

Российский физиологический журнал им. И.М. Сеченова, 2022, T. 108, № 6, стр. 745-761
Влияние оксида азота на электрическую активность тройничного нерва и состояние тучных клеток твердой мозговой оболочки крысы
К. С. Королёва 1, *, С. О. Свитко 1, Д. А. Нурмиева 1, О. Ш. Гафуров 1, А. Д. Буглинина 1, Г. Ф. Ситдикова 1
1 Казанский (Приволжский) федеральный университет
Казань, Россия
* E-mail: k.s.koroleva@yandex.ru
Поступила в редакцию 18.04.2022
После доработки 12.05.2022
Принята к публикации 12.05.2022
- EDN: CWLASG
- DOI: 10.31857/S0869813922060048
Аннотация
Оксид азота (NO) – представитель семейства газомедиаторов, участвующих в регуляции различных биологических процессов. Нитроглицерин, донор NO, широко используется для моделирования мигрени как у человека, так и у животных. Однако роль периферических нейрональных структур в эффектах NO практически не изучена. Целью работы являлось выявление эффектов NO на электрическую активность тройничного нерва и состояние тучных клеток оболочек головного мозга крыс. В работе использовали электрофизиологический метод регистрации потенциалов действия (ПД) тройничного нерва крыс, иннервирующего твердую мозговую оболочку, в препарате получерепа крысы. Для анализа электрической активности использовали метод кластеризации, позволяющий разбить ПД на группы с близкими характеристиками, генерируемые отдельными волокнами. Морфологию тучных клеток оценивали путем окрашивания оболочек головного мозга крыс толуидиновым синим. Субстрат синтеза NO – L-аргинин дозозависимо увеличивал электрическую активность тройничного нерва, и этот эффект отменялся на фоне блокатора синтеза NO – L-NAME (100 мкМ). Экзогенный донор NO – нитропруссид натрия (НПН 200 мкМ) вызывал усиление частоты ПД, тогда как инактивированный светом НПН не оказывал влияния на частоту ПД. Использование кластерного анализа выявило, что НПН вызывал сначала увеличение частоты ПД низкой амплитуды, распространяющиеся с низкой скоростью в волокнах С-типа, отвечающих на капсаицин; затем наблюдалось появление высокоамплитудных ПД, распространяющихся в Аδ-волокнах. Блокатор растворимой гуанилатциклазы – ODQ (10 мкМ) предотвращал НПН-вызванное увеличение электрической активности. При этом инкубация препарата в НПН не оказывала влияния на морфологию тучных клеток. Полученные данные свидетельствуют о том, что как экзогенный, так и эндогенный NO усиливают электрическую активность тройничного нерва путем активации гуанилатциклазы, оказывая вклад в периферические нейрональные механизмы возникновения боли при мигрени.
Оксид азота (NO) – газообразная сигнальная молекула, вовлеченная в регуляцию широкого спектра физиологических процессов и клеточных функций в организме человека [1–5]. NO продуцируется внутриклеточно в результате окисления L-аргинина с образованием NO и L-цитруллина с помощью фермента NO-синтазы (NOS), которая существует в виде трех изоформ: нейрональная NOS (nNOS), эндотелиальная NOS (eNOS) и индуцибельная NOS (iNOS). nNOS и eNOS являются конститутивными и активируются в ответ на повышение внутриклеточной концентрации Ca2+ и его последующего связывания с кальмодулином [6]. nNOS преимущественно экспрессируется в нейронах и обнаруживается как в центральной, так и в периферической нервной системе [6, 7]. В менингеальных оболочках eNOS была обнаружена в эндотелии средней менингеальной артерии, nNOS – в тонких разветвлениях нервных волокон, располагающихся вдоль артерии, при этом только небольшая доля нейронов тройничного ганглия является NOS-позитивной [8].
NO играет важную роль в регуляции тонуса кровеносных сосудов, в том числе и церебральных, а также в механизмах ноцицепции [6]. Это позволяет рассматривать NO в качества “ключевого звена” в формировании мигрени [9], в основе которой лежат сосудистые и нейрональные изменения [10, 11]. Действительно, донор NO – нитроглицерин используется для моделирования мигрени у человека, а также в экспериментальных моделях мигрени у животных, а специфические ингибиторы NO-синтаз проявляют обезболивающие эффекты у испытуемых, страдающих мигренью [12].
Растворимая гуанилатциклаза является основным рецептором для NO в организме, хотя NO также может иметь независимые от гуанилатциклазы эффекты [13], такие как изменение возбудимости нейронов за счет активации или ингибирования ионных каналов [14], стимуляция синтеза и высвобождения кальцитонин-ген-связанного пептида (CGRP) [15–17], а также влияние на состояние тучных клеток [18–20]. Кроме того, NO может взаимодействовать с другой газообразной молекулой, образующейся в тройничном ганглии, сероводородом (H2S) с образованием нитрозила, способного активировать TRPA1-рецепторы, увеличивая высвобождение CGRP [21, 22].
Достоверно известно, что боль во время приступа мигрени вызывается сенсибилизацией/активацией периферических отростков тройничного нерва, находящихся в менингеальных тканях, где формируется начальный ноцицептивный сигнал [23, 24]. Однако данные о влиянии донора NO на активность афферентов оболочки головного мозга неоднозначны. Показано, что NO может как увеличивать, так и снижать чувствительность ноцицепторов в зависимости от исходного уровня возбудимости [25].
Известно, что активация иммунной системы оказывает вклад в патогенез мигрени [26]. В твердой мозговой оболочке наблюдается многочисленное скопление тучных клеток, которые локализованы вблизи сосудов и нервных волокон тройничного нерва [27, 28]. Тучные клетки содержат и могут высвобождать провоспалительные медиаторы, которые вносят вклад в поддержание длительной ноцицептивной импульсации в афферентах тройничного нерва во время приступа мигрени [29–31]. Согласно некоторым данным, тучные клетки экспрессируют NO-синтазы и сами продуцируют NO [32–34].
Целью настоящей работы являлся анализ роли оксида азота в регуляции возбудимости периферических структур твердой мозговой оболочки, участвующих в генерации ноцицептивных сигналов. Для этого мы исследовали эффекты субстрата синтеза NO – L-аргинина и экзогенного донора NO – нитропруссида натрия (НПН) на электрическую активность тройничного нерва крысы. Мы также оценили роль гуанилатциклазы в эффектах НПН и состояние тучных клеток твердой оболочки головного мозга при действии НПН.
МЕТОДЫ ИССЛЕДОВАНИЯ
Электрофизиологический метод регистрации потенциалов действия тройничного нерва
Эксперименты проводились на самцах крыс линии Wistar возраста Р40–45 (Р – день рождения). Все экспериментальные протоколы соответствовали этическим нормам по гуманному обращению с животными, принятым в Казанском Федеральном Университете и одобрены Локальным этическим комитетом КФУ (протокол 33 от 25.11.2021). Для изучения электрических сигналов, возникающих в периферическом отростке тройничного нерва, мы использовали препарат изолированного черепа крысы, представляющий из себя половину черепной коробки с извлеченным головным мозгом [35–37]. После декапитации черепная коробка крысы тщательно очищалась от наружных черепных мышц. Череп разделялся сагиттальным разрезом на две половины, мозг аккуратно извлекался с помощью пинцета из черепной коробки, оставляя нетронутой твердую мозговую оболочку (dura mater). В твердой оболочке мозга сохраняли отростки тройничного нерва (nervus spinosus), иннервирующие среднюю менингеальную артерию.
Препарат постоянно перфузировался физиологическим раствором следующего состава (в мМ): NaCl 120, KCl 2.5, CaCl2 2, MgCl2 1, глюкоза 11, Na2HPO4 1, NaHCO3 24, рН 7.2–7.4 с постоянной аэрацией карбогеном (O2 95%/CO2 5%). Регистрирующие электроды были изготовлены из боросиликатных заготовок фирмы “Sutter instrument”, диаметр кончика электрода составлял 150 мкм. Перед началом эксперимента стеклянный электрод фиксировался в держателе с манипулятором, подключенному к предусилителю электрических сигналов. Под визуальным контролем из твердой мозговой оболочки выделяли периферический отросток тройничного нерва, nervus spinosus (V1-ветвь тройничного нерва), иннервирующих среднюю менингеальную артерию, и помещали его в стеклянный регистрирующий электрод. Далее осуществляли запись потенциалов действия (ПД) при помощи усилителя DAM 80 (World Precision Instruments, Sarasota, FL, США). Вначале регистрировалась базовая электрическая активность тройничного нерва. Затем в область расхождения средней менингеальной артерии апплицировали интересующие нас вещества и записывали их действие на протяжении 20–40 мин, после чего проводили отмывку. В экспериментах использовали: нитропруссид натрия (НПН) в концентрациях 100 и 200 мкМ, НПН, инактивированный светом в концентрации 200 мкМ; L-аргинин в концентрациях 100, 200 и 300 мкМ. Каждая исследуемая концентрация НПН и L-аргинина тестировалась на новом препарате. Запись ПД при воздействии тестируемых соединений осуществлялась на протяжении 20 мин. Также был использован 1H-[1, 2, 4]Oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) – ингибитор гуанилатциклазы, 10 мкМ; в котором препарат инкубировался на протяжении 20 мин, после чего осуществлялась запись ПД в растворе с добавлением НПН (200 мкМ) на фоне ODQ (20 мин). N(G)-Nitro-L-arginine (L-NAME), 100 мкМ, в котором препарат инкубировался на протяжении 30 мин, после чего в раствор добавлялся L-аргинин (300 мкМ) на 20 мин. Для активации ноцицептивных пептидергических С-волокон в конце эксперимента апплицировали капсаицин в концентрации 1 мкМ. Сигналы были оцифрованы на ПК с использованием платы NI PCI6221 (National Instruments, Austin, TX, США). Сигналы визуализировались и анализировались с использованием программного обеспечения WinEDR v.3.2.7 (University of Strathclyde, Glasgow, Великобритания). Частота рассчитывалась как количество ПД за 5 мин. Контролем для сравнения в каждом эксперименте служил пятиминутный отрезок записи перед подачей исследуемого вещества.
Кластерный анализ потенциалов действия
Экспериментальные данные записывались и проходили первоначальную фильтрацию с помощью усилителя в полосе 100–3000 Гц. Оцифрованные данные фильтровались с помощью фильтра Чебышева типа II. Для обнаружения сигналов выбирался интервал 20 с без ПД в начале каждого эксперимента, где рассчитывалась дисперсия шума. Поиск ПД осуществлялся по превышению амплитуды больше пяти стандартных отклонений базового шума. Амплитуда каждого найденного сигнала была нормализована по базовому шуму и выражена в относительных единицах (о.е.). Для каждого найденного ПД определялись амплитудно-временные параметры такие как: амплитуда положительной и отрицательной фазы, время нарастания; длительности фаз спайка; время спада. Для анализа активности отдельных нервных волокон проводили разделение ПД на группы – кластеризацию в соответствии с амплитудно-временными параметрами. Для кластеризации использовали метод KlustaKwik [38]. Проведение кластеризации, построение графиков и гистограмм были выполнены с помощью программного обеспечения MATLAB (MathWorks, Natick, MA, США).
Исследование дегрануляции тучных клеток
Исследование дегрануляции тучных клеток осуществлялось с помощью гистологического метода окрашивания твердой оболочки мозга крыс (Р35–40) толуидиновым синим [39]. Интактные черепа были помещены в раствор Кребса с исследуемыми растворами (НПН в концентрации 100 и 200 мкМ) на 30 мин в полной темноте для предотвращения диссоциации НПН, после чего получерепа помещались в параформальдегид (4%-ный раствор) на 12 ч. Перед выделением мозговых оболочек черепа промывали в фосфатно-солевом буферном растворе следующего состава (мМ): 137 NaCl, 2.7 KCl, 10 Na2HPO4, 1.8 K2HPO4. Выделенные оболочки фиксировали на предметном стекле. Окрашивание толуидиновым синим длилось 10 мин, затем фиксированные препараты омывались дистиллированной водой и обезвоживались этиловым спиртом (95–99%). Съемка осуществлялась при 20× увеличении. Степень дегрануляции оценивали визуально, расчет велся в % от общего количества клеток (не менее 100).
Статистическая обработка
Статистический анализ проводили с помощью программного обеспечения Origin Pro (OriginLab Corporation, Northampton, MA, США), Matlab (The MathWorks, Inc., Natick, MA, США) и GraphPad Prism 4.0 (GraphPad Software, Inc., San Diego, CA, США). Нормальность распределения выборки определяли при помощи F-теста Фишера и критерия Шапиро–Уилка. Достоверность различий оценивали с помощью критерия Вилкоксона для связанных выборок. Различия считали статистически значимыми при р < 0.05; n указывает количество животных. В тексте данные представлены как среднее и SE, где SE — стандартная ошибка. Критерий хи-квадрат использовался для определения значимости между количественным распределением кластеров электрической активности нерва при воздействии экзогенного донора NO.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Многочисленные данные указывают на то, что головные боли при мигрени возникают в результате активации окончаний тройничного нерва в мозговых оболочках с последующей сенсибилизацией нейронов [10, 40]. Для анализа роли эндогенного NO в регуляции активности тройничного нерва использовали субстрат NO-синтазы – L-аргинин. L-аргинин в концентрации 100 мкМ не вызывал изменения частоты ПД в течение 20 мин действия (рис. 1). Частота ПД в контроле составила 421.6 ± 135.7 ПД за 5 мин и к 15-й мин действия L-аргинина – 417.6 ± 107.6 ПД за 5 мин (n = 4, p = 1; рис. 1a, c). L-аргинин в концентрации 200 мкМ вызывал увеличение частоты ПД с 292.1 ± 73.1 ПД за 5 мин в контроле до 661.6 ± 150.8 ПД за 5 мин к 15-й мин (n = 4; p = 0.04) и к 20-й мин частота ПД составила 439.8 ± 128.1 ПД за 5 мин (n = 4; p = 0.18), прирост частоты составил 138 ± 26% к концу 15-й мин (n = 4; рис. 1a, c). Максимальное увеличение частоты ПД наблюдалось при использовании L-аргинина в концентрации 300 мкМ, которое составило 357.4 ± 64.4 ПД за 5 мин, а к 15-й мин (в контроле – 150.2 ± 35.6 ПД за 5 мин) прирост составил 182 ± 84% (n = 4; рис. 1b, c).
Рис. 1.
Влияние L-аргинина на частоту потенциалов действия в тройничном нерве крысы. a – пример записи ПД в тройничном нерве крысы в контроле и после аппликации L-аргинина (L-arg) в концентрациях 100, 200 и 300 мкМ и L-arg (300 мкМ) на фоне ингибитора NO-синтазы, L-NAME (100 мкМ); b – изменение частоты ПД (число ПД за 5 мин) в абсолютных значениях во времени при аппликации L-arg в концентрации 300 мкМ; c – средняя частота ПД за 5 мин (%) при аппликации L-arg в концентрациях 100, 200 и 300 мкМ и L-arg на фоне L-NAME (100 мкМ) по сравнению с контрольными значениями, принятыми за 100% (*p < 0.05; парный тест Вилкоксона).
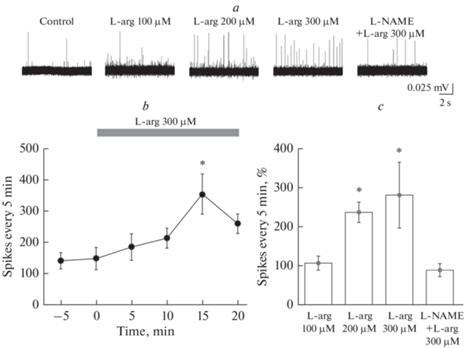
Блокатор NO-синтазы L-NAME не изменял базовую частоту ПД в афферентах тройничного нерва. В контроле частота ПД составила 306.8 ± 73.1 ПД за 5 мин, к 15-й мин инкубации в L-NAME частота ПД составила 365.8 ± 105.1 ПД за 5 мин (p = 0.72); к 20-й мин – 369.4 ± 55.2 ПД за 5 мин (n = 4; p = 0.52). L-аргинин (300 мкМ) на фоне L-NAME не вызывал увеличения частоты ПД в течение всего времени аппликации: частота ПД к 15-й мин составила – 264.6 ± 33.7 ПД за 5 мин (n = 4; p = 0.18; рис. 1a, c).
Донор NO, НПН в концентрации 100 мкМ не оказывал влияния на электрическую активность тройничного нерва (p > 0.05; n = 5; рис. 2a, c). В контроле частота составила 454.8 ± 111.4 ПД за 5 мин, после добавления НПН к 15-й мин – 672.8 ± ± 163.4 ПД за 5 мин (n = 5, p = 0.29). Увеличение концентрации НПН до 200 мкМ вызывало двукратное повышение частоты ПД с 260.4 ± 64.8 ПД за 5 мин в контроле до 552.5 ± 99.3 ПД к 10-й мин (p = 0.04); 532.7 ± 84.8 ПД к 15-й мин (p = 0.01) и 476.7 ± 154.8 ПД за 5 мин к 20-й мин инкубации (p = 0.15; n = 4; рис. 2b). Прирост частоты ПД составил 109 ± 42% (рис. 2c). Поскольку НПН при диссоциации образует дисульфидные соединения и цианид-ионы [9], нами был использован инактивированный на свету НПН. Инактивированный светом НПН (Ин) в концентрации 200 мкМ не изменял активность тройничного нерва (частота ПД в контроле составила 716.7 ± 220.9 ПД за 5 мин; в присутствии НПН (Ин) – 972.5 ± 298.3 ПД за 5 мин к 10-й мин аппликации (p = 0.62); 876.7 ± 287.6 ПД за 5 мин к 15-й мин (p = = 0.37); 840.1 ± 196.6 ПД за 5 мин к 20-й мин; p = 0.37; n = 4; рис. 2a, c).
Рис. 2.
Влияние нитропруссида натрия (SNP) на частоту потенциалов действия в тройничном нерве крысы. a – пример записи ПД в тройничном нерве крысы в контроле, при аппликации SNP в концентрациях 100 и 200 мкМ, а также инактивированного светом (SNP (N)) и SNP (200 мкМ) на фоне ODQ (10 мкМ); b – динамика изменения средней частоты ПД за 5 мин при аппликации SNP в концентрации 200 мкМ; c – средняя частота ПД за 5 мин в %, возникающих при аппликации SNP в концентрациях 100 и 200 мкМ, инактивированного светом (SNP (N)) и SNP на фоне ODQ (10 мкМ) по отношению контрольным значениям, принятым за 100% (*p < 0.05; парный тест Вилкоксона).
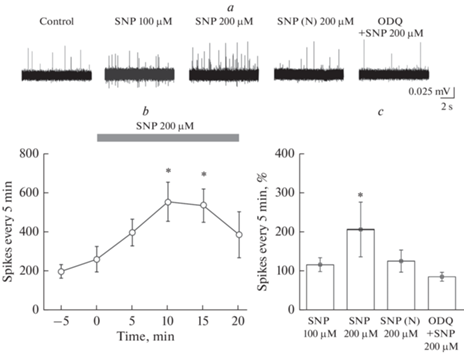
Для выявления роли растворимой гуанилатциклазы в эффектах NO использовали ингибитор ODQ в концентрации 10 мкМ. Инкубация препарата в ODQ в течение 20 мин не приводила к изменениям частоты ПД (n = 4; р = 0.87). Базовая частота ПД в тройничном нерве составила 187.5 ± 86.2 ПД за 5 мин и 172.5 ± 30.1 ПД за 5 мин после 20-минутной инкубации в ODQ (10 мкМ) (n = 4; р = 0.87). Последующее добавление НПН (200 мкМ) не вызывало достоверного изменения частоты ПД и к 10-й мин аппликации частота ПД составила 145.2 ± 47.3 ПД за 5 мин и к 15-й мин 129.7 ± 50.8 ПД за 5 мин (p = 0.62); 20 мин 127.1 ± 40.3 ПД за 5 мин (n = 4; p = 0.12; рис. 2c).
Регистрируемые нами ПД отличались амплитудно-временными параметрами, такими как амплитуда, время роста, постоянная спада и др. Например, амплитуда ПД могла составлять от 35 до 120 мкВ. Такое широкое варьирование амплитудно-временных параметров можно объяснить уникальными свойствами различных нервных волокон и особенностями регистрации ПД с помощью подсасывающего электрода. Используемый нами метод кластеризации [36] позволил разбить регистрируемые ПД на группы или кластеры с близкими характеристиками, которые, как было предположено, сформированы одним волокном. Поэтому считается, что активность возникновения ПД в кластере отражает активность возникновения ПД в отдельном нервном волокне. Применение агониста TRPV1-рецепторов, капсаицина (1 мкМ), позволило выявить пептидергические капсаицин-чувствительные С-волокна [41, 42]. С использованием кластерного анализа нами было получено следующее распределение кластеров при аппликации НПН и капсаицина: отвечающие только на аппликацию НПН (рис. 3a(a)); НПН и капсаицина (рис. 3a(b)); только капсаицина (рис. 3a(c)) и не отвечающие (рис. 3a(d)).
Рис. 3.
Кластерный анализ электрической активности тройничного нерва при действии донора NO нитропруссида натрия. a – Примеры частотной активности различных кластеров при аппликации 200 мкМ SNP и 1 мкМ капсаицина (Caps). a (a, b) – кластеры с выраженным ответом на SNP 200 мкМ; a (c, d) – кластеры без выраженного ответа на SNP 200 мкМ. b – Круговая диаграмма распределения кластеров по типам ответов на SNP и Caps (в процентах к общему количеству кластеров): а – SNP 100 мкМ; b – SNP 200 мкМ; c – Изменение частоты ПД низкой, средней и высокой амплитуды в контроле и на фоне SNP 200 мкМ (*p < 0.05).
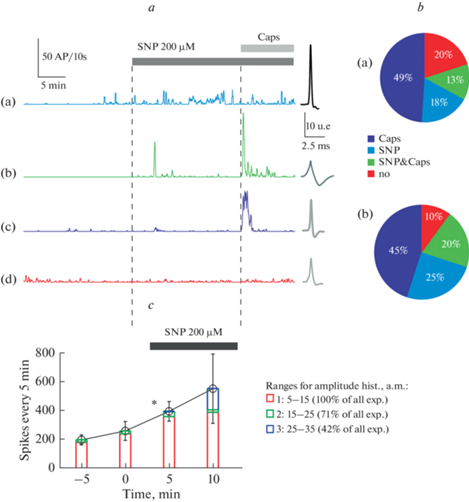
Аппликация НПН в концентрации 100 мкМ приводила к повышению частоты ПД в 18% кластерах. В 13% кластеров наблюдалось увеличение частоты ПД в ответ и на НПН, и на капсаицин (рис. 3b(a), n = 7). Повышение концентрации НПН до 200 мкМ привело к увеличению доли кластеров, отвечающих только на НПН (25% кластеров) и одновременно отвечающих на НПН и капсаицин (20% кластеров) (рис. 3b(b), n = 7). В обеих сериях экспериментов около 50% кластеров отвечали только на капсаицин и процент “молчащих” кластеров составил 20% при действии НПН 100 мкМ и 10% при действии НПН 200 мкМ (рис. 3b). Сравнение количественного распределения кластеров электрической активности нерва показало, что при воздействии более высоких концентрациях НПН (200 мкМ) наблюдается не только увеличение частоты, но и увеличение общего числа кластеров. В контроле общее число кластеров составило 52, после добавления НПН 200 мкМ число кластеров составило 74 (chi = 3.98; р = 0.04; n = 7). При воздействии НПН 100 мкМ общее число кластеров составило 71, сопоставимо с количеством в контроле (81 кластер, n = 7). Кроме того, было показано достоверное увеличение количества кластеров “отвечающих” на НПН 200 мкМ (42 кластеров, по сравнению с НПН 100 (14 кластеров); chi = 9.78; p = 0.01; n = 7).
Далее был проведен анализ вклада ПД различной амплитуды в эффекты НПН. Для этого были выделены кластеры низкоамплитудных (рис. 3c, красная колонка), среднеамплитудных (рис. 3c, зеленая колонка) и высокоамплитудных (рис. 3c, синяя колонка) ПД. Оказалось, что основной вклад в активность тройничного нерва вносят низкоамплитудные ПД. Аппликация НПН в концентрации 100 мкМ не оказала заметного влияния на распределение ПД различной амплитуды, тогда как НПН 200 мкМ вызывал усиление частоты ПД низкой амплитуды, так и кластеров высокоамплитудных ПД (рис. 3c).
Исследование роли донора оксида азота на состояние тучных клеток
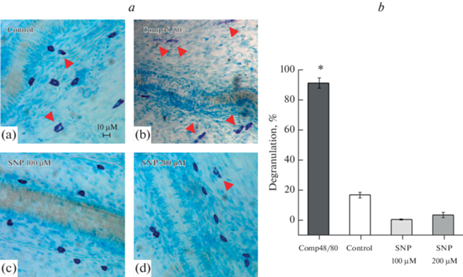
Тучные клетки в изобилии присутствуют в твердой мозговой оболочке, окружая сосуды и нервные окончания, а состояние тучных клеток может оказывать влияние на возбудимость нейрональной мембраны вследствие выделения различных активных молекул [29, 30, 43]. Для исследования эффектов NO на состояние тучных клеток интактные оболочки мозга инкубировали в растворе НПН в концентрациях 100 и 200 мкМ в течение 30 мин. В качестве положительного контроля был использован Compоund 48/80 (10 мкг/мл), дегранулирующий тучные клетки [44]. Инкубация оболочек головного мозга крысы в растворе, содержащем Compоund 48/80 (10 мкг/мл), в течение 30 мин приводила к увеличению числа дегранулированных тучных клеток, которое составило 91.1 ± 3.4% (n = 5; рис. 4a(b), b). Инкубация оболочек в НПН не приводила к изменению морфологии тучных клеток, и количество дегранулированных клеток не превышало контрольные значения. В контрольной группе число дегранулированных тучных клеток составило 16.6 ± 1.34% (n = 5; рис. 4a(a), b), в группе НПН 100 мкМ было зафиксировано около 1% дегранулированных клеток (n = 5; рис. 4a(c), b), при увеличении концентрации НПН до 200 мкМ число дегранулированных клеток составило 3.6 ±1.7% (n = 4; рис. 4a(d), b).
Рис. 4.
Влияние нитропруссида натрия на морфологию тучных клеток. a – оригинальные снимки окрашенных оболочек головного мозга крыс толуидиновым синим в контроле (n = 5); после инкубации в Compound 48/80 (10 мкг/мл; n = 5); SNP (100 мкМ; n = 5); SNP (200 мкМ; n = 4). b – Уровень дегрануляции тучных клеток в контроле, при действии Compound 48/80, SNP в концентрациях 100 и 200 мкМ (*p < 0.05; U-критерий Манна–Уитни).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Изучение периферических механизмов мигрени имеет фундаментальное значение для разработки новых методов лечения, характеризующихся направленным действием непосредственно в место возникновения болевого сигнала. Твердая оболочка головного мозга, сосуды, множество чувствительных нервных окончаний тройничного нерва, а также тучные клетки формируют тригемино-васкулярную систему. Афференты тройничного нерва отвечают за сбор, анализ и обработку ноцицептивной информации от черепных структур, являются местом возникновения исходного болевого сигнала и могут быть потенциальной мишенью для терапевтического воздействия с целью профилактики и/или лечения мигрени [45–47]. Одной из эндогенных молекул, играющих ключевую роль в развитии мигрени, является NO, оказывающий сосудорасширяющие эффекты, что сопровождается выделением провоспалительных медиаторов в оболочках головного мозга, таких как гистамин, брадикинин, серотонин, CGRP, вещество P, нейрокинин A и простагландины, АТФ [48–51] и лежит в основе немедленной головной боли, вызванной донорами NO [52].
Согласно современным представлениям, NO может также непосредственно влиять на возбудимость нейрональных мембран, поэтому в нашей работе было проанализировано влияние экзогенного и эндогенного NO на частоту импульсации афферентов тройничного нерва, увеличение которой можно соотнести с возникновением субъективного ощущения боли. Поэтому импульсацию тройничного нерва также называют ноцицептивной, а эффекты повышения частоты ПД тройничного нерва при действии альгогенов – проноцицептивными [53].
Для анализа эффектов эндогенно образующегося NO на возбудимость афферентов тройничного нерва использовали субстрат синтеза NO – L-аргинин, который дозозависимо повышал электрическую активность тройничного нерва, и этот эффект блокировался использованием ингибитора NO-синтазы L-NAME. Полученные данные позволяют предположить, что NO образуется эндогенно в области иннервации тройничного нерва и повышает возбудимость его афферентов. Действительно, в тригемино-васкулярной системе эндогенный NO может образовываться с помощью eNOS, экспрессирующейся в эндотелиальных клетках артерий твердой мозговой оболочки [54], и nNOS периваскулярных нервных волокон [55, 56]. Эндогенными стимулами для NOS являются провоспалительные цитокины, CGRP, активация НМДА-рецепторов, увеличение концентрации внутриклеточного кальция [8]. Ранее нами было показано наличие функциональных НМДА-рецепторов в нейронах тройничного ганглия, как в соме, так и в окончаниях тройничного нерва [57]. Активация НМДА-рецепторов во время приступа мигрени может способствовать усилению синтеза NO за счет притока Ca2+. Кроме того, распространяющаяся корковая депрессия, являющаяся электрофизиологическим коррелятом мигрени с аурой, может способствовать выделению NO, вызывающему не только сосудистые, но и нейрональные эффекты [8, 58–60].
Участие эндогенного NO в патогенезе мигрени было подтверждено введением специфических ингибиторов NO-синтаз, которые показали свою эффективность во время лечения острого приступа мигрени [61–65]. L-NAME ослаблял CGRP-вызванную вазодилатацию [61], ингибировал как текущую активацию нейронов [64], так и вызванную экспрессию Fos-иммунореактивных белков в тригемино-цервикальном комплексе [66].
Экзогенный донор NO – НПН вызывал сходные с L-аргинином эффекты, вызывая повышение электрической активности тройничного нерва. Механизм высвобождения NO из НПН связан с взаимодействием с сульфгидрил-содержащими соединениями, такими как глутатион и цистеин. В результате этого взаимодействия происходит образование дисульфидных соединений, цианид-ионов и S-нитрозотиолов. S-нитрозитиолы in vivo являются хранилищем NO, из которых происходит его дальнейшее высвобождение посредством механизма переноса электронов, в который вовлечена тиоловая группа [67]. Инактивированный светом НПН не оказывал влияния на частоту ПД тройничного нерва, это указывает на то, что именно высвобождающийся NO как эффекторная молекула реализует свои эффекты при аппликации НПН.
Использование кластерного анализа позволило показать, что часть эффектов НПН опосредована активацией капсаицин-чувствительных С-волокон. Действительно, анализ распределения амплитуд ПД во времени показал, что 200 мкМ НПН приводил сначала к увеличению частоты ПД низкой амплитуды, которые распространяются с низкой скоростью в немиелинизированных С волокнах, затем наблюдалось появление высокоамплитудных ПД, распространяющихся быстрее, по-видимому, генерирующихся в Аδ волокнах [68]. Кроме того, примерно половина волокон, отвечающих на НПН, также отвечала на аппликацию капсаицина, специфического агониста TRPV1-рецепторов, экспрессирующихся в пептидергических волокнах С-типа [41, 42].
Эффекты NO могут опосредоваться как цГМФ-зависимыми, так и цГМФ-независимыми механизмами [8]. В нашем исследовании эффекты НПН полностью предотвращались ингибитором гуанилатциклазы ODQ, что указывает на ведущую роль гуанилатциалзы в эффектах НПН. Активация гуанилатциклазы приводит к синтезу цГМФ и активации протеинкиназы G [65, 69], которая через процессы фосфорилирования может влиять на активность ионных каналов, рецепторов и играет важную роль в гомеостазе Ca2+. Основной мишенью PKG в нервных окончаниях являются Са2+-активируемые K+-каналы большой проводимости (BK-каналы) [70]. BK-каналы экспрессируются в связанных с мигренью структурах, таких как черепные артерии, тройничный ганглий, ядро тройничного нерва и играют существенную роль в регуляции тонуса сосудов и возбудимости нейронов [71]. Активация BK-каналов способствует поддержанию длительной нейрональной активности за счет уменьшения времени инактивации Na+-каналов. Конечным результатом является усиление входа Ca2+, приводящее к высвобождению нейропептидов [70]. Кроме того, стимуляция растворимой гуанилатциклазы с помощью VL-102 непосредственно может повышать экспрессию и высвобождение CGRP из нейронов тройничного ганглия [72, 73]. Участие гуанилатциклазы в проноцицептивных эффектах подтверждается также исследованиями, в которых ODQ оказывал антиноцицептивные эффекты после интратекальной инъекции в моделях воспалительной и нейропатической боли [74–77].
Проноцицептивное действие NO может осуществляться и на уровне тучных клеток, локализующихся в твердой мозговой оболочке вблизи кровеносных сосудов и афферентов тройничного нерва как у людей [78, 79], так и у грызунов [80–82], образуя нейроиммунный синапс [29, 30, 43, 81]. В этой структуре активные вещества, выделяемые тучными клетками, могут активировать соседние ноцицептивные волокна, а соединения, высвобождаемые из активных волокон, в свою очередь, могут дегранулировать тучные клетки [83]. Недавно показано, что нитроглицерин при введении in vivo приводил к значительному увеличению дегрануляции тучных клеток [20, 84]. В нашей работе инкубация препарата в растворе, содержащем донор NO, не приводила к дегрануляции тучных клеток в оболочках головного мозга крысы, что согласуется с предыдущими исследованиями, где в условиях ex vivo экзогенные доноры NO не изменяли состояние тучных клеток [20]. По-видимому, при системном введении донора NO происходит выделение провоспалительных соединений (АТФ, CGRP, субстанции Р) из эндотелиальных клеток сосудов и нервных окончаний, которые вызывают дегрануляцию тучных клеток [85, 88]. Поэтому в условиях in vitro требуется больше времени для реализации эффекта NO на тучные клетки.
Известно, что нитроглицерин имеет двухфазные эффекты: кратковременная вазодилятация, которая, по-видимому, опосредует головную боль непосредственно после инфузии, особенно ярко выраженную у пациентов с хронической мигренью [86]. У этих же пациентов впоследствии наблюдается головная боль через несколько часов после введения нитроглицерина [8, 87].
По-видимому, нейрональная активация при действии NO, которая была показана в нашем исследовании, приводит к выделению CGRP и АТФ, а также к усилению синтеза NO эндотелиальными клетками и его высвобождению в межклеточное пространство [52–54]. Увеличение концентрации NO и провоспалительных медиаторов (в частности АТФ, серотонин, CGRP) вызывает сенситизацию афферентов тройничного нерва [55, 56], что может лежать в основе длительных проноцицептивных эффектов введения нитроглицерина [92].
Таким образом, полученные нами результаты свидетельствуют о том, что эндогенный и экзогенный донор NO оказывают проноцицептивное действие, вызывая активацию афферентов тройничного нерва крысы, увеличивая активность как пептидергических С-волокон, так и Аδ волокон, не изменяя состояния тучных клеток. Основной мишенью действия NO является растворимая гуанилатциклаза, активация которой, как известно, может приводить к синтезу цГМФ и к усилению активности протеинкиназы G, способной фосфорилировать ионные каналы и рецепторы, а также к усилению высвобождения CGRP из пептидергических нервных окончаний. Полученные данные раскрывают нейрональные механизмы участия NO в патогенезе мигрени и обосновывают возможность использования блокаторов NO для купирования боли при остром приступе мигрени.
Список литературы
Hermann A, Sitdikova GF, Weiger TM (2013) Gasotransmitters: Physiology and pathophysiology. Gasotransmitters Physiol Pathophysiol 1–204. https://doi.org/10.1007/978-3-642-30338-8
Ситдикова ГФ, Яковлев АВ, Герасимова ОВ, Яковлева ОВ (2014) Газообразные посредники – оксид азота, монооксид углерода и сероводород – как новый класс сигнальных молекул межклеточных коммуникаций. В кн: Избранные главы фундаментальной и трансляционной медицины. Ред Жданов РИ Казань. Изд-во Казанск универ. 97–112. [Sitdikova GF, Yakovlev AV, Gerasimova OV, Yakovleva OV (2014) Gaseous mediators – nitric oxide, carbon monoxide and hydrogen sulfide – as a new class of signaling molecules of intercellular communications. In book: Selected Chapters of Fundamental and Translational Medicine. Zhdanov RI (ed) Kazan. Kazan Univer Press. 97–112. (In Russ)].
Кузнецова ВЛ, Соловьева АГ (2015) Оксид азота: свойства, биологическая роль, механизмы действия. Современные проблемы науки и образования 4: 462–462. [Kuznetsova VL, Solovieva AG (2015) Nitric oxide: properties, biological role, mechanisms of action. Modern Problems Science Education 4: 462–462. (In Russ)].
Метельская ВА, Гуманова НГ (2005) Оксид азота: роль в регуляции биологических функций, методы определения в крови человека. Лаб мед 7: 19–24. [Metelskaya VA, Gumanova NG (2005) Nitric oxide: role in the regulation of biological functions, methods of determination in human blood. Lab Med 7: 19–24. (In Russ)].
Львова ОА, Орлова АЕ, Гусев ВВ, Ковтун ОП, Чегодаев ДА (2010) К вопросу о роли оксида азота в норме и при патологии нервной системы. Системная интеграция в здравоохранении 4: 20–35. [Lvova OA, Orlova AE, Gusev VV, Kovtun OP, Chegodaev DA (2010) To the question of the role of nitric oxide in normal and pathological conditions of the nervous system. System Integrat Healthcare 4: 20–35. (In Russ)].
Moncada S, Higgs EA (2006) Nitric oxide and the vascular endothelium. Handb Exp Pharmacol 176: 213–254. https://doi.org/10.1007/3-540-32967-6_7
Chachlaki K, Garthwaite J, Prevot V (2017) The gentle art of saying NO: How nitric oxide gets things done in the hypothalamus. Nat Rev Endocrinol 13: 521–535. https://doi.org/10.1038/nrendo.2017.69
Pradhan AA, Bertels Z, Akerman S (2018) Targeted Nitric Oxide Synthase Inhibitors for Migraine. Neurotherapeutics 15: 391–401. https://doi.org/10.1007/s13311-018-0614-7
Olesen J (2010) Nitric Oxide-Related Drug Targets in Headache. Neurotherapeutics 7: 183–190. https://doi.org/10.1016/j.nurt.2010.03.006
Goadsby PJ (2007) Recent advances in understanding migraine mechanisms, molecules and therapeutics. Trends Mol Med 13: 39–44. https://doi.org/10.1016/j.molmed.2006.11.005
Goadsby PJ (2009) Pathophysiology of Migraine. Neurol Clin 27: 335–360. https://doi.org/10.1016/j.ncl.2008.11.012
Ashina M, Hansen JM, Á Dunga BO, Olesen J (2017) Human models of migraine-short-Term pain for long-Term gain. Nat Rev Neurol 13: 713–724. https://doi.org/10.1038/nrneurol.2017.137
Bryan NS, Bian K, Murad F (2009) Discovery of the nitric oxide signaling pathway and targets for drug development. Front Biosci 14: 1–18. https://doi.org/10.2741/3228
Bonnet C, Hao J, Osorio N, Donnet A, Penalba V, Ruel J, Delmas P (2019) Maladaptive activation of Nav1.9 channels by nitric oxide causes triptan-induced medication overuse headache. Nat Commun 10: 1–13. https://doi.org/10.1038/s41467-019-12197-3
Bellamy J, Bowen EJ, Russo AF, Durham PL (2006) Nitric oxide regulation of calcitonin gene-related peptide gene expression in rat trigeminal ganglia neurons. Eur J Neurosci 23: 2057–2066. https://doi.org/10.1111/j.1460-9568.2006.04742.x
Capuano A, De Corato A, Lisi L, Tringali G, Navarra P, Dello Russo C (2009) Proinflammatory-activated trigeminal satellite cells promote neuronal sensitization: Relevance for migraine pathology. Mol Pain 5: 1–13. https://doi.org/10.1186/1744-8069-5-43
Messlinger K, Lennerz JK, Eberhardt M, Fischer MJM (2012) CGRP and NO in the trigeminal system: Mechanisms and role in headache generation. Headache 52: 1411–1427. https://doi.org/10.1111/j.1526-4610.2012.02212.x
Coleman JW (2002) Nitric oxide: A regulator of mast cell activation and mast cell-mediated inflammation. Clin Exp Immunol 129: 4–10. https://doi.org/10.1046/j.1365-2249.2002.01918.x
Forsythe P, Gilchrist M, Kulka M, Befus AD (2001) Mast cells and nitric oxide: Control of production, mechanisms of response. Int Immunopharmacol 1: 1525–1541. https://doi.org/10.1016/S1567-5769(01)00096-0
Pedersen SH, Ramachandran R, Amrutkar DV, Petersen S, Olesen J, Jansen-Olesen I (2015) Mechanisms of glyceryl trinitrate provoked mast cell degranulation. Cephalalgia 35: 1287–1297. https://doi.org/10.1177/0333102415574846
Eberhardt M, Dux M, Namer B, Miljkovic J, Cordasic N, Will C, Kichko TI, De La Roche J, Fischer M, Suárez SA, Bikiel D, Dorsch K, Leffler A, Babes A, Lampert A, Lennerz JK, Jacobi J, Martí MA, Doctorovich F, Högestätt ED, Zygmunt PM, Ivanovic-Burmazovic I, Messlinger K, Reeh P, Filipovic MR (2014) H2S and NO cooperatively regulate vascular tone by activating a neuroendocrine HNO-TRPA1-CGRP signalling pathway. Nat Commun 5: 1–17. https://doi.org/10.1038/ncomms5381
Dux M, Will C, Vogler B, Filipovic MR, Messlinger K (2016) Meningeal blood flow is controlled by H2S-NO crosstalk activating a HNO-TRPA1-CGRP signalling pathway. Br J Pharmacol 173: 431–445. https://doi.org/10.1111/bph.13164
Zhang X, Levy D, Kainz V, Noseda R, Jakubowski M, Burstein R (2011) Activation of central trigeminovascular neurons by cortical spreading depression. Ann Neurol 69: 855–865. https://doi.org/10.1002/ana.22329
Yam MF, Loh YC, Tan CS, Adam SK, Manan NA, Basir R (2018) General pathways of pain sensation and the major neurotransmitters involved in pain regulation. Int J Mol Sci 19. https://doi.org/10.3390/ijms19082164
Levy D, Strassman AM (2004) Modulation of dural nociceptor mechanosensitivity by the nitric oxide-cyclic GMP signaling cascade. J Neurophysiol 92: 766–772. https://doi.org/10.1152/jn.00058.2004
Edvinsson L, Haanes KA, Warfvinge K (2019) Does inflammation have a role in migraine? Nat Rev Neurol 15: 483–490. https://doi.org/10.1038/s41582-019-0216-y
Undem BJ, Riccio MM, Weinreich D, Ellis JL, Myers AC (1995) Neurophysiology of mast cell-nerve interactions in the airways. Int Arch Allergy Immunol 107: 199–201. https://doi.org/10.1159/000236976
Spanos C, Pang X, Ligris K, Letourneau R, Alferes L, Alexacos N, Sant GR, Theoharides TC (1997) Stress-induced bladder mast cell activation: Implications for interstitial cystitis. J Urol 157: 669–672. https://doi.org/10.1016/S0022-5347(01)65247-9
Theoharides TC, Donelan J, Kandere-Grzybowska K, Konstantinidou A (2005) The role of mast cells in migraine pathophysiology. Brain Res Rev 49: 65–76. https://doi.org/10.1016/j.brainresrev.2004.11.006
Levy D, Burstein R, Kainz V, Jakubowski M, Strassman AM (2007) Mast cell degranulation activates a pain pathway underlying migraine headache. Pain 130: 166–176. https://doi.org/10.1016/j.pain.2007.03.012
Levy D (2009) Migraine pain, meningeal inflammation, and mast cells. Curr Pain Headache Rep 13: 237–240. https://doi.org/10.1007/s11916-009-0040-y
Gilchrist M, McCauley SD, Befus AD (2004) Expression, localization, and regulation of NOS in human mast cell lines: Effects on leukotriene production. Blood 104: 462–469. https://doi.org/10.1182/blood-2003-08-2990
McCauley SD, Gilchrist M, Befus AD (2005) Nitric oxide: A major determinant of mast cell phenotype and function. Mem Inst Oswaldo Cruz 100: 11–14. https://doi.org/10.1590/S0074-02762005000900003
Forsythe P, Gilchrist M, Kulka M, Befus AD (2001) Mast cells and nitric oxide: Control of production, mechanisms of response. Int Immunopharmacol 1: 1525–1541. https://doi.org/10.1016/S1567-5769(01)00096-0
De Col R, Messlinger K, Carr RW (2012) Repetitive activity slows axonal conduction velocity and concomitantly increases mechanical activation threshold in single axons of the rat cranial dura. J Physiol 590: 725–736. https://doi.org/10.1113/jphysiol.2011.220624
Zakharov A, Vitale C, Kilinc E, Koroleva K, Fayuk D, Shelukhina I, Naumenko N, Skorinkin A, Khazipov R, Giniatullin R (2015) Hunting for origins of migraine pain: Cluster analysis of spontaneous and capsaicin-induced firing in meningeal trigeminal nerve fibers. Front Cell Neurosci 9: 287. https://doi.org/10.3389/fncel.2015.00287
Shatillo A, Koroleva K, Giniatullina R, Naumenko N, Slastnikova AA, Aliev RR, Bart G, Atalay M, Gu C, Khazipov R, Davletov B, Grohn O, Giniatullin R (2013) Cortical spreading depression induces oxidative stress in the trigeminal nociceptive system. Neuroscience 253: 341–349. https://doi.org/10.1016/j.neuroscience.2013.09.002
Shimada A, Cairns BE, Vad N, Ulriksen K, Pedersen AML, Svensson P, Baad-Hansen L (2013) Headache and mechanical sensitization of human pericranial muscles after repeated intake of monosodium glutamate (MSG). J Headache Pain 14: 1–9. https://doi.org/10.1186/1129-2377-14-2
Gusel’nikova VV, Sukhorukova EG, Fedorova EA, Polevshchikov A V., Korzhevskii DE (2015) A Method for the Simultaneous Detection of Mast Cells and Nerve Terminals in the Thymus in Laboratory Mammals. Neurosci Behav Physiol 45: 371–374. https://doi.org/10.1007/s11055-015-0084-x
Levy D (2010) Migraine pain and nociceptor activation – Where do we stand? Headache 50: 909–916. https://doi.org/10.1111/j.1526-4610.2010.01670.x
Julius D, Basbaum AI (2001) Molecular mechanisms of nociception. Nature 413: 203–210. https://doi.org/10.1038/35093019
Moran MM, McAlexander MA, Bíró T, Szallasi A (2011) Transient receptor potential channels as therapeutic targets. Nat Rev Drug Discov 10: 601–620. https://doi.org/10.1038/nrd3456
Theoharides TC, Spanos C, Pang X, Alferes L, Ligris K, Letourneau R, Rozniecki JJ, Webster E, Chrousos GP (1995) Stress-induced intracranial mast cell degranulation: A corticotropin-releasing hormone-mediated effect. Endocrinology 136: 5745–5750. https://doi.org/10.1210/endo.136.12.7588332
Schemann M, Kugler EM, Buhner S, Eastwood C, Donovan J, Jiang W, Grundy D (2012) The Mast Cell Degranulator Compound 48/80 Directly Activates Neurons. PLoS One 7: e52104. https://doi.org/10.1371/journal.pone.0052104
Bartsch T, Goadsby PJ (2002) Stimulation of the greater occipital nerve induces increased central excitability of dural afferent input. Brain 125: 1496–1509. https://doi.org/10.1093/brain/awf166
Bove GM, Moskowitz MA (1997) Primary afferent neurons innervating guinea pig dura. J Neurophysiol 77: 299–308. https://doi.org/10.1152/jn.1997.77.1.299
Burstein R, Yamamura H, Malick A, Strassman AM (1998) Chemical stimulation of the intracranial dura induces enhanced responses to facial stimulation in brain stem trigeminal neurons. J Neurophysiol 79: 964–982. https://doi.org/10.1152/jn.1998.79.2.964
Moskowitz MA (1984) The neurobiology of vascular head pain. Ann Neurol 16: 157–168. https://doi.org/10.1002/ana.410160202
Moskowitz MA, Macfarlane R (1993) Neurovascular and molecular mechanisms in migraine headaches. Cerebrovasc Brain Metab Rev 5: 159–177.
Bolay H, Reuter U, Dunn AK, Huang Z, Boas DA, Moskowitz MA (2002) Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat Med 8: 136–142. https://doi.org/10.1038/nm0202-136
Karatas H, Erdener SE, Gursoy-Ozdemir Y, Lule S, Eren-Koçak E, Sen ZD, Dalkara T (2013) Spreading depression triggers headache by activating neuronal Panx1 channels. Science (80) 339: 621. https://doi.org/10.1126/science.1231897
Thomsen LL (1997) Investigations into the role of nitric oxide and the large intracranial arteries in migraine headache. Cephalalgia 17: 873–895. https://doi.org/10.1046/j.1468-2982.1997.1708873.x
Edvinsson L, Villalón CM, Maassenvandenbrink A (2012) Basic mechanisms of migraine and its acute treatment. Pharmacol Ther 136: 319–333. https://doi.org/10.1016/j.pharmthera.2012.08.011
Edvinsson L, Mulder H, Goadsby PJ, Uddman R (1998) Calcitonin gene-related peptide and nitric oxide in the trigeminal ganglion: Cerebral vasodilatation from trigeminal nerve stimulation involves mainly calcitonin gene-related peptide. J Auton Nerv Syst 70: 15–22. https://doi.org/10.1016/S0165-1838(98)00033-2
Toda N (1993) Mediation by nitric oxide of neurally-induced human cerebral artery relaxation. Experientia 49: 51–53. https://doi.org/10.1007/BF01928789
Ayajiki K, Fujioka H, Okamura T, Toda N (2001) Relatively selective neuronal nitric oxide synthase inhibition by 7-nitroindazole in monkey isolated cerebral arteries. Eur J Pharmacol 423: 179–183. https://doi.org/10.1016/S0014-2999(01)01068-8
Guerrero-Toro C, Koroleva K, Ermakova E, Gafurov O, Abushik P, Tavi P, Sitdikova G, Giniatullin R (2022) Testing the Role of Glutamate NMDA Receptors in Peripheral Trigeminal Nociception Implicated in Migraine Pain. Int J Mol Sci 23: 1529. https://doi.org/10.3390/ijms23031529
Colonna DM, Meng W, Deal DD, Busija DW (1994) Nitric oxide promotes arteriolar dilation during cortical spreading depression in rabbits. Stroke. https://europepmc.org/article/med/7526490. Accessed 17 Apr 2022
Colonna DM, Meng W, Deal DD, Gowda M, Busija DW (1997) Neuronal NO promotes cerebral cortical hyperemia during cortical spreading depression in rabbits. Am J Physiol Hear Circ Physiol 272. https://doi.org/10.1152/ajpheart.1997.272.3.h1315
Meng W, Colonna DM, Tobin JR, Busija DW (1995) Nitric oxide and prostaglandins interact to mediate arteriolar dilation during cortical spreading depression. Am J Physiol Hear Circ Physiol 269. https://doi.org/10.1152/ajpheart.1995.269.1.h176
Akerman S, Williamson DJ, Kaube H, Goadsby PJ (2002) Nitric oxide synthase inhibitors can antagonize neurogenic and calcitonin gene-related peptide induced dilation of dural meningeal vessels. Br J Pharmacol 137: 62–68. https://doi.org/10.1038/sj.bjp.0704842
Lassen LH, Ashina M, Christiansen I, Ulrich V, Olesen J (1997) Nitric oxide synthase inhibition in migraine. Lancet 349: 401–402. https://doi.org/10.1016/S0140-6736(97)80021-9
Ashina M, Lassen LH, Bendtsen L, Jensen R, Olesen J (1999) Effect of inhibition of nitric oxide synthase on chronic tension-type headache: A randomised crossover trial. Lancet 353: 287–289. https://doi.org/10.1016/S0140-6736(98)01079-4
De Col R, Koulchitsky SV, Messlinger KB (2003) Nitric oxide synthase inhibition lowers activity of neurons with meningeal input in the rat spinal trigeminal nucleus. Neuroreport 14: 229–232. https://doi.org/10.1097/00001756-200302100-00014
Olesen J (2010) Nitric Oxide-Related Drug Targets in Headache. Neurotherapeutics 7: 183–190. https://doi.org/10.1016/j.nurt.2010.03.006
Hoskin KL, Bulmer DCE, Goadsby PJ (1999) Fos expression in the trigeminocervical complex of the cat after stimulation of the superior sagittal sinus is reduced by L-NAME. Neurosci Lett 266: 173–176. https://doi.org/10.1016/S0304-3940(99)00281-5
Grossi L, D’Angelo S (2005) Sodium nitroprusside: Mechanism of NO release mediated by sulfhydryl-containing molecules. J Med Chem 48: 2622–2626. https://doi.org/10.1021/jm049857n
MacIver MB, Tanelian DL (1993) Structural and functional specialization of Aδ and C fiber free nerve endings innervating rabbit corneal epithelium. J Neurosci 13: 4511–4524. https://doi.org/10.1523/jneurosci.13-10-04511.1993
Neeb L, Reuter U (2008) Nitric Oxide in Migraine. CNS Neurol Disord – Drug Targets 6: 258–264. https://doi.org/10.2174/187152707781387233
Klyachko VA, Ahern GP, Jackson MB (2001) cGMP-mediated facilitation in nerve terminals by enhancement of the spike afterhyperpolarization. Neuron 31: 1015–1025. https://doi.org/10.1016/S0896-6273(01)00449-4
Al-Karagholi MAM, Ghanizada H, Waldorff Nielsen CA, Skandarioon C, Snellman J, Lopez-Lopez C, Hansen JM, Ashina M (2021) Opening of BKCa channels causes migraine attacks: a new downstream target for the treatment of migraine. Pain 162: 2512–2520. https://doi.org/10.1097/j.pain.0000000000002238
Ben Aissa M, Tipton AF, Bertels Z, Gandhi R, Moye LS, Novack M, Bennett BM, Wang Y, Litosh V, Lee SH, Gaisina IN, Thatcher GRJ, Pradhan AA (2018) Soluble guanylyl cyclase is a critical regulator of migraine-associated pain. Cephalalgia 38: 1471–1484. https://doi.org/10.1177/0333102417737778
Bertels Z, Pradhan AAA (2019) Emerging Treatment Targets for Migraine and Other Headaches. Headache 59: 50–65. https://doi.org/10.1111/head.13585
Ferreira J, Santos ARS, Calixto JB (1999) The role of systemic, spinal and supraspinal L-arginine-nitric oxide-cGMP pathway in thermal hyperalgesia caused by intrathecal injection of glutamate in mice. Neuropharmacology 38: 835–842. https://doi.org/10.1016/S0028-3908(99)00006-4
Kawamata T, Omote K (1999) Activation of spinal N-methyl-D-aspartate receptors stimulates a nitric oxide/cyclic guanosine 3',5'-monophosphate/glutamate release cascade in nociceptive signaling. Anesthesiology 91: 1415–1424. https://doi.org/10.1097/00000542-199911000-00035
Tao YX, Johns RA (2002) Activation and up-regulation of spinal cord nitric oxide receptor, soluble guanylate cyclase, after formalin injection into the rat hind paw. Neuroscience 112: 439–446. https://doi.org/10.1016/S0306-4522(02)00075-1
Song XJ, Wang ZB, Gan Q, Walters ET (2006) cAMP and cGMP contribute to sensory neuron hyperexcitability and hyperalgesia in rats with dorsal root ganglia compression. J Neurophysiol 95: 479–492. https://doi.org/10.1152/jn.00503.2005
Artico M, Cavallotti C (2001) Catecholaminergic and acetylcholine esterase containing nerves of cranial and spinal dura mater in humans and rodents. Microsc Res Tech 53: 212–220. https://doi.org/10.1002/jemt.1085
Varatharaj A, MacK J, Davidson JR, Gutnikov A, Squier W (2012) Mast cells in the human dura: Effects of age and dural bleeding. Child’s Nerv Syst 28: 541–545. https://doi.org/10.1007/s00381-012-1699-7
Dimlich RVW, Keller JT, Strauss TA, Fritts MJ (1991) Linear arrays of homogenous mast cells in the dura mater of the rat. J Neurocytol 20: 485–503. https://doi.org/10.1007/BF01252276
Rozniecki JJ, Dimitriadou V, Lambracht-Hall M, Pang X, Theoharides TC (1999) Morphological and functional demonstration of rat dura mater mast cell-neuron interactions in vitro and in vivo. Brain Res 849: 1–15. https://doi.org/10.1016/S0006-8993(99)01855-7
Strassman AM, Weissner W, Williams M, Ali S, Levy D (2004) Axon Diameters and Intradural Trajectories of the Dural Innervation in the Rat. J Comp Neurol 473: 364–376. https://doi.org/10.1002/cne.20106
Dimitriadou V, Rouleau A, Trung Tuong MD, Newlands GJF, Miller HRP, Luffau G, Schwartz JC, Garbarg M (1997) Functional relationships between sensory nerve fibers and mast cells of dura mater in normal and inflammatory conditions. Neuroscience 77: 829–839. https://doi.org/10.1016/S0306-4522(96)00488-5
Kilinc E, Tore F, Dagistan Y, Bugdayci G (2020) Thymoquinone Inhibits Neurogenic Inflammation Underlying Migraine Through Modulation of Calcitonin Gene-Related Peptide Release and Stabilization of Meningeal Mast Cells in Glyceryltrinitrate-Induced Migraine Model in Rats. Inflammation 43: 264–273. https://doi.org/10.1007/s10753-019-01115-w
Ferrari LF, Levine JD, Green PG (2016) Mechanisms mediating nitroglycerin-induced delayed-onset hyperalgesia in the rat. Neuroscience 317: 121–129. https://doi.org/10.1016/j.neuroscience.2016.01.005
Olesen J, Iversen HK, Thomsen L (1993) Nitric oxide supersensitivity: a possible molecular mechanism of migraine pain. Neuro Report. 4(8): 1027–1030. https://doi.org/10.1097/00001756-199308000-00008
Headache Classification Committee of the International Headache Society. The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia. 2013 Jul; 33(9): 629–808. https://doi.org/10.1177/0333102417738202
Jacobs B, Dussor G (2016) Neurovascular contributions to migraine: Moving beyond vasodilation. Neuroscience 338: 130–144. https://doi.org/10.1016/j.neuroscience.2016.06.012
Li J, Vause C V, Durham PL (2008) Calcitonin gene-related peptide stimulation of nitric oxide synthesis and release from trigeminal ganglion glial cells. Brain Res 1196: 22–32. https://doi.org/10.1016/j.brainres.2007.12.028
Messlinger K, Russo AF (2019) Current understanding of trigeminal ganglion structure and function in headache. Cephalalgia 39: 1661–1674. https://doi.org/10.1177/0333102418786261
Iyengar S, Johnson KW, Ossipov MH, Aurora SK (2019) CGRP and the Trigeminal System in Migraine. Headache 59: 659–681. https://doi.org/10.1111/head.13529
Ashina M (2012) Vascular changes have a primary role in migraine. Cephalalgia 32 (5): 428–430. https://doi.org/10.1177/0333102412438978
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова