18 (55%) |
| VEGFR2 (n = 32) | 0 | 6 (19%) | 17 (53%) | 8 (25%) | 1 (3%) |
| VEGFR3 (n = 33) | 1 (3%) | 5 (15%) | 12 (36%) | 13 (40%) | 2 (6%) |
| FGF2 (n = 33) | 0 | 4 (12%) | 15 (46%) | 13 (39%) | 1 (3%) |
| TSP-1 (n = 17) | 3 (18%) | 7 (41%) | 7 (41%) | 0 | 0 |
| PDGF-B (n = 33) | 0 | 0 | 18 (55%) | 11 (33%) | 4 (12%) |
0 – отсутствие экспрессии или отдельные иммунореактивные клетки; 1 – единичные небольшие скопления иммунореактивных клеток; 2 – иммунореактивные клетки занимают в совокупности до 15% площади среза; 3 – иммунореактивные клетки занимают в совокупности от 15 до 30% площади среза; 4 – иммунореактивные клетки занимают в совокупности более 30% площади среза.
Анализ экспрессии ангиогенных факторов продемонстрировал отчетливое преобладание экспрессии VEGFD над прочими оцененными факторами роста (р < 0.001). Выраженная иммунореактивность к VEGFD наблюдалась во всех типах клеток, включая макрофаги и гигантские многоядерные клетки, ГМК/миофибробласты бляшки и ГМК средней оболочки, ЭК каротидного синуса и новообразованных сосудов бляшки (табл. 3). Следует также подчеркнуть распространенный характер иммунореактивности – в подавляющем большинстве бляшек наблюдалась субтотальная экспрессия маркера клеточными популяциями бляшки (табл. 4, рис. 2d).
Рис. 2.
Экспрессия ангиогенных факторов и их рецепторов в кальцинозной атеросклеротической бляшке каротидного синуса (тип VII). Масштабный отрезок соответствует 20 мкм.

В меньшей по сравнению с VEGFD, но также в значительной степени была выражена экспрессия VEGFA, FGF2 и PDGF-B (рис. 2a, 2e и 2f соответственно). Распространенность и интенсивность экспрессии данных факторов роста значимо между собой не различались (р < 0.05). Положительная цитоплазматическая иммунореактивность, как и в случае VEGFD, наблюдалась в макрофагах и гигантских многоядерных клетках, ГМК/миофибробластах бляшки и средней оболочки, ЭК новообразованных сосудов бляшки и каротидного синуса. При этом экспрессия маркеров наиболее стабильно выявлялась в макрофагах, тогда как в ГМК и ЭК значительно варьировала в разных бляшках – от отдельных иммунореактивных клеток и небольших их очаговых скоплений до субтотальной экспрессии клеточными популяциями бляшки. Наиболее выраженная экспрессия VEGFA, FGF2 и PDGF-B в ГМК и ЭК отмечалась в бляшках с многочисленными крупными скоплениями макрофагов, также экспрессирующих данные маркеры. Кроме того, при иммуногистохимической реакции наблюдалось окрашивание волокнистого компонента бляшки – преимущественно слабое очаговое в случае VEGFA, выраженное и распространенное в случае FGF2, а также выраженное и распространенное, но ассоциированное в наибольшей степени с эластическими волокнами, в случае PDGF-B.
Содержание в бляшке VEGFA, VEGFD и FGF2 прямо коррелировало между собой (р < 0.05), в особенности VEGFA с двумя другими факторами (p < 0.01). Кроме того, экспрессия PDGF-B слабо коррелировала с экспрессией FGF2 (p < 0.05), при этом в бляшках с многочисленными скоплениями PDGF-B+ клеток было значимо больше FGF2 иммунопозитивных клеток (р < 0.05). Результаты корреляционного анализа представлены в табл. 5.
Таблица 5.
Корреляционная матрица степени выраженности неоваскуляризации и экспрессии регуляторов ангиогенеза и их рецепторов в атеросклеротической бляшке каротидного синуса человека
| Коэффициент корреляции, R | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| VEGFA | VEGFB | VEGFC | VEGFD | FGF2 | TSP-1 | PDGF-В | VEGFR2 | VEGFR3 | Количество CD34+ сосудов в 1 см2 бляшки | |
| VEGFA | 1.00 | 0.26 | 0.18 | 0.62** | 0.51** | 0.43 | 0.29 | 0.39* | 0.56** | 0.48** |
| VEGFB | 0.26 | 1.00 | 0.42* | 0.17 | 0.35* | 0.46 | –0.07 | 0.32 | 0.41* | 0.34 |
| VEGFC | 0.18 | 0.42* | 1.00 | 0.20 | 0.21 | 0.79** | 0.34 | 0.13 | 0.13 | 0.24 |
| VEGFD | 0.62** | 0.17 | 0.20 | 1.00 | 0.42* | 0.31 | 0.37* | 0.33 | 0.42* | 0.51** |
| FGF2 | 0.51** | 0.35* | 0.21 | 0.42* | 1.00 | 0.20 | 0.37* | 0.72** | 0.58** | 0.51** |
| TSP-1 | 0.43 | 0.46 | 0.79** | 0.31 | 0.20 | 1.00 | 0.56* | 0.00 | 0.45 | 0.19 |
| PDGF-В | 0.29 | –0.07 | 0.34 | 0.37* | 0.37* | 0.56* | 1.00 | 0.32 | 0.24 | 0.45** |
| VEGFR2 | 0.39* | 0.32 | 0.13 | 0.33 | 0.72** | 0.00 | 0.32 | 1.00 | 0.60** | 0.59** |
| VEGFR3 | 0.56** | 0.41* | 0.13 | 0.42* | 0.58** | 0.45 | 0.24 | 0.60** | 1.00 | 0.38* |
| Количество CD34+ сосудов в 1 см2 бляшки | 0.48** | 0.34 | 0.24 | 0.51** | 0.51** | 0.19 | 0.45** | 0.59** | 0.38* | 1.00 |
В наименьшей степени среди активаторов ангиогенеза в бляшке экспрессировались VEGFB и VEGFC (p < 0.001). Слабая или умеренная иммунореактивность к VEGFB (рис. 2b) отмечалась главным образом в макрофагах и отдельных ЭК или ГМК. Среди иммунореактивных к VEGFC клеток также преобладали макрофаги, однако экспрессия в ЭК и ГМК встречалась чаще и имела более выраженную интенсивность (табл. 3, рис. 2c), а доля иммунореактивных клеток больше, хотя различие в выраженности экспрессии данных факторов не достигало статистической значимости.
Экспрессия TSP-1 оценена только в 17 бляшках. Среди иммунореактивных клеток преобладали макрофаги, как правило формирующие немногочисленные небольшие скопления (табл. 4). Слабая экспрессия также наблюдалась в единичных ГМК и ЭК (табл. 3). Количество TSP-1 в бляшках было значимо ниже, чем прочих проанализированных регуляторов ангиогенеза (p < 0.001 в случае VEGFA, VEGFD, FGF2 и PDGF-B; р < 0.05 в случае VEGFC), за исключением VEGFB (p = 0.5), и коррелировала с уровнем VEGFC (p < 0.001) и PDGF-B (p < 0.05) (табл. 5).
Помимо ангиогенных факторов была оценена экспрессия рецепторов к сосудистым эндотелиальным факторам роста. VEGFR1 выявлялся в бляшке в следовых количествах в макрофагах, поэтому было исследовано только 5 бляшек. В бляшках преобладала экспрессия VEGFR2 и VEGFR3, коррелировавшая между собой (p < 0.001, табл. 5). Иммунореактивность к обоим рецепторам наблюдалась в макрофагах, ГМК бляшки и средней оболочки, а также ЭК (рис. 1g, 1h), однако характер и интенсивность экспрессии различались (табл. 3). В то время как VEGFR2 активно экспрессировался во всех типах клеток, причем наиболее выражена экспрессия была в ГМК/миофибробластах, иммунореактивность к VEGFR3 главным образом отмечалась в макрофагах, значительно реже в ГМК и в отдельных бляшках в ЭК, причем экспрессия наблюдалась в отдельных клетках эндотелиальной выстилки новообразованного сосуда. С экспрессией обоих рецепторов в бляшках прямо коррелировало содержание в ней FGF2 (p < 0.001), причем с VEGFR2 в большей степени, чем с VEGFR3 (табл. 5). Также отмечена прямая зависимость между экспрессией VEGFA и VEGFR3 (p < 0.001, табл. 5).
Содержание проанализированных факторов роста и их рецепторов значимо не различалось в бляшках разного типа, в зависимости от объема атероматоза, кальцификатов, степени выраженности воспаления (размера скоплений CD68+ макрофагов). В то же время отмечена прямая зависимость между плотностью расположения новообразованных сосудов в бляшке (количеством сосудов в 1 см2 бляшки) и содержанием в ней VEGFA, VEGFD, FGF2, PDGF-B, VEGFR2 (р < 0.01) и VEGFR3 (p < 0.05, табл. 5).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Ангиогенез представляет собой сложный процесс, регулируемый про- и антиангиогенными факторами, которые включают динамическое взаимодействие между многочисленными клетками, факторами роста и внеклеточным матриксом [15]. Ключевым фактором запуска ангиогенеза считается VEGFA, который при взаимодействии с VEGFR2 вызывает пролиферацию, миграцию и инвазию ЭК в окружающие ткани, способствует привлечению воспалительных клеток, а также стимулирует синтез матриксных металлопротеиназ, необходимых для деградации базальной мембраны и внеклеточного матрикса, обязательного этапа ангиогенеза [16]. VEGFA является наиболее изученным ангиогенным фактором в семействе сосудистых эндотелиальных факторов роста [3]. Связывание VEGFA с VEGFR2 также приводит к выраженному повышению проницаемости эндотелия [16], вследствие чего он также известен как фактор сосудистой проницаемости.
VEGFA в норме экспрессируется преимущественно в ЭК и перицитах, однако в условиях гипоксии и воспаления также отмечена его продукция моноцитами/макрофагами и ГМК [17]. Согласно ранее проведенным исследованиям, VEGFA в большом количестве представлен в бляшках сонных артерий на поздних стадиях развития атеросклероза, где он выявлялся в ЭК, ГМК и макрофагах [18]. Мы также отметили выраженную экспрессию VEGFA в бляшках каротидного синуса, при этом иммунореактивность наиболее стабильно наблюдалась в макрофагах, тогда как в ГМК и ЭК варьировала в широких пределах, но увеличивалась с повышением количества иммунореактивных макрофагов.
Прямая зависимость между уровнем VEGFA и выраженностью неоваскуляризации продемонстрирована в многочисленных работах при широком спектре различных заболеваний, включая атеросклероз [17]. Кроме того, применение моноклональных антител к VEGFA приводило к уменьшению неоваскуляризации и размера бляшки [13], подчеркивая значимость фактора в ангиогенезе при атеросклерозе. Полученные нами данные подтверждают описанную закономерность – выраженность экспрессии VEGFA коррелировала с количеством микрососудов в бляшке. Активация VEGFA в бляшках приводит к формированию высокопроницаемых, структурно неполноценных микрососудов, которые способствуют усилению воспаления, окислительного стресса и, в целом, прогрессированию атеросклероза [16]. В этой связи неудивительно, что VEGFA рассматривался как многообещающая мишень терапевтического воздействия для блокады ангиогенеза в бляшке. Тем не менее, спектр побочных эффектов, выявленных в клинических исследованиях при онкологических и офтальмологических заболеваниях [17], исключает использование блокаторов VEGFA/VEGFR2 для длительного лечения атеросклероза.
Экспрессия VEGFR2 в проанализированных бляшках также была выражена в значительной степени, наблюдалась в ЭК, макрофагах и особенно ГМК, а также коррелировала с количеством микрососудов. По данным литературы, VEGFR2 преимущественно выявляется в ЭК, но не в макрофагах или ГМК, опосредуя передачу сигнала VEGFA [16]. Тем не менее, экспрессия VEGFR2 в ГМК была отмечена в немногочисленных исследованиях. В частности, в одном исследовании на культуре ГМК продемонстрировано резкое повышение экспрессии VEGFR2 под действием гипоксии, что в сочетании с повышенным уровнем VEGFA приводило к активации пролиферации ГМК [19]. Кроме того, экспрессия рецептора была зафиксирована в ГМК аорты кроликов WHHL (генетически гиперлипемичные кролики Ватанаба) [20]. Таким образом, персистирующая резко выраженная гипоксия в бляшке каротидного синуса на поздних стадиях развития атеросклероза может стимулировать экспрессию VEGFR2 в ГМК с последующей их пролиферацией, что, возможно, объясняет выявленную нами экспрессию рецептора в ГМК и требует дополнительного подтверждения.
В моноцитах/макрофагах и ГМК наряду с ЭК чаще описывается экспрессия другого рецептора VEGFA – VEGFR1 [16]. VEGFR1 имеет в 10 раз более высокое сродство к VEGFA, чем VEGFR2, однако результирующий сигнал является слабым [17]. Растворимая форма рецептора связывает VEGFA, предотвращая его связывание с полноразмерной формой VEGFR1 и VEGFR2 [21]. Кроме того, гетеродимеры VEGFR1/VEGFR2 блокируют передачу сигнала VEGFA, предотвращая VEGFR2 гомодимерную активность [12]. В этой связи VEGFR1 известен своей негативной регуляторной ролью в ангиогенезе. В проведенном исследовании VEGFR1 выявлялся в бляшках в следовых количествах, что могло дополнительно стимулировать ангиогенез и ускорить прогрессирование атеросклероза вследствие усиления сигнала VEGFA/VEGFR2. Имеются данные о деградации рецептора при связывании с липопротеинами низкой плотности [15], широко представленными в бляшках, что может отчасти объяснить отмеченный в нашем исследовании столь низкий уровень VEGFR1.
Мы также проанализировали содержание в бляшках других членов семейства VEGF, а именно VEGFB, VEGFC и VEGFD, с неожиданным результатом – из всех проанализированных сосудистых эндотелиальных факторов роста, включая VEGFA, отмечено ранее не описанное отчетливое преобладание VEGFD с субтотальной выраженной экспрессией маркера всеми типами клеточных популяций в большинстве исследованных бляшек.
VEGFD очень близок по структуре и рецепторной специфичности к VEGFC, и основным его биологическим эффектом при взаимодействии с VEGFR3 ранее считался лимфангиогенез [17]. Тем не менее, было продемонстрировано, что VEGFD является эффективным ангиогенным фактором, реализующим свое действие через посредство VEGFR2 или гетеродимеров VEGFR2/VEGFR3 либо опосредованно, повышая экспрессию других факторов роста, включая VEGFA [16]. Полученные нами данные о наличии корреляции между уровнями VEGFD и VEGFA в бляшке могут косвенно подтверждать ранее отмеченную другими авторами взаимосвязь двух факторов. Прямая зависимость степени выраженности неоваскуляризации в бляшке и экспрессии в ней VEGFD также может указывать на активное участие VEGFD в регуляции ангиогенеза при атеросклерозе.
Большинство опубликованных результатов исследований по механизму действия VEGFD посвящено его роли в канцерогенезе, в том числе в аспекте патологического ангио- и лимфангиогенеза, а также в патогенезе других заболеваний, включая сердечно-сосудистые [16, 22]. Данные о роли VEGFD в атерогенезе и прогрессировании атеросклероза, в том числе в неоваскуляризации бляшки, единичны. В частности, на кроличьей модели сахарного диабета в атеросклеротических бляшках каротидного синуса продемонстрировано, что экспрессия VEGFD в атеросклеротических бляшках была повышена и наблюдалась в ЭК, ГМК и макрофагах [20, 23], это согласуется с полученными нами данными по характеру экспрессии маркера. Проатерогенное действие VEGFD также показано на модели каротидной гиперплазии у кроликов, у которых введение в адвентицию аденовируса, кодирующего VEGFD, приводило к утолщению интимы [1]. Помимо ангио- и лимфангиогенного действия, сигнальный путь VEGFD/VEGFR3 вовлечен в стимуляцию миграции и пролиферации миофибробластов, а также стимуляцию синтеза коллагена 1-го типа в культуре миофибробластов сердца, способствуя фиброзу [24]. VEGFD также обладает антиоксидантной активностью и регулирует липидный обмен [16].
В целом, имеющиеся в литературе данные свидетельствует о потенциально значительной роли VEGFD в атерогенезе и неоваскуляризации, что требует дополнительного изучения и делает его привлекательной мишенью терапевтического воздействия при атеросклерозе, в особенности с учетом широкого спектра уже разработанных препаратов, направленных на активацию или подавление VEGFD/VEGFR3 сигнального пути. Препараты данного типа успешно прошли доклинические исследования и применяются в клинических исследованиях I/IIa фазы при онкологических, офтальмологических (влажная возрастная макулярная дегенерация) и сердечно-сосудистых заболеваниях (рефрактерная стенокардия) [22].
По нашим данным, содержание VEGFC в бляшках было значимо ниже, чем VEGFD и VEGFA. VEGFC считается ключевым фактором лимфангиогенеза, реализующим свое действие через взаимодействие с VEGFR3 [16]. Тем не менее, VEGFC может связываться с VEGFR2, хотя и с более низкой афинностью, чем с VEGFR3, и запускать ангиогенез [16]. Отмечена обратная связь между ангиогенезом и лимфангиогенезом, а именно значимое снижение ангиогенеза при активном развитии лимфатической сети и наоборот. Поэтому ангиогенез, опосредуемый VEGFC, преимущественно происходил вдали от очагов лимфангиогенеза [16]. Кроме того, получены данные о возможности запуска ангиогенеза при связывании VEGFC с VEGFR3 за счет стимуляции экспрессии PDGF-B, вовлеченного в активацию ЭК [25], или под действием атерогенной микроРНК MiR-27b, которая усиливала экспрессию VEGFC с последующей стимуляцией пролиферации и миграции клеток и ангиогенеза в культуре HMEC-1 [26]. В дополнение к регуляции ангио-/лимфангиогенеза и воспаления, VEGFC, как и VEGFD, обладает фиброгенной активностью – непосредственно способствует пролиферации, миграции и синтезу коллагена фибробластами [16].
Обнаруженное нами высокое содержание в бляшках каротидного синуса VEGFR3 – основного рецептора VEGFC и VEGFD, сопоставимое с уровнем VEGFR2, указывало на его потенциально значимую роль в ангиогенезе и атерогенезе. VEGFR3 преимущественно экспрессируется в лимфатических ЭК, стимулируя их дифференцировку, пролиферацию, миграцию и выживаемость при связывании с VEGFC и VEGFD, тем самым стимулируя лимфангиогенез [16]. Маркер также отмечен в макрофагах [16]. Мы получили сходные данные по экспрессии рецептора – в атеросклеротической бляшке VEGFR3 главным образом выявлялся в макрофагах, значительно реже в ГМК и лишь в единичных ЭК, которые также экспрессировали CD34 (сосудистые ЭК). При этом экспрессия VEGFR3, согласно полученным нами результатам, коррелировала с содержанием в бляшке VEGFR2 и VEGFA. О наличии модулирующей роли VEGFR3 в передаче сигнала VEGFA/VEGFR2, а также тесной связи VEGFR2 и VEGFR3 свидетельствовали результаты ряда ранее проведенных исследований [27, 28]. В частности, отмечено взаимодействие VEGFR2 и VEGFR3 по системе обратной связи – VEGFR2 стимулировал экспрессию VEGFR3, который затем подавлял экспрессию VEGFR2 [27]. VEGFA, VEGFC и VEGFD стимулировали образование гетеродимеров VEGFR2/VEGFR3, которые активируют пролиферацию ЭК, способствуя новообразованию сосудов [28]. При этом гетеродимеры могут активировать ангиогенез даже при блокаде VEGFR2, вероятно, вследствие усиления тирозинкиназной активности VEGFR3 при связывании с VEGFR2 [27].
В наименьшей степени среди сосудистых эндотелиальных факторов роста в бляшках был представлен VEGFB, который выявлялся главным образом в макрофагах и единичных ГМК и ЭК. Тем не менее, экспрессия VEGFB значимо не отличалась от экспрессии VEGFC. VEGFB является гомологом VEGFA и реализует свое действие только через взаимодействие с VEGFR1 [16]. Он широко экспрессируется в различных типах клеток, включая ГМК сосудов и ЭК, и обладает слабой ангиогенной активностью [16]. Проангиогенный эффект VEGFB может быть связан с подавлением апоптоза/стимуляцией выживаемости клеток или с вытеснением VEGFA из комплекса с VEGFR1, что может усилить передачу сигнала VEGFA/VEGFR2 [29]. Наиболее значимая роль VEGFB в атерогенезе и неоваскуляризации бляшки может быть связана с недавно выявленными его антиатерогенными эффектами – сильным антиоксидантным действием и регуляцией метаболизма клеток [29]. Тем не менее, VEGFR1, необходимый для реализации эффекта VEGFB, выявлялся в бляшках в следовых количествах.
Помимо классических (семейство VEGF) был оценен уровень альтернативного ангиогенного фактора – FGF2, который является сильным активатором ангиогенеза [11]. Ранее проведенные исследования продемонстрировали, что FGF2 является сильным митогеном для различных клеток, включая ЭК [10]; стимулирует пролиферацию и структурную организацию ЭК в тубулярные структуры, а также рост, дифференцировку и выживание ассоциированных с кровеносными сосудами клеток [30]; оказывает антифиброзное действие [31]. При этом FGF2 регулирует ангиогенез опосредованно, в том числе за счет активации высвобождения других ангиогенных факторов, и относится к “непрямым” ангиогенным факторам [6, 10, 32].
Нами выявлена выраженная экспрессия FGF2 в исследованных бляшках каротидного синуса, которая по степени и характеру значимо не отличалась от экспрессии VEGFA и коррелировала с ней, а также с экспрессией VEGFR2 и VEGFR3. Полученные данные согласуются с результатами опубликованных работ, в которых указан синергетический эффект FGF2 и VEGFA, обусловливающий стимуляцию миграции ЭК, перицитов и ГМК [32], а также корреляция экспрессии VEGFA/VEGFR2 и FGF2/FGFR1 [6]. В ряде исследований также продемонстрирована сложная и тесная взаимосвязь между сигнальными путями FGF2 и VEGFA в ходе ангиогенеза. В частности, для поддержания экспрессии VEGFR2 необходима стимуляция ЭК FGF2 [10]; FGF2 регулирует сплайсинг VEGFR1 с активацией образования растворимых форм рецептора, которые поддерживают градиент VEGFA для навигации образующихся сосудов [10]; FGF2 стимулирует синтез VEGFA [32].
Исследования членов семейства FGF при атеросклерозе немногочисленны, а полученные результаты зачастую противоречивы, хотя исследователи сходятся во мнении относительно повышенного содержания и критической роли членов FGF в атеросклеротических бляшках [33]. К примеру, имеются данные о ключевой роли FGF2 в неоваскуляризации бляшки [3, 6] и об отсутствии экспрессии FGF2 в бляшках [3].
PDGF-B – наиболее хорошо охарактеризованный фактор роста для привлечения периваскулярных клеток и стабилизации новообразованной сосудистой сети [11, 30]. ЭК в составе развивающегося сосуда секретируют PDGF-B, который связывается с гепарансульфат-протеогликанами внеклеточного матрикса и поверхности клеток, тем самым создавая градиент, необходимый для навигации перицитов в направлении развивающегося сосуда [34], нарушение которого приводило к образованию высокопроницаемых сосудов. Также показано, что VEGFA стимулирует экспрессию рецептора PDGF, тем самым регулируя миграцию и пролиферацию фибробластов и ГМК, а также образование внеклеточного матрикса [35].
Описанное в литературе связывание PDGF-B, FGF2 и VEGFA с внеклеточным матриксом создает депо данных факторов роста в межклеточном веществе [34, 36], что может объяснить иммунореактивность волокнистого компонента бляшки к вышеуказанным факторам, обнаруженное в проведенном нами исследовании, и подчеркивает возможность резкого повышения ангиогенного сигнала при их высвобождении из депо под действием протеаз. Экспрессия PDGF-B описана в многочисленных клетках, включая ЭК, ГМК и макрофаги [37], что также было отмечено и нами. Дисфункция сигнального пути PDGF-B выявляется при широком спектре заболеваний, включая атеросклероз, при котором отмечено повышение экспрессии фактора [38]. Ряд авторов также указывает на сходный характер экспрессии PDGF-B и VEGFA [18], что также подтверждали результаты нашего исследования.
PDGF-B, как и FGF2, считается “непрямым” ангиогенным фактором [6]. Показано, что он может стимулировать экспрессию VEGFA и FGF2 [35], при этом на кроличьей модели атеросклероза недавно был продемонстрирован синергетический эффект PDGF-B и FGF2 в части привлечения перицитов, стабилизации новообразованной сосудистой сети и уменьшении площади сосудистой сети [11]. Несмотря на то, что FGF2 и PDGF-B считаются важными регуляторами ангиогенеза в бляшке, их точная роль в регуляции и координации этого сложного процесса остается во многом слабо изученной [1, 6]. В последние годы получены данные о том, что в бляшках на поздних стадиях развития атеросклероза была снижена экспрессия рецептора PDGF-B, несмотря на относительно высокий уровень экспрессии самого фактора [6]. Кроме того, описана возможность образования нефункциональных гетеродимеров VEGFR2/PDGFR-бета в перицитах под действием VEGFA [39]. Окисленные липопротеины низкой плотности при краткосрочной инкубации с ГМК индуцировали активацию PDGFR-бета, тогда как длительная инкубация приводила к подавлению фосфорилирования рецептора [6].
Мы оценивали экспрессию рецепторов только сосудистых эндотелиальных факторов роста, что ограничивает возможности интерпретации результатов активации сигнальных путей других факторов. При высоком уровне PDGF-В и FGF2 (с учетом их синергетического эффекта) мы зачастую наблюдали в бляшках многочисленные тонкостенные сосуды с нарушенной целостностью эндотелиальной выстилки без слоя перицитов и периваскулярными кровоизлияниями разной давности. В совокупности это свидетельствует о незрелой структуре новообразованных сосудов с повышением проницаемости их стенки, что может быть связано с нарушением передачи сигнала PDGF-B вследствие снижения экспрессии, стимуляции деградации его рецептора или подавления фосфорилирования рецептора под действием VEGFA/VEGFR2, липопротеинов низкой плотности и других возможных факторов на поздних стадиях развития атеросклероза.
TSP-1 считается сильным ингибитором ангиогенеза, действие которого может быть реализовано различными путями при атеросклерозе: подавление экспрессии VEGFA, снижение уровня липопротеинов низкой плотности, клиренс матриксных металлопротеиназ, индукция апоптоза ЭК, подавление их миграции и пролиферации [40]. Экспрессия TSP-1 выявляется в клетках различных типов, включая ЭК, макрофаги и ГМК [9], что подтверждалось в проведенном исследовании. TSP-1 модулирует клеточный ответ на факторы внешней среды, конкурентно связываясь с клеточными рецепторами, компонентами внеклеточного матрикса, протеолитическими ферментами и растворимыми факторами [36]. Данные по экспрессии и значимости TSP-1 при атеросклерозе и неоваскуляризации бляшки немногочисленны. Ряд авторов указывает на повышение экспрессии маркера в атеросклеротической бляшке [41], что может быть связано с его активацией в ответ на действие факторов роста (PDGF-B, FGF2) и гипоксии [9]. Тем не менее, содержание в бляшках TSP-1 по нашим данным было низким, а интенсивность экспрессии слабой. Значимость TSP-1 в ангиогенезе в бляшке неоднозначна, поскольку помимо антиангиогенного действия фактор также позиционируется как проатерогенный, стимулирующий миграцию и пролиферацию ГМК с формированием секреторного фенотипа, в особенности под влиянием глюкозы [41]. Неоднозначная роль TSP-1 в атерогенезе и неоваскуляризации бляшки, а также его тесная связь с ключевыми регуляторными сигнальными путями ангиогенеза при атеросклерозе свидетельствует о необходимости дополнительного изучения механизмов его действия в комплексе с другими ангиогенными факторами.
Нами также была оценена взаимосвязь различных ангиогенных факторов со структурными компонентами бляшки, однако выявить какую-либо зависимость (за исключением неоваскуляризации), в том числе с выраженностью воспалительной реакции (содержание CD68+ макрофагов) нам не удалось. Возможной причиной такого результата может быть небольшой объем выборки, в особенности малое количество стабильных бляшек. Кроме того, мы не выделяли различные подтипы макрофагов и не оценивали содержание в бляшке провоспалительных цитокинов, которые играют значимую роль в ангиогенезе.
Таким образом, в проведенном исследовании продемонстрирован резко выраженный проангиогенный профиль экспрессии различных регуляторных факторов ангиогенеза в атеросклеротических бляшках каротидного синуса на поздних стадиях развития атеросклероза со значимым преобладанием VEGFD иммунореактивности на фоне резкого дефицита антиангиогенного фактора TSP-1. Высокое содержание в бляшках VEGFA, VEGFR2/3, FGF2 и PDGF-B, а также относительно низкий уровень VEGFC/VEGFB и следовые количества VEGFR1 на фоне высокопроницаемой новообразованной сосудистой сети свидетельствует о резком смещении равновесия в сторону стимуляторов ангиогенеза и неполноценности механизма стабилизации новообразованной сосудистой сети.
С учетом сложной системы взаимодействия между различными ангиогенными факторами и их рецепторами, а также наличием многочисленных локальных модуляторов процесса при атеросклерозе, необходимо дальнейшее изучение регуляции неоваскуляризации атеросклеротических бляшек с обязательным комплексным анализом разнонаправленных звеньев регуляции ангиогенеза, в том числе на клеточных и животных моделях атеросклероза. В этой связи стоит обратить особое внимание на механизм действия VEGFD, гиперэкспрессия которого была впервые нами продемонстрирована в бляшках каротидного синуса наряду с выраженной экспрессией его рецепторов (VEGFR2/VEGFR3). Широкий спектр эффектов VEGFD, отмеченный при различных патологиях, указывает на его возможно значимую роль как в ангиогенезе, так и атерогенезе, что делает его многообещающей мишенью терапевтического воздействия при атеросклерозе.
Список литературы
Camaré C, Pucelle M, Nègre-Salvayre A, Salvayre R (2017) Angiogenesis in the atherosclerotic plaque. Redox Biol 12: 18–34. https://doi.org/10.1016/j.redox.2017.01.007
Sedding DG, Boyle EC, Demandt JAF, Sluimer JC, Dutzmann J, Haverich A, Bauersachs J (2018) Vasa Vasorum Angiogenesis: Key Player in the Initiation and Progression of Atherosclerosis and Potential Target for the Treatment of Cardiovascular Disease. Front Immunol 9: 706. https://doi.org/10.3389/fimmu.2018.00706
Van der Veken B, De Meyer GR, Martinet W (2016) Intraplaque neovascularization as a novel therapeutic target in advanced atherosclerosis. Expert Opin Ther Targets 20: 1247–1257. https://doi.org/10.1080/14728222.2016.1186650
Sluimer JC, Kolodgie FD, Bijnens APJJ, Maxfield K, Pacheco E, Kutys B, Duimel H, Frederik PM, van Hinsbergh VWM, Virmani R, Daemen MJAP (2009) Thin-Walled Microvessels in Human Coronary Atherosclerotic Plaques Show Incomplete Endothelial Junctions. J Am Coll Cardiol 53: 1517–1527. https://doi.org/10.1016/j.jacc.2008.12.056
Parma L, Baganha F, Quax PHA, de Vries MR (2017) Plaque angiogenesis and intraplaque hemorrhage in atherosclerosis. Eur J Pharmacol 816: 107–115. https://doi.org/10.1016/j.ejphar.2017.04.028
Mao Y, Liu X, Song Y, Zhai C, Zhang L (2018) VEGF-A/VEGFR-2 and FGF-2/FGFR-1 but not PDGF-BB/PDGFR-β play important roles in promoting immature and inflammatory intraplaque angiogenesis. PLoS One 13: e0201395. https://doi.org/10.1371/journal.pone.0201395
Jeziorska M, Woolley DE (1999) Neovascularization in early atherosclerotic lesions of human carotid arteries: Its potential contribution to plaque development. Hum Pathol 188: 189–196. https://doi.org/10.1016/S0046-8177(99)90245-9
Kashiwazaki D, Koh M, Uchino H, Akioka N, Kuwayama N, Noguchi K, Kuroda S (2018) Hypoxia accelerates intraplaque neovascularization derived from endothelial progenitor cells in carotid stenosis. J Neurosurg 131: 884–891. https://doi.org/10.3171/2018.4.JNS172876
Adams JC, Lawler J (2004) The thrombospondins. Int J Biochem Cell Biol 36: 961–968. https://doi.org/10.1016/j.biocel.2004.01.004
Jia T, Jacquet T, Dalonneau F, Coudert P, Vaganay E, Exbrayat-Héritier C, Vollaire J, Josserand V, Ruggiero F, Coll J-L, Eymin B (2021) FGF-2 promotes angiogenesis through a SRSF1/SRSF3/SRPK1-dependent axis that controls VEGFR1 splicing in endothelial cells. BMC Biol 19: 173.https://doi.org/10.1186/s12915-021-01103-3
Mao Y, Liu XQ, Song Y, Zhai CG, Xu XL, Zhang L, Zhang Y (2020) Fibroblast growth factor-2/platelet-derived growth factor enhances atherosclerotic plaque stability. J Cell Mol Med 24: 1128–1140. https://doi.org/10.1111/jcmm.14850
Simons M, Gordon E, Claesson-Welsh L (2016) Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol 17: 611–625. https://doi.org/10.1038/nrm.2016.87
Perrotta P, Emini Veseli B, Van der Veken B, Roth L, Martinet W, De Meyer GRY (2019) Pharmacological strategies to inhibit intra-plaque angiogenesis in atherosclerosis. Vascul Pharmacol 112: 72–78. https://doi.org/10.1016/j.vph.2018.06.014
Stary HC (2000) Natural History and Histological Classification of Atherosclerotic Lesions. Arterioscler Thromb Vasc Biol 20: 1177–1178. https://doi.org/10.1161/01.ATV.20.5.1177
Camaré C, Pucelle M, Nègre-Salvayre A, Salvayre R (2017) Angiogenesis in the atherosclerotic plaque. Redox Biol 12: 18–34. https://doi.org/10.1016/j.redox.2017.01.007
Zhou Y, Zhu X, Cui H, Shi J, Yuan G, Shi S, Hu Y (2021) The Role of the VEGF Family in Coronary Heart Disease. Front Cardiovasc Med 8: 738325. https://doi.org/10.3389/fcvm.2021.738325
Melincovici CS, Boşca AB, Şuşman S, Mărginean M, Mihu C, Istrate M, Moldovan IM, Roman AL, Mihu CM (2018) Vascular endothelial growth factor (VEGF) – key factor in normal and pathological angiogenesis. Rom J Morphol Embryol 59: 455–467. http://www.ncbi.nlm.nih.gov/pubmed/30173249
Pelisek J, Well G, Reeps C, Rudelius M, Kuehnl A, Culmes M, Poppert H, Zimmermann A, Berger H, Eckstein H-H (2012) Neovascularization and angiogenic factors in advanced human carotid artery stenosis. Circ J 76: 1274–1282. https://doi.org/10.1253/circj.cj-11-0768
Chanakira A, Dutta R, Charboneau R, Barke R, Santilli SM, Roy S (2012) Hypoxia differentially regulates arterial and venous smooth muscle cell proliferation via PDGFR-β and VEGFR-2 expression. Am J Physiol Heart Circ Physiol 302: H1173–H1184. https://doi.org/10.1152/ajpheart.00411.2011
Roy H, Bhardwaj S, Babu M, Kokina I, Uotila S, Ahtialansaari T, Laitinen T, Hakumaki J, Laakso M, Herzig K-H, Ylä-Herttuala S (2006) VEGF-A, VEGF-D, VEGF receptor-1, VEGF receptor-2, NF-kappaB, and RAGE in atherosclerotic lesions of diabetic Watanabe heritable hyperlipidemic rabbits. FASEB J 20: 2159–2161. https://doi.org/10.1096/fj.05-5029fje
Claesson-Welsh L (2016) VEGF receptor signal transduction – A brief update. Vascul Pharmacol 2001: re21. https://doi.org/10.1126/stke.2001.112.re21
Stacker SA, Achen MG (2018) Emerging Roles for VEGF-D in Human Disease. Biomolecules 8: 1. https://doi.org/10.3390/biom8010001
Rutanen J, Leppänen P, Tuomisto TT, Rissanen TT, Hiltunen MO, Vajanto I, Niemi M, Häkkinen T, Karkola K, Stacker SA, Achen MG, Alitalo K, Ylä-Herttuala S (2003) Vascular endothelial growth factor-D expression in human atherosclerotic lesions. Cardiovasc Res 59: 971–979. https://doi.org/10.1016/s0008-6363(03)00518-2
Zhao T, Zhao W, Meng W, Liu C, Chen Y, Bhattacharya SK, Sun Y (2016) Vascular endothelial growth factor-D mediates fibrogenic response in myofibroblasts. Mol Cell Biochem 413: 127–135. https://doi.org/10.1007/s11010-015-2646-1
Engelmann D, Mayoli-Nüssle D, Mayrhofer C, Fürst K, Alla V, Stoll A, Spitschak A, Abshagen K, Vollmar B, Ran S, Pützer BM (2013) E2F1 promotes angiogenesis through the VEGF-C/VEGFR-3 axis in a feedback loop for cooperative induction of PDGF-B. J Mol Cell Biol 5: 391–403. https://doi.org/10.1093/jmcb/mjt035
Liu J, Sun F, Wang X, Bi Q (2020) miR-27b promotes angiogenesis and skin repair in scalded rats through regulating VEGF-C expression. Lasers Med Sci 35: 1577–1588. https://doi.org/10.1007/s10103-020-02991-7
Zarkada G, Heinolainen K, Makinen T, Kubota Y, Alitalo K (2015) VEGFR3 does not sustain retinal angiogenesis without VEGFR2. Proc Natl Acad Sci U S A 112: 761–766. https://doi.org/10.1073/pnas.1423278112
Nilsson I, Bahram F, Li X, Gualandi L, Koch S, Jarvius M, Söderberg O, Anisimov A, Kholová I, Pytowski B, Baldwin M, Ylä-Herttuala S, Alitalo K, Kreuger J, Claesson-Welsh L (2010) VEGF receptor 2/-3 heterodimers detected in situ by proximity ligation on angiogenic sprouts. EMBO J 29: 1377–1388. https://doi.org/10.1038/emboj.2010.30
Chen R, Lee C, Lin X, Zhao C, Li X (2019) Novel function of VEGF-B as an antioxidant and therapeutic implications. Pharmacol Res 143: 33–39. https://doi.org/10.1016/j.phrs.2019.03.002
Fallah A, Sadeghinia A, Kahroba H, Samadi A, Heidari HR, Bradaran B, Zeinali S, Molavi O (2019) Therapeutic targeting of angiogenesis molecular pathways in angiogenesis-dependent diseases. Biomed Pharmacother 110: 775–785. https://doi.org/10.1016/j.biopha.2018.12.022
Dolivo DM, Larson SA, Dominko T (2017) Fibroblast Growth Factor 2 as an Antifibrotic: Antagonism of Myofibroblast Differentiation and Suppression of Pro-Fibrotic Gene Expression. Cytokine Growth Factor Rev 38: 49–58. https://doi.org/10.1016/j.cytogfr.2017.09.003
Presta M, Dell’Era P, Mitola S, Moroni E, Ronca R, Rusnati M (2005) Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev 16: 159–178. https://doi.org/10.1016/j.cytogfr.2005.01.004
Chen P, Qin L, Li G, Tellides G, Simons M (2016) Smooth muscle FGF/TGFβ cross talk regulates atherosclerosis progression. EMBO Mol Med 8: 712–728. https://doi.org/10.15252/emmm.201506181
Tillie RJHA, Theelen TL, van Kuijk K, Temmerman L, de Bruijn J, Gijbels M, Betsholtz C, Biessen EAL, Sluimer JC (2021) A Switch from Cell-Associated to Soluble PDGF-B Protects against Atherosclerosis, despite Driving Extramedullary Hematopoiesis. Cells 10: 1746. https://doi.org/10.3390/cells10071746
Laddha AP, Kulkarni YA (2019) VEGF and FGF-2: Promising targets for the treatment of respiratory disorders. Respir Med 156: 33–46. https://doi.org/10.1016/j.rmed.2019.08.003
Margosio B, Marchetti D, Vergani V, Giavazzi R, Rusnati M, Presta M, Taraboletti G (2003) Thrombospondin 1 as a scavenger for matrix-associated fibroblast growth factor 2. Blood 102: 4399–4406. https://doi.org/10.1182/blood-2003-03-0893
Heldin CH, Westermark B (1999) Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev 79: 1283–1316. https://doi.org/10.1152/physrev.1999.79.4.1283
Papadopoulos N, Lennartsson J (2018) The PDGF/PDGFR pathway as a drug target. Mol Aspects Med 62: 75–88. https://doi.org/10.1016/j.mam.2017.11.007
Greenberg JI, Shields DJ, Barillas SG, Acevedo LM, Murphy E, Huang J, Scheppke L, Stockmann C, Johnson RS, Angle N, Cheresh DA (2008) A role for VEGF as a negative regulator of pericyte function and vessel maturation. Nature 456: 809–813. https://doi.org/10.1038/nature07424
Greenaway J, Lawler J, Moorehead R, Bornstein P, Lamarre J, Petrik J (2007) Thrombospondin-1 inhibits VEGF levels in the ovary directly by binding and internalization via the low density lipoprotein receptor-related protein-1 (LRP-1). J Cell Physiol 210: 807–818. https://doi.org/10.1002/jcp.20904
Ganguly R, Sahu S, Ohanyan V, Haney R, Chavez RJ, Shah S, Yalamanchili S, Raman P (2017) Oral chromium picolinate impedes hyperglycemia-induced atherosclerosis and inhibits proatherogenic protein TSP-1 expression in STZ-induced type 1 diabetic ApoE-/- mice. Sci Rep 7: 45279. https://doi.org/10.1038/srep45279
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова