

Российский физиологический журнал им. И.М. Сеченова, 2022, T. 108, № 9, стр. 1077-1093
Нейроэндокринный контроль гиперглутаматергических состояний при патологиях мозга: влияние глюкокортикоидов
1 Институт высшей нервной деятельности и нейрофизиологии РАН
Москва, Россия
2 Научно-практический психоневрологический центр им. З.П. Соловьева
Москва, Россия
* E-mail: nata_gul@ihna.ru
Поступила в редакцию 09.08.2022
После доработки 12.08.2022
Принята к публикации 15.08.2022
- EDN: EKURCX
- DOI: 10.31857/S0869813922090102
Аннотация
Нейроэндокринный контроль, опосредованный глюкокортикоидами, важен для поддержания нормального функционирования мозга и баланса между системами возбуждения и торможения. Глюкокортикоиды регулируют состояние глутаматергической системы мозга как непосредственно, через рецепторы на глутаматергических синапсах, так и опосредованными путями. Нарушение функционирования гипоталамо-гипофизарно-надпочечниковой оси и ее неспособность оптимально регулировать глутаматергическую синаптическую пластичность приводит к развитию нейропсихических заболеваний, в патогенезе которых ключевую роль могут играть гиперглутаматергические состояния. Нарушение глюкокортикоидного контроля глутаматергических процессов лежит в основе когнитивных и эмоциональных расстройств, эпилепсии и ряда других церебральных патологий, являясь общим базовым механизмом развития многих болезней мозга и их коморбидностей. В связи с этим исследование механизмов взаимодействия гипоталамо-гипофизарно-надпочечниковой оси и глутаматергической системы мозга имеет приоритетное трансляционное значение.
ВВЕДЕНИЕ
Реакция гипоталамо-гипофизарно-надпочечниковой оси (ГГНО) является ключевым событием нейроэндокринного ответа на любое значимое для организма событие (стресс). Основными компонентами этой каскадной реакции являются секреция гипоталамического кортикотропин-рилизинг гормона, стимулирующего высвобождение гипофизарного адренокортикотропного гормона (АКТГ), который в свою очередь вызывает высвобождение глюкокортикоидных гормонов (ГК) из коры надпочечников (кортикостерона у большинства грызунов; кортизола у человека) [1]. Поступившие в кровь ГК, наряду с периферическим, реализуют центральное действие, взаимодействуя со специфическими рецепторами в мозге. Многообразный спектр эффектов ГК на метаболизм тканей нервной системы был отмечен достаточно давно и не вызывал удивления исследователей, учитывая большое биохимическое и морфологическое разнообразие различных типов клеток, присутствующих в центральной и периферической нервной системах [2]. Обширные исследования, проведенные в течение последних десятилетий, предоставили убедительные доказательства того, что ГК обладают способностью регулировать развитие, выживание и гибель нейронов. При этом стали очевидными парадоксальные особенности ГК, которые могут быть критически вовлечены как в нейродегенеративные, так и в нейропротекторные процессы [3].
ГЛЮКОКОРТИКОИДЫ – КЛЮЧЕВЫЕ НЕЙРОЭНДОКРИННЫЕ РЕГУЛЯТОРЫ ПЛАСТИЧНОСТИ МОЗГА
Нормальная активность ГГНО, обеспечивающая ритмичное высвобождение ГК, необходима для гомеостаза организма и преодоления неблагоприятных условий среды. Действуя через специфические внутриклеточные рецепторы в головном мозге и на периферии, ГК регулируют поведение, а также метаболическую, сердечно-сосудистую, иммунную и нейроэндокринную активности. В дополнение к метаболическим эффектам, ведущим к выработке необходимого количества энергии, ГК, помимо иммуносупрессивного действия, может стимулировать иммунную систему при циркадианном повышении их уровня, обеспечивая острые защитные реакции. Отрицательная обратная связь, реализуемая ГК, включает множество механизмов, приводящих к ограничению активации ГГНО и предотвращению вредных последствий чрезмерной выработки ГК. Циркадианное и вызванное умеренным острым стрессом повышение уровня ГК необходимо для выживания нейронов, облегчения глутаматергической нейротрансмиссии и образования возбуждающих синапсов, а также индукции ранних генов [4].
В нервной системе ГК взаимодействуют с клеточными рецепторами, глюкокортикоидными (ГР) и минералокортикоидными (МР), которые существуют в цитоплазматической/ядерной и мембранной формах. Трансдукция сигнала через мембранные рецепторы реализует быстрые негеномные эффекты ГК, в частности на возбудимость и активность нейронов в гиппокампе, гипоталамусе, амигдале и префронтальной коре, и таким образом влияет на когнитивные функции, адаптивное поведение и нейроэндокринную систему. Цитоплазматические/ядерные рецепторы участвуют в более медленных и продолжительных изменениях, связанных с экспрессией генов. Несмотря на то, что рецепторы ГК экспрессированы в каждой клетке нервной системы, их состав и уровень экспрессии варьируется, в результате чего различные типы клеток по-разному реагируют на активацию этих рецепторов. ГК индуцируют структурную пластичность в нейронах, шванновских клетках, микроглии, олигодендроцитах и астроцитах, а также влияют на нейротрансмиссию, в частности изменяя высвобождение и обратный захват глутамата.
Для осуществления контроля функции мозга посредством ГГНО принципиально важна дифференциальная локализация различных рецепторов ГК в мозге. ГР экспрессируются в большинстве структур и регионов мозга, а МР экспрессированы в основном в тех регионах мозга, которые имеют решающее значение для формирования памяти и эмоций, в первую очередь в структурах лимбической системы, гиппокампе, миндалине и префронтальной коре [5]. МР обладают высокой аффинностью и поэтому связаны с ГК при низком уровне гормонов, а сродство ГР к ГК на порядок ниже, поэтому активация этих рецепторов происходит при высоком уровне ГК. Быстрые эффекты ГК реализуются преимущественно через сигнал, проводимый мембранными МР, а медленные структурно-функциональные перестройки клеток мозга регулируются в основном геномными эффектами через ГР. Таким образом, благодаря трансдукция сигнала ГК через МР и ГР клетки мозга способны адекватно отвечать на значимые события и реализовать быстрые и продолжительные пластические изменения. Это дает возможность всем клеткам мозга, и в первую очередь нейронам, адекватно отвечать на действие разнообразных стрессорных факторов.
Сигнальная трансдукция, осуществляемая через рецепторы ГК, играет ключевую роль в интеграции ответа и взаимодействия ключевых систем (в т.ч. нейромедиаторных, метаболических, нейроиммунной, нейротрофиновой и др.) при поддержании и настройке нейропластичности. В итоге дифференцированная рецепция ГК в разных структурах и клетках мозга контролирует практически все активности нервной системы, включая обучение и память, регулируя их на субклеточном, клеточном и сетевом уровнях [1, 6, 7]. В последние годы расшифрованы многие механизмы влияния ГК на синаптическую функцию и синаптическую пластичность, лежащие в основе когнитивных функций и эмоций [8]. Если представить эффекты ГК максимально упрощенно, можно констатировать, что ГК модулируют кодирование информации в мозге через сигнальные механизмы, опосредованные МР, а консолидацию и процессинг информации через ГР. ГК изменяют динамику активности нейронов, приводя к изменениям, зависящим от контекста, включая как возбуждение, так и торможение, и в норме эти эффекты обеспечивают и поддерживают реакции организма, связанные с решаемой задачей. ГК обеспечивают диверсификацию доступных источников энергии, которые могут использоваться для поддержания активности мозга, однако чрезмерные концентрации ГК ухудшают выработку энергии (АТФ) и окисление в митохондриях. Таким образом, в соответствии с концепцией аллостатической нагрузки и перегрузки, ГК оказывают как адаптивное, так и дезадаптивное действие [9]. В связи с этим дисфункция ГГНО и рецепторов ГК нарушает адекватную реакцию организма на значимые факторы, и это лежит в основе развития неврологических и психических заболеваний.
За последние 30 лет появляется все больше свидетельств того, что ключевыми мишенями ГК в мозге являются лимбические структуры, в первую очередь гиппокамп, структура с максимальной плотностью рецепторов ГК. Считается, что кумулятивное воздействия ГК определяет скорость потери нейронов в стареющем гиппокампе, поэтому стресс может ускорить дегенерацию гиппокампа при старении. Более того, при обстоятельствах, когда воздействие ГК недостаточно для повреждения нейронов, ГК снижают способность нейронов выживать при экстремальных воздействиях, таких как гипоксия–ишемия, судороги или гипогликемия [10]. В связи с тем, что именно гиппокамп является ключевой структурой, опосредующей реализацию обучения и памяти, а также эмоций, и с учетом регуляции ГГНО гиппокампом, вызванное ГК повреждение гиппокампа, в т.ч. его атрофия, может играть важную роль в патогенезе целого ряда нервно-психических расстройств [11, 12].
ГЛУТАМАТЕРГИЧЕСКАЯ СИСТЕМА МОЗГА: РЕГУЛЯЦИЯ ГЛЮКОКОРТИКОИДАМИ
Универсальный межнейронный интерфейс, синапс, является ключевым элементом пластичности мозга. Глутамат – наиболее распространенный нейромедиатор в ЦНС, рецепторы которого присутствуют на более чем 90% нейронов и 40% синапсов [13]. В настоящее время в ЦНС идентифицировано более 20 рецепторов глутамата (ионотропных, представляющим собой ионный канал, и метаботропных, трансдукция сигнала через которые опосредована вторичными посредниками), причем каждый отдельный рецептор имеет несколько подтипов. Функциональными элементами, обеспечивающими глутаматную нейротрансмиссию, являются пре- и постсинаптические нейроны вместе с глиальными клетками, которые образуют так называемый “трехсторонний синапс” (tripartite synapse) [14]. Трехсторонний синапс функционирует как через метаботропные, так и через ионотропные рецепторы (эти рецепторы и их функции подробно описаны в статье [15]). Глутамат синтезируется из глутамина глутаминазой в пресинаптических нейронах, и затем транспортируется к синаптическим окончаниям. В пресинапсе глутамат накапливается в везикулах везикулярными транспортерами глутамата (VGLUT) 1–3 и высвобождается при деполяризации. При этом передача сигналов осуществляется через ионотропные и метаботропные глутаматные рецепторы, расположенные в разных компартментах трехстороннего синапса. Передача сигнала прекращается путем захвата глутамата из синаптической щели высокоэффективными транспортерами возбуждающих аминокислот (EAAT), которые преимущественно расположены на астроцитах. В астроците глутаминсинтетаза превращает глутамат в глутамин, который может затем попадать во внеклеточное пространство и пресинапс в процессе, называемом циклом глутамат/глутамин. Глутаматные рецепторы и транспортеры вместе действуют как критические регуляторы глутаматергической системы за счет модуляции силы сигнала и внеклеточных концентраций глутамата. Именно тщательная регуляция внеклеточного глутамата имеет решающее значение для предотвращения постоянной активации рецепторов, которая может привести к эксайтотоксичности и гибели нейронов.
Ионотропные рецепторы, NMDA (N-метил D-аспартат) и AMPA (α-амино-3-гидрокси-5-метил-4-изоксазолпропионовая кислота) типов колокализованы на постсинаптической мембране, функционально зависят друг от друга во время деполяризации мембраны, но взаимодействуют с глутаматом независимо. Высокая плотность NMDA-рецепторов, экспрессируемых на нейронах и астроцитах во всем мозге и особенно в гиппокампе, способствует обучению, памяти и синаптической пластичности [16, 17], но гиперактивация рецепторов приводит к нейродегенерации. Считают, что такая двойственность обусловлена субклеточной локализацией NMDA-рецепторов: их синаптическая активация вызывает кальций-опосредованные благоприятные изменения транскрипции, в то время как экстрасинаптическая активация запускает сигнальные пути, которые снижают устойчивость к стрессу и стимулируют нейродегенерацию [18]. Предполагается, что AMPA-рецепторы играют важную роль в синаптической пластичности, обеспечивая необходимый уровень деполяризации для деблокирования NMDA-рецепторов от магниевого блока, и косвенно увеличивают поступления кальция в нейрон. Ионотропные глутаматные рецепторы опосредуют быстрые нейронные коммуникации, в то время как в результате трансдукции сигнала через метаботропные рецепторы реализуются более медленные процессы. Метаботропные рецепторы разных типов, экспрессирующиеся как пресинаптически, так и постсинаптически, по-разному модулируют глутаматергическую трансмиссию, принимая участие в реализации феноменов синаптической пластичности, ассоциированных с эмоциональным поведением и когнитивными функциями [18].
Настройка глутаматергической трансмиссии является важным механизмом коммуникации между нейронами, поскольку глутаматергическая система играет решающую роль в синаптической стабильности и пластичности. В этой связи особое внимание привлекает глутаматергическая система гиппокампа, ключевой структуры, участвующей в переработке когнитивной и эмоциональной информации и селективно чувствительной к действию различных стрессорных факторов [1]. Решающее значение как ГК, так и глутамата, для жизнеспособности клеток гиппокампа было отмечено в 90-е годы, когда было показано, что экспрессия в гиппокампе генов, контролируемых рецепторами ГК, необходима в основном для взаимодействия с другими факторами транскрипции (например, CREB, AP-1), хотя не исключалось связывание с чувствительными к гормонам элементами ГК-специфичных генов [19].
В последние годы показано, что хронический избыток ГК вызывает специфический синаптический дефицит в гиппокампе. Важнейшую роль в этом играют возбуждающие глутаматергические синапсы, ключевые участники синаптической передачи, синаптической пластичности и поведенческой адаптации [20, 21]. Физиологический стресс активирует глутаматергические нейроны в гиппокампе, а хронический стресс вызывает дисгомеостаз внеклеточного глутамата. Этот дисгомеостаз усиливается под действием избытка ГК, приводя к изменениям когнитивного и эмоционального поведения. При этом эффекты ГК на глутаматергический синапс зависят от типа и продолжительности воздействия, определяющего уровни и динамику ГК, регион-специфичны, зависят от возраста и пола [21].
В обзоре [22] мы представили общую схему известных в настоящее время механизмов синаптических эффектов ГК, опосредованных МР и ГР, в глутаматергическом синапсе на основе результатов опубликованных ранее исследований [1, 6, 21, 23–26]. Схема предполагает, что в основе высокой пластичности глутаматергических синапсов лежит большое число мишеней прямого или опосредованного влияния ГК, наличие которых и формирует множественность механизмов, вовлеченных в реакцию глутаматергической трансмиссии на ГК. Это дает возможность адекватной реакции организма в каждой конкретной ситуации. Большое число потенциальных механизмов сигнальной трансдукции определяется наличием как мембранных, так и цитоплазматических форм рецепторов ГК с разным сродством (МР и ГР) в глутаматергическом синапсе. Они обеспечивают широкий репертуар путей регуляции синаптической пластичности, реализуемых как немедленно негеномными механизмами, осуществляющими деполяризацию синаптических мембран, так и отложенно за счет включения геномных механизмов.
Согласно предложенной схеме, негеномные механизмы опосредованы мембранными рецепторами ГК. В пресинапсе ГК связываются с высокоаффинными МР, регулируя через них возбудимость и высвобождение везикул с глутаматом. Частота высвобождения глутамата контролируется опосредованным G-белком сигнальным путем ERK1/2 (extracellular signal-regulated kinase 1/2). Связывание ГК с локализованными в постсинапсе мембранными МР подавляет активацию калиевых каналов, а с мембранными постсинаптическими ГР – ингибирует кальциевые каналы L- и N-типа, а также протеинкиназу С, снижая приток кальция в клетку. Связывание ГК с мембранными постсинаптическими ГР опосредованно (через протеинкиназу А), ингибирует ионотропные NMDA-рецепторы и усиливает гиперполяризацию, регулируя расположенные в постсинаптической области ионотропные рецепторы гамма-аминомасляной кислоты ГАМКA. Считается, что геномные механизмы действия ГК на глутаматергическую трансмиссию реализуются главным образом в постсинапсе и в основном трансдукцией сигнала через цитоплазматические ГР, хотя имеются сведения и об участии постсинаптических цитоплазматических МР в реализации длительных эффектов ГК. Свободно диффундируя через клеточную мембрану, ГК связываются в цитоплазме с МР и ГР, вызывая димеризацию рецепторов при участии белков теплового шока. Гомодимер ГР или МР транслоцируется в ядро, где связывается со специфическими участками ДНК glucocorticoid response elements (GRE). С областью GRE, с которой связан гомодимер рецептора ГК, связываются кофакторы и гистон-модифицирующие элементы (histone-modifying elements), в результате чего, в зависимости от природы факторов, связавшихся с этим участком, происходит изменение (инициация или ингибирование) транскрипции, экспрессии генов и синтеза белка. Описаны, хотя и расцениваются как некоторое исключение, геномные эффект ГК через мембранный ГР, который в определенных условиях может транслоцироваться в ядро и вызывать трансрепрессию или запускать ERK1/2-зависимую транскрипцию генов. Наиболее важные для глутаматергической трансмиссии индуцированные ГК геномные эффекты включают модуляцию экспрессии субъединиц NMDA- и AMPA-рецепторов, а также метаболизма и транспорта глюкозы за счет изменения активности ее мембранных транспортеров [22].
Наряду с ионотропными АМРА- и NMDA-рецепторами, функция других глутаматных рецепторов (mGluR1, KA1, GluR6 и GluR7) также регулируется ГК [22]. Эффекты ГК на пластичность, опосредованную глутаматергическими синапсами, продемонстрированы не только в гиппокампе, но и в других структурах мозга, ключевых для когнитивной функции и эмоционального состояния, в первую очередь лимбической системы. Такие данные получены для префронтальной и лобной коры, миндалины и гипоталамуса. Важно, что в различных регионах мозга и даже ядрах и областях одной структуры регуляция глутаматергической трансмиссии ГК может различаться по дозовой и временной зависимости и иметь противоположные эффекты. При этом регион-специфичные эффекты могут касаться различных субъединиц глутаматных рецепторов [22].
Еще один потенциальный механизм контроля ГК функционирования глутаматергического синапса является влияние этих гормонов на физиологию и функционирование астроцитов. В первую очередь это влияние включает ингибирование транспорта глюкозы, снижение синтеза гликогена и снижение поглощения глутамата [27]. Кроме того, возможны косвенные эффекты кортикостероидов на астроциты. ГК снижают уровень серотонина в нейронах, что влияет на функционирование астроцитов, изменяя активность цитокинов и транспорт ГАМК.
Нейротрофический фактор головного мозга (BDNF) представляет собой нейротрофин, в изобилии экспрессируемый в центральной нервной системе. Он способствует длительному усилению синаптической эффективности, ассоциированной со специфическими процессами обучения и памяти. Одна из ключевых молекул, модулирующих пластичность мозга, BDNF по различным механизмам модулирует глутаматергическую трансмиссию, изменяя экспрессию и активность как ионотропных, так и метаботропных глутаматных рецепторов, а также транспортеров (см. обзор [28]). В ряде исследований продемонстрированы корреляции между экспрессией/функцией BDNF и гормонами надпочечников (главным образом ГК и дегидроэпиандростероном [29]), что может указывать на BDNF-опосредованные механизмы регуляции глутаматергической трансмиссии ГК, которые до настоящего времени остаются недостаточно изученными.
Очевидно, что прямое взаимодействие ГК и глутаматергического синапса через рецепторы ГК не исчерпывает возможные механизмы взаимодействия глутаматергической трансмиссии и ГГНО. Установлено участие возбуждающих аминокислот в контроле высвобождения АКТГ [30]. Активация ионотропных глутаматных рецепторов оказывает стимулирующее действие на высвобождение АКТГ, в то время как роль метаботропных рецепторов еще детально не изучена. Глутаматергическая регуляция высвобождения АКТГ имеет очевидное значение для реакции на стресс. Интересно, что некоторые антидепрессанты, такие как тианептин, которые модулируют высвобождение АКТГ, влияют и на глутаматергическую систему мозга.
ГИПЕРГЛУТАМАТЕРГИЧЕСКИЕ СОСТОЯНИЯ ПРИ ПАТОЛОГИЯХ МОЗГА И ГЛЮКОКОРТИКОИДЫ
В оптимально функционирующем мозге возбуждающие и тормозные сигналы регулируются и сбалансированы. Нарушение баланса возбуждения и торможения нарушает передачу сигналов и, как следствие, приводит к патологическим изменениям поведения, когнитивных и двигательных функций. Дисбаланс между основными возбуждающим и тормозным медиаторами соответственно глутаматом и γ-аминомасляной кислотой (ГАМК) приводит к нарушениям на уровне синапсов и в итоге к нейродегенерации.
Клеточный гомеостаз глутамата имеет первостепенное значение для нормального функционирования мозга и зависит от сложного метаболического взаимодействия между нейронами и астроцитами [31]. Глиальные клетки, и прежде всего астроциты как компонент глутаматергического синапса, играют важную роль в поддержании этого баланса в норме и нарушениях его в патологических ситуациях [32]. Накапливается все больше доказательств, что патофизиология нервно-психических расстройств, включая расстройства настроения и памяти, связана с нарушением функции и регуляции глутаматергической системы, поэтому гомеостаз глутамата имеет решающее значение для снижения риска различных неврологических и психических заболеваний. Нарушение регуляции глутамата показано как в доклинических моделях неврологических и психических расстройств, так и в клинике [33, 34]. За последние десятилетия получено достаточно фактов о том, что стрессорные события, действуя через ГК и их рецепторы, вызывают целый комплекс изменений глутаматергического сигналинга в лимбической системе мозга, в первую очередь гиппокампе и фронтальной коре, влияющих на когнитивные и эмоциональные процессы [35]. Эти изменения затрагивают пресинаптическое высвобождение глутамата, мембранный трафик и деградацию постсинаптических глутаматных рецепторов, структуру шипиков и сети цитоскелета, а также эпигенетический контроль экспрессии генов. Модификации подвергается и область постсинаптической плотности, сложного субклеточного домена, важного при постсинаптической трансдукции сигналов для точной передачи информации от нейрона к нейрону и реализации синаптической пластичности. Глутаматергическая синаптическая дисфункция, влияющая на морфологию постсинаптической плотности и сигнальные события, была описана при многих нейродегенеративных расстройствах [36].
Эксайтотоксичность – это термин, который описывает токсическое действие возбуждающих нейротрансмиттеров, в первую очередь глутамата. Усиленная или длительная активация глутаматных рецепторов в гиперглутаматергических состояниях (т.е. в ситуациях, сопровождающихся избыточной глутаматергической трансмиссией) запускает каскад, который приводит к развитию нейротоксичности, а в итоге – к потере функции нейронов и гибели клеток. Считается, что опосредованная NMDA-рецепторами эксайтотоксичность является центральным звеном в патогенезе многих заболеваний мозга как неврологических, так и психических, в основе которых лежат нейродегенеративные процессы. Граница между нормальной физиологической функцией и эксайтотоксичностью в значительной степени контролируется астроцитами, поскольку они могут модулировать уровни глутамата в синаптической щели, выводя глутамат и обеспечивая его последующую рециркуляцию через глутамат – глутаминовый цикл [37]. Молекулярный механизм, который вызывает эксайтотоксичность, включает изменения в метаболизме глутамата и кальция, дисфункцию переносчиков глутамата и нарушение работы рецепторов глутамата, в частности NMDA-рецепторов. Эксайтотоксичность можно также рассматривать как следствие или причину других клеточных явлений, в т.ч. митохондриальной дисфункции и окислительного стресса. Поступление кальция через экстрасинаптические NMDA-рецепторы связано с перегрузкой кальцием митохондрий нейронов. Митохондрии, помимо своей роли в производстве АТФ в клетке, участвуют в гомеостазе кальция, действуя как буферная органелла, поэтому нарушение митохондриального кальциевого гомеостаза ассоциировано с гибелью нейронов либо в результате запуска апоптоза, либо в результате открытия митохондриальной поры [38].
Учитывая ключевую роль глутаматергической трансмиссии в нормальном функционировании мозга, неудивительно, что практически во всех случаях, когда исследования осуществлялись на экспериментальном (модели заболеваний мозга) или клиническом материале от пациентов, были показаны нарушения глутаматергической системы. Эти нарушения имеют свою специфику в зависимости от конкретного патологического состояния, но, как правило, ассоциированы с изменениями в ГГНО, которая, как показано выше, осуществляет контроль глутаматергической трансмиссии. Интересно, что связанные изменения глутаматергической системы мозга и активности ГГНО наблюдаются и при метаболических изменениях в целом организме. Например, диета с высоким содержанием жира в эксперименте на грызунах выявила повышение уровня кортикостерона, аспартата и глутамата на фоне изменений пластичности, опосредованной NMDA-рецепторами, т.е. гиперглутаматергическая активность в гиппокампе была ассоциирована с дисфункцией ГГНО [39].
Экстраординарные возможности синаптической пластичности в гиппокампе определяют его ключевую роль в обучении и памяти, а также регуляции эмоционального состояния, однако оборотной стороной такой пластичности является селективная чувствительность к повреждениям, опосредованным ГК (в частности, из-за высокой плотности кортикостероидных рецепторов). Принято считать, что именно эта особенность гиппокампа лежит в основе возникновения и развития когнитивных и эмоциональных расстройств при старении и нейродегенеративных заболеваниях, а прогрессирующее повреждение нейронов гиппокампа за счет гиперглутаматергической трансмиссии, в свою очередь, может быть причиной непрерывной активации ГГНО и повышенной экспрессии вазопрессина и кортикотропин-рилизинг-гормона в гипоталамусе [41–43]. Избыток ГК вызывает также увеличение количества и изменение фенотипа микроглии, способствующие нарушению глутаматергической трансмиссии и потенциально усугубляющие нарушения синаптической пластичности и памяти [44], а нарушение функционирования астроцитов вносит вклад в развитие глутаматной эксайтотоксичности за счет избыточного тока глутамата через мембранные ионотропные и метаботропные рецепторы [45].
Особый интерес представляет взаимодействие глутаматергической системы и ГК в патогенезе болезни Альцгеймера (БА), наиболее распространенной и активно изучаемой формы деменции. Подавляющее большинство случаев БА не имеют известной генетической причины, что делает жизненно важным выявление факторов окружающей среды, участвующих в возникновении и прогрессировании заболевания. Стресс может быть решающим фактором, способствующим развитию БА, что подтверждается огромным числом исследований, хотя механизмы, лежащие в основе этой связи, остаются не до конца ясными [46, 47]. Хронический психосоциальный стресс все чаще признается фактором риска развития спорадической БА. При БА отмечены нарушения функционирования ГГНО, а экспериментальные и клинические данные указывают на то, что при старении и БА гиперсекреция ГК ассоциирована с дисфункцией гиппокампа [48]. Нарушение регуляции ГГНО и повышенный базальный уровень кортизола, которые обнаруживаются у пациентов с БА, вносят значительный вклад в патогенез заболевания [49, 50]. Наблюдаемая гиперсекреция ГК механистически согласуется с предсказанным теоретически ухудшением состояния гиппокампа при БА и последующим снижением способности ГК ингибировать ГГНО. Показано, что умеренная гиперкортизолемия, которая проявляется уже на ранних стадиях БА, связана с гиперчувствительностью надпочечников к адренокортикотропному гормону [51]. Интересно, что депрессия является продромальной и составной частью БА, она также может быть триггером для начинающейся БА. Общими механизмами БА и депрессии, которые обеспечивают также коморбидность этих заболеваний, являются нарушение секреции ГК, изменения в глутаматергической трансмиссии, в первую очередь в лимбических структурах, а также развитие нейровоспалительных процессов [1, 52].
Клинические исследования показали, что гиперактивность неокортекса и гиппокампа характерна для пациентов на ранних стадиях заболевания БА, прогрессируя до гипоактивности на более поздних стадиях нейродегенерации. Факторы, способствующие аберрантной возбудимости нейронов, включают аномальные уровни внутриклеточного Ca2+ и глутамата. Интересно, что гипервозбудимость может быть прогностическим маркером когнитивной дисфункции [53].
На мышах линии APP/PS1, имеющих мутации, характерные для БА, получены важные данные о гиперглутаматергическом сигналинге в гиппокампе в связи с БА-подобными симптомами. Животные изначально когнитивно нормальны, но имеют повышенное высвобождение глутамата в гиппокампе в возрасте 2–4 мес. Показано, что гиперглутаматергическое состояние в гиппокампе этих животных формируется до накопления амилоидных бляшек. В возрасте 6–8 мес. начинают проявляться когнитивные нарушения и накопление амилоидных бляшек в мозге, а очевидная невропатология БА и когнитивные нарушения проявляются в возрасте 10–12 мес. Введение антиглутаматергического препарата рилузола (бензотиазольный антиконвульсант, ингибирующий избыточную глутаматергическую трансмиссию в синапсах за счет блокирования натриевых каналов) в возрасте 2–6 мес., предотвращает снижение когнитивных способностей и восстанавливает нормальный уровень глутаматергической нейротрансмиссии [54]. Это одно из наиболее убедительных доклинических доказательств, подтверждающих непосредственную ассоциацию симптоматики БА с гиперглутаматергическим состоянием [55].
Нарушение синаптической пластичности в связи с изменением функционирования ионотропных и метаботропных глутаматных рецепторов и потеря дендритов происходят на ранних этапах БА [56]. Синаптопатия, сопровождающая БА, ассоциирована с индуцируемым бета-амилоидным пептидом дисбалансом между синаптическими и экстрасинаптическими NMDA-рецепторами и сниженным числом поверхностных AMPA-рецепторов [57].
Сопоставление данных, полученных от пациентов с БА, с функциональными исследованиями на животных моделях заболевания показывает, что на ранних стадиях заболевания накопление токсичных агрегатов бета-амилоида, особенно димеров и низкомолекулярных олигомеров, нарушает обратный захват глутамата, что приводит к его внеклеточному накоплению, вызывающему деполяризацию нейронов. Это вызывает гиперактивацию нейронов и может способствовать их повреждению и дегенерации нейронов из-за нейротоксичности глутамата [58, 59]. Метаболизм глутамата в митохондриях тесно связан с БА. Нарушение баланса метаболизма глутамата в мозге ассоциировано не только с изменением захвата глутамата, его циркуляции, но и с внутриклеточным митохондриальным транспортом и митохондриальным метаболизмом глутамата [60]. Интересно, что изменения уровня глюкозы и глутамата в мозге, которые могут быть следствием дисфункции ГГНО, предшествуют появлению амилоидных бляшек [61].
Высокий уровень кортизола может оказывать нейротоксическое воздействие на гиппокамп, способствуя окислительному стрессу и токсичности бета-амилоидного пептида. Нейроны гиппокампа являются одними из первых нейрональных клеток, которые дегенерируют в головном мозге пациентов, страдающих БА. Эти нейроны уязвимы к окислительному стрессу, важному звену патогенеза БА. ГК усиливают окислительную гибель клеток, индуцируемую бета-амилоидным пептидом и глутаматом [62, 63], тем самым реализуя пагубное воздействие на когнитивные способности и дальнейшее развитие патологии БА в результате повышенного уровня кортизола.
Установлено, что стресс и его нейрохимические и эндокринные медиаторы вызывают изменения в глутаматных синапсах и сетях, а это, в свою очередь, изменяет психические состояния. Многочисленные данные, полученных на животных моделях депрессии, показали, что секреция ГК при различных типах стресса усиливает высвобождение и трансмиссию сигнала глутамата в лимбических и кортикальных областях мозга и оказывает мощные структурные эффекты, вызывая ремоделирование дендритов, сокращение числа синапсов и сокращение объема структур мозга, подобные тем, которые наблюдаются у пациентов с депрессией [64]. Доклинические исследования на моделях депрессии показали, что ГК оказывают решающее влияние на возбудимость и функцию нейронов, особенно в кортикальных и лимбических областях. Это влияние осуществляется через глутаматергические синапсы как за счет негеномных, так и более медленных, геномных механизмов, запускаемых ГК. Устойчивые изменения глутаматной трансмиссии могут играть ключевую роль в долгосрочных структурных/функциональных изменениях, связанных с расстройствами настроения у пациентов [65]. Иными словами, в настоящее время признается, что глутаматергическая система является основным медиатором психической патологии, и это отражает сдвиг парадигмы от моноаминовой гипотезы депрессии к гипотезе глутамат-зависимой нейропластичности. Гипотеза нейропластичности предполагает, что объемные изменения, постоянно обнаруживаемые в лимбических и кортикальных областях у пациентов с депрессией, в значительной степени обусловлены ремоделированием дендритов и потерей шипиков, вызванным ГК, и что гиперглутаматергические состояния играют ключевую роль в индукции неадаптивных клеточных эффектов, в свою очередь ответственных за неблагоприятные структурные изменения. Косвенным подтверждением гипотезы является тот факт, что препараты, используемые для терапии расстройств настроения/тревоги (антидепрессанты), предотвращают усиленное высвобождения глутамата [66].
Опосредованная глутаматными рецепторами усиленная возбуждающая нейротрансмиссия обычно ассоциируется с эпилепсией мезиальной височной доли со склерозом гиппокампа [67]. Гиперглутаматергическая активность в гиппокампе таких пациентов происходит на фоне повышенного уровня ГК (кортизола) и предполагает как непосредственное, так и опосредованное влияние ГК на глутаматергический синапс. Например, при эпилепсии височной доли изменение триптофан-кинуренинового пути в гиппокампе пациентов способствует формированию с гиперглутаматергического состояния [68], при этом активность этого пути находится под контролем ГК [69]. Важно отметить, что дисфункция глутаматергической системы в лимбических областях мозга, в первую очередь в гиппокампе, ассоциированная с нарушениями функционирования ГГНО, является ключевым звеном коморбидности эпилепсии и депрессии (пациенты с депрессией подвергаются большему риску развития эпилепсии и наоборот) [70, 71].
Еще одним примером заболевания мозга, в патогенезе которого прослеживается дисфункция взаимодействия ГГНО и глутаматергической системы, является шизофрения. Характерная особенность шизофрении – нарушение регуляции ГГНО и системы воспалительного ответа [72]. Полагают, что эти нарушения связаны с изменениями в развитии нервной системы в некоторых областях мозга, таких как гиппокамп, и это может затрагивать главным образом глутаматергические пути, в т.ч. за счет дисфункции NMDA-рецепторов. Одним из подтверждений участия глутаматергической дисрегуляция в патофизиологии шизофрении является способность антагонистов NMDA-рецептора, таких как кетамин, индуцировать шизофреноподобное поведение. Это может быть связано с их предполагаемым нейропатологическим действием на ГАМКергические интернейроны, которое может привести к гиперглутаматергическому состоянию. Предполагается, что при шизофрении, наряду с NMDA-рецепторами в ряде регионов мозга нарушено функционирование метаботропных глутаматных рецепторов группы II [73, 74]. Показано, что таламо-кортикальная гиперглутаматергическая трансмиссия при рефрактерной шизофрении связана с нарушениями NMDA-рецепторов и метаботропных глутаматных рецепторов группы III [75]. Окислительный стресс, который играет ключевую роль в патофизиологии шизофрении, ассоциирован с избытком свободных радикалов в результате гиперглутаматергического состояния [76].
Состояния, связанные с патологиями мозга в результате потребления алкоголя или наркотических соединений (как формирование зависимости, так и абстиненция), также ассоциированы с изменениями глутаматергической системы и функции ГГНО. Например, тяжесть синдрома отмены алкоголя, сопровождающегося гиперглутаматергическим состоянием [77], а также повреждение мозга и когнитивные нарушения связаны со степенью нарушения гомеостаза глутамата в мозге [78]. ГК играют важную роль в формировании зависимости от этанола, в частности в повышении пластичности глутаматергических синапсов, связанном с его потреблением, путем влияния на экспрессию эндогенных полиаминов и полиамин-чувствительных субъединиц NMDA-рецепторов [79]. При этом взаимодействия между ГК, полиаминами и NMDA-рецепторами важны как для развития зависимости от этанола, так и для возникновения поведенческих и невропатологических последствий, связанных с синдромом отмены. В формировании гиперглутаматергического состояния при хроническом потреблении алкоголя принимают участие и метаботропные глутаматные рецепторы mGluR1 и mGluR5 [80]. Показано ключевое участие ГГНО в формировании кокаиновой зависимости [81], при которой также отмечаются нарушения регуляции глутаматергической сигнализации. Например, самовведение кокаина и его отмена у крыс вызывает изменения структурной пластичности в прелимбической коре, соответствующие раннему гипоглутаматергическому состоянию, которое затем сменяется гиперглутаматергическим состоянием. Интересно, что мемантин, который временно блокирует NMDA-рецепторы и защищает нейроны от чрезмерной стимуляции избыточным синаптическим глутаматом, способен ослаблять наркотическую зависимость [82].
ЗАКЛЮЧЕНИЕ
Нейроэндокринный контроль, осуществляемый ГГНО, в т.ч. за счет секреции ГК, принципиально важен для поддержания нормального функционирования мозга и баланса между системами возбуждения и торможения. ГК регулируют состояние глутаматергической системы мозга как непосредственно через рецепторы на глутаматергических синапсах, так и опосредованными путями. Неспособность ГГНО адекватно регулировать глутаматергическую синаптическую пластичность приводит к развитию нейропсихических заболеваний, в патогенезе которых ключевую роль могут играть гиперглутаматергические состояния, вызывающие эксайтотоксичность (рис. 1).
Рис. 1.
Общая схема ассоциации ГК с гиперглутаматергическими состояниями при патологиях мозга (пояснения в тексте).
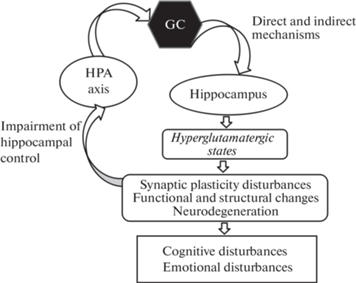
Как показано на примерах, приведенных в предыдущей главе, нарушение опосредованного ГР контроля глутаматергических процессов лежит в основе когнитивных и эмоциональных расстройств, эпилепсии и многих других церебральных патологий, т.е. представляет общий базовый механизм для многих болезней мозга и их коморбидностей. Исследование механизмов взаимодействия ГГНО и глутаматергической системы мозга имеет приоритетное трансляционное значение, поскольку знание этих механизмов может послужить основой эффективного предотвращения и терапии как неврологических (в т.ч. нейродегенеративных), так и психических заболеваний.
Список литературы
Gulyaeva NV (2019) Biochemical Mechanisms and Translational Relevance of Hippocampal Vulnerability to Distant Focal Brain Injury: The Price of Stress Response. Biochemistry (Mosc) 84: 1306–1328. https://doi.org/10.1134/S0006297919110087
Meyer JS (1985) Biochemical effects of corticosteroids on neural tissues. Physiol Rev 65: 946–1020. https://doi.org/10.1152/physrev.1985.65.4.946
Bolshakov AP, Tret’yakova LV, Kvichansky AA, Gulyaeva NV (2021) Glucocorticoids: Dr. Jekyll and Mr. Hyde of Hippocampal Neuroinflammation. Biochemistry (Mosc) 86: 156–167. https://doi.org/10.1134/S0006297921020048
Uchoa ET, Aguilera G, Herman JP, Fiedler JL, Deak T, de Sousa MB (2014) Novel aspects of glucocorticoid actions. J Neuroendocrinol 26: 557–572. https://doi.org/10.1111/jne.12157
de Kloet ER, Meijer OC, de Nicola AF, de Rijk RH, Joëls M (2018) Importance of the brain corticosteroid receptor balance in metaplasticity,cognitive performance and neuro-inflammation. Front Neuroendocrinol 49: 124–145. https://doi.org/10.1016/j.yfrne.2018.02.003
Prager EM, Johnson LR (2009) Stress at the synapse: signal transduction mechanisms of adrenal steroids at neuronal membranes. Sci Signal 2(86): re5. https://doi.org/10.1126/scisignal.286re5
Gulyaeva NV (2017) Molecular Mechanisms of Neuroplasticity: An Expanding Universe. Biochemistry (Mosc) 82: 237–242. https://doi.org/10.1134/S0006297917030014
Xiong H, Krugers HJ (2015) Tuning hippocampal synapses by stress-hormones: Relevance for emotional memory formation. Brain Res 1621: 114–120. https://doi.org/10.1016/j.brainres.2015.04.010
Jaszczyk A, Juszczak GR (2021) Glucocorticoids, metabolism and brain activity. Neurosci Biobehav Rev 126: 113–145. https://doi.org/10.1016/j.neubiorev.2021.03.007
Sapolsky RM (1993) Potential behavioral modification of glucocorticoid damage to the hippocampus. Behav Brain Res 57: 175–182. https://doi.org/10.1016/0166-4328(93)90133-b
Höschl C, Hajek T (2001) Hippocampal damage mediated by corticosteroids–a neuropsychiatric research challenge. Eur Arch Psychiatry Clin Neurosci 251 Suppl 2:II81–88. https://doi.org/10.1007/BF03035134
Gulyaeva N (2019) Functional Neurochemistry of the Ventral and Dorsal Hippocampus:Stress, Depression, Dementia and Remote Hippocampal Damage. Neurochem Res 44: 1306–1322. https://doi.org/10.1007/s11064-018-2662-0
Cox MF, Hascup ER, Bartke A, Hascup KN (2022) Friend or Foe? Defining the Role of Glutamate in Aging and Alzheimer’s Disease. Front Aging 3: 929474. https://doi.org/10.3389/fragi.2022.929474
Lalo U, Koh W, Lee CJ, Pankratov Y (2021) The Tripartite Glutamatergic Synapse. Neuropharmacology 199: 108758. https://doi.org/10.1016/j.neuropharm.2021.108758
Findley CA, Bartke A, Hascup KN, Hascup ER (2019) Amyloid Beta-Related Alterations to Glutamate Signaling Dynamics during Alzheimer’s Disease Progression. ASN Neuro 11. https://doi.org/10.1177/1759091419855541
Barco A, Bailey CH, Kandel ER (2006) CommonMolecular Mechanisms in Explicit and Implicit Memory. J Neurochem 97 (6): 1520–1533. https://doi.org/10.1111/J.1471-4159.2006.03870.X
Lee MC, Ting KK, Adams S, Brew BJ, Chung R, Guillemin GJ (2010) Characterisation of the Expression of NMDA Receptors in HumanAstrocytes. PLoS One 5 (11): e14123. https://doi.org/10.1371/JOURNAL.PONE.0014123
Hardingham GE, Bading H (2010). Synaptic versus Extrasynaptic NMDA Receptor Signalling: Implications for Neurodegenerative Disorders. Nat Rev Neurosci 11: 682–696. https://doi.org/10.1038/nrn2911
Vreugdenhil E, de Jong J, Schaaf MJ, Meijer OC, Busscher J, Vuijst C, de Kloet ER (1996) Molecular dissection of corticosteroid action in the rat hippocampus. Application of the differential display techniques. J Mol Neurosci 7(2): 135–146. https://doi.org/10.1007/BF02736793
Kessels HW, Malinow R (2009) Synaptic AMPA receptor plasticity and behaviour. Neuron 61: 340–350. https://doi.org/10.1016/j.neuron.2009.01.015
Timmermans W, Xiong H, Hoogenraad CC, Krugers HJ (2013) Stress and excitatory synapses: from health to disease. Neuroscience 248: 626–636. https://doi.org/10.1016/j.neuroscience.2013.05.043
Gulyaeva NV (2021) Glucocorticoid Regulation of the Glutamatergic Synapse: Mechanisms of Stress-Dependent Neuroplasticity. J Evol Biochem Phys 57: 564–576. https://doi.org/10.1134/S0022093021030091
Joëls M, Pasricha N, Karst H (2013) The interplay between rapid and slow corticosteroid actions in brain. Eur J Pharmacol 719: 44–52. https://doi.org/10.1016/j.ejphar.2013.07.015
Joëls M, de Kloet ER (2017) 30 Years of the mineralocorticoid receptor: The brain mineralocorticoid receptor: a saga in three episodes. J Endocrinol 234: T49–T66. https://doi.org/10.1530/JOE-16-0660
Le Menuet D, Lombès M (2014) The neuronal mineralocorticoid receptor: from cell survival to neurogenesis. Steroids 91: 11–19.https://doi.org/10.3389/fendo.2016.00066
Joëls M, Sarabdjitsingh RA, Karst H (2012) Unraveling the time domains of corticosteroid hormone influences on brain activity: rapid, slow, and chronic modes. Pharmacol Rev 64: 901–938. https://doi.org/10.1124/pr.112.005892
Pretorius E, Marx J (2004) Direct and indirect effects of corticosteroids on astrocyte function. Rev Neurosci15(3): 199–207. https://doi.org/10.1515/revneuro.2004.15.3.199
Gulyaeva NV (2017) Interplay between Brain BDNF and Glutamatergic Systems: A Brief State of the Evidence and Association with the Pathogenesis of Depression. Biochemistry (Mosc) 82: 301–307. https://doi.org/10.1016/j.ejphar.2013.07.015
Pluchino N, Russo M, Santoro AN, Litta P, Cela V, Genazzani AR (2013) Steroid hormones and BDNF. Neuroscience 239: 271–279. https://doi.org/10.1016/j.neuroscience.2013.01.025
Jezova D (2005) Control of ACTH secretion by excitatory amino acids: functional significance and clinical implications. Endocrine 28(3): 287–294. https://doi.org/10.1385/ENDO:28:3:287
Andersen JV, Markussen KH, Jakobsen E, Schousboe A, Waagepetersen HS, Rosenberg PA, Aldana BI (2021) Glutamate metabolism and recycling at the excitatory synapse in health and neurodegeneration. Neuropharmacology 196: 108719. https://doi.org/10.1016/j.neuropharm.2021.108719
Sood A, Preeti K, Fernandes V, Khatri DK, Singh SB (2021) Glia: A major player in glutamate-GABA dysregulation-mediated neurodegeneration. J Neurosci Res 99: 3148–3189. https://doi.org/10.1002/jnr.24977
Abulseoud OA, Alasmari F, Hussein AM, Sari Y (2022) Ceftriaxone as a Novel Therapeutic Agent for Hyperglutamatergic States: Bridging the Gap Between Preclinical Results and Clinical Translation. Front Neurosci 16: 841036. https://doi.org/10.3389/fnins.2022.841036
Fairless R, Bading H, Diem R (2021) Pathophysiological Ionotropic Glutamate Signalling in Neuroinflammatory Disease as a Therapeutic Target. Front Neurosci 15: 741280. https://doi.org/10.3389/fnins.2021.741280
Yuen EY, Wei J, Yan Z (2017) Molecular and Epigenetic Mechanisms for the Complex Effects of Stress on Synaptic Physiology and Cognitive Functions. Int J Neuropsychopharmacol 20: 948–955. https://doi.org/10.1093/ijnp/pyx052
Moraes BJ, Coelho P, Fão L, Ferreira IL, Rego AC (2021) Modified Glutamatergic Postsynapse in Neurodegenerative Disorders. Neuroscience 454: 116–139. https://doi.org/10.1016/j.neuroscience.2019.12.002
Armada-Moreira A, Gomes JI, Pina CC, Savchak OK, Gonçalves-Ribeiro J, Rei N, Pinto S, Morais TP, Martins RS, Ribeiro FF, Sebastião AM, Crunelli V, Vaz SH (2020) Going the Extra (Synaptic) Mile: Excitotoxicity as the Road Toward Neurodegenerative Diseases. Front Cell Neurosci 14: 90. https://doi.org/10.3389/fncel.2020.00090
Mira RG, Cerpa W (2021) Building a Bridge Between NMDAR-Mediated Excitotoxicity and Mitochondrial Dysfunction in Chronic and Acute Diseases. Cell Mol Neurobiol 41(7): 1413–1430. https://doi.org/10.1007/s10571-020-00924-0
Lim SI, Song KH, Yoo CH, Woo DC, Choe BY (2018) High-fat diet-induced hyperglutamatergic activation of the hippocampus in mice: A proton magnetic resonance spectroscopy study at 9.4T. Neurochem Int 114: 10–17. https://doi.org/10.1016/j.neuint.2017.12.007
Гуляева НВ (2020) Физиологический континуум пластичности и патологии нервной системы. Интеграт физиол 1: 294–302. [Gulyaeva NV (2020) Physiological continuum of plasticity and pathology of the nervous system. Integrat Physiol 1: 294–302. (In Russ)]. https://doi.org/10.33910/2687-1270-2020-1-4-294-302
Lupien SJ, Nair NP, Brière S, Maheu F, Tu MT, Lemay M, McEwen BS, Meaney MJ (1999) Increased cortisol levels and impaired cognition in human aging: implication for depression and dementia in later life. Rev Neurosci10: 117–139. https://doi.org/10.1515/revneuro.1999.10.2.11710658955
Magri F, Cravello L, Barili L, Sarra S, Cinchetti W, Salmoiraghi F, Micale G, Ferrari E (2006) Stress and dementia: the role of the hypothalamicpituitary-adrenal axis. Aging Clin Exp Res 18(2): 167–170. https://doi.org/10.1007/BF03327435
Justice NJ (2018) The relationship between stress and Alzheimer’s disease. Neurobiol Stress 8: 127–133. https://doi.org/10.1016/j.ynstr.2018.04.002
Sanguino-Gómez J, Buurstede JC, Abiega O, Fitzsimons CP, Lucassen PJ, Eggen BJL, Lesuis SL, Meijer OC, Krugers HJ. (2022) An emerging role for microglia in stress-effects on memory. Eur J Neurosci 55: 2491–2518. https://doi.org/10.1111/ejn.15188
Kim E, Otgontenger U, Jamsranjav A, Kim SS (2020) Deleterious Alteration of Glia in the Brain of Alzheimer’s Disease. Int J Mol Sci 21(18): 6676. https://doi.org/10.3390/ijms21186676
Escher CM, Sannemann L, Jessen F (2019) Stress and Alzheimer’s disease. J Neural Transm (Vienna) 126(9): 1155–1161. https://doi.org/10.1007/s00702-019-01988-z
Lyons CE, Bartolomucci A (2020) Stress and Alzheimer’s disease: A senescence link? Neurosci Biobehav Rev 115: 285–298. https://doi.org/10.1016/j.neubiorev.2020.05.010
Polleri A, Gianelli MV, Murialdo G (2002) Dementia: a neuroendocrine perspective. J Endocrinol Invest 25: 73–83. https://doi.org/10.1007/BF03343964
Saelzler UG, Verhaeghen P, Panizzon MS, Moffat SD (2021) Intact circadian rhythm despite cortisol hypersecretion in Alzheimer’s disease: A meta-analysis. Psychoneuroendocrinology 132: 105367. https://doi.org/10.1016/j.psyneuen.2021.105367
Milligan Armstrong A, Porter T, Quek H, White A, Haynes J, Jackaman C, Villemagne V, Munyard K, Laws SM, Verdile G, Groth D (2021) Chronic stress and Alzheimer’s disease: the interplay between the hypothalamic-pituitary-adrenal axis, genetics and microglia. Biol Rev Camb Philos Soc 96(5): 2209–2228. https://doi.org/10.1111/brv.12750
Notarianni E (2017) Cortisol: Mediator of association between Alzheimer’s disease and diabetes mellitus? Psychoneuroendocrinology 81: 129–137. https://doi.org/10.1016/j.psyneuen.2017.04.008
Herbert J, Lucassen PJ (2016) Depression as a risk factor for Alzheimer’s disease: Genes, steroids, cytokines and neurogenesis - What do we need to know? Front Neuroendocrinol 41: 153–171. https://doi.org/10.1016/j.yfrne.2015.12.001
Targa Dias Anastacio H, Matosin N, Ooi L (2022) Neuronal hyperexcitability in Alzheimer’s disease: what are the drivers behind this aberrant phenotype? Transl Psychiatry 12(1): 257. https://doi.org/10.1038/s41398-022-02024-7
Hascup KN, Findley CA, Britz J, Esperant-Hilaire N, Broderick SO, Delfino K, Tischkau S, Bartke A, Hascup ER (2021) Riluzole attenuates glutamatergic tone andcognitive decline in AβPP/PS1 mice. J Neurochem 156(4): 513–523. https://doi.org/10.1111/jnc.15224
Gulyaeva NV (2021) Hippocampal hyperglutamatergic signaling matters: Early targeting glutamate neurotransmission as a preventive strategy in Alzheimer’s disease: An Editorial Highlight for “Riluzole attenuates glutamatergic tone and cognitive decline in AβPP/PS1 mice”. J Neurochem 156: 399–402. https://doi.org/10.1111/jnc.15238
Srivastava A, Das B, Yao AY, Yan R (2020) Metabotropic Glutamate Receptors in Alzheimer’s Disease Synaptic Dysfunction: Therapeutic Opportunities and Hope for the Future. J Alzheimers Dis 78): 1345–1361. https://doi.org/10.3233/JAD-201146
Babaei P (2021) NMDA and AMPA receptors dysregulation in Alzheimer’s disease. Eur J Pharmacol 908: 174310. https://doi.org/10.1016/j.ejphar.2021.174310
Conway ME (2020) Alzheimer’s disease: targeting the glutamatergic system. Biogerontology 21(3): 257–274. https://doi.org/10.1007/s10522-020-09860-4
Zott B, Konnerth A (2022) Impairments of glutamatergic synaptic transmission in Alzheimer’s disease. Semin Cell Dev Biol.S1084-9521(22)00080-5. https://doi.org/10.1016/j.semcdb.2022.03.013
Song J, Yang X, Zhang M, Wang C, Chen L (2021) Glutamate Metabolism in Mitochondria is Closely Related to Alzheimer’s Disease. J Alzheimers Dis 84: 557–578. https://doi.org/10.3233/JAD-210595
Bukke VN, Archana M, Villani R, Romano AD, Wawrzyniak A, Balawender K, Orkisz S, Beggiato S, Serviddio G, Cassano T (2020) The Dual Role of Glutamatergic Neurotransmission in Alzheimer’s Disease: From Pathophysiology to Pharmacotherapy. Int J Mol Sci 21: 7452. https://doi.org/10.3390/ijms21207452
Behl C (1998) Effects of glucocorticoids on oxidative stress-induced hippocampal cell death: implications for the pathogenesis of Alzheimer’s disease. Exp Gerontol 33: 689–696. https://doi.org/10.1016/s0531-5565(98)00019-9
Ouanes S, Popp J (2019) High Cortisol and the Risk of Dementia and Alzheimer’s Disease: A Review of the Literature. Front Aging Neurosci 11: 43. https://doi.org/10.3389/fnagi.2019.00043
Sanacora G, Treccani G, Popoli M (2012) Towards a glutamate hypothesis of depression: an emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology 62(1): 63–77. https://doi.org/10.1016/j.neuropharm.2011.07.036
Musazzi L, Treccani G, Popoli M (2015) Functional and structural remodeling of glutamate synapses in prefrontal and frontal cortex induced by behavioral stress. Front Psychiatry 6:60. https://doi.org/10.3389/fpsyt.2015.00060
Musazzi L, Racagni G, Popoli M (2011) Stress, glucocorticoids and glutamate release: effects of antidepressant drugs. Neurochem Int 59(2): 138–149. https://doi.org/10.1016/j.neuint.2011.05.002
Banerjee J, Dey S, Dixit AB, Tripathi M, Doddamani R, Sharma MC, Chandra PS (2020) α7 nicotinic receptors contributes to glutamatergic activity in the hippocampus of patients with mesial temporal lobe epilepsy with hippocampal sclerosis (MTLE- HS). J Neural Transm (Vienna) 127(10): 1441–1446. https://doi.org/10.1007/s00702-020-02239-2
Dey S, Banerjee Dixit A, Tripathi M, Doddamani RS, Sharma MC, Lalwani S, Chandra PS, Banerjee J (2021) Altered hippocampal kynurenine pathway metabolism contributes to hyperexcitability in human mesial temporal lobe epilepsy-hippocampal sclerosis. Br J Pharmacol 178(19): 3959–3976. https://doi.org/10.1111/bph.15534
Jamshed L, Debnath A, Jamshed S, Wish JV, Raine JC, Tomy GT, Thomas PJ, Holloway AC (2022) An Emerging Cross-Species Marker for Organismal Health: Tryptophan-Kynurenine Pathway. Int J Mol Sci 23: 6300. https://doi.org/10.3390/ijms23116300
Kanner AM (2009) Depression and epilepsy: do glucocorticoids and glutamate explain their relationship? Curr Neurol Neurosci Rep 9: 307–312. https://doi.org/10.1007/s11910-009-0046-1
Gulyaeva NV (2021) Stress-Associated Molecular and Cellular Hippocampal Mechanisms Common for Epilepsy and Comorbid Depressive Disorders. Biochemistry (Mosc) 86: 641–656. https://doi.org/10.1134/S0006297921060031
Altamura AC, Boin F, Maes M (1999) HPA axis and cytokines dysregulation in schizophrenia: potential implications for the antipsychotic treatment. Eur Neuropsychopharmacol 10: 1–4. https://doi.org/10.1016/s0924-977x(99)00017-6
Martínez-Pinteño A, García-Cerro S, Mas S, Torres T, Boloc D, Rodríguez N, Lafuente A, Gassó P, Arnaiz JA, Parellada E (2020) The positive allosteric modulator of the mGlu2 receptor JNJ-46356479 partially improves neuropathological deficits and schizophrenia-like behaviors in a postnatal ketamine mice model. J Psychiatr Res 126: 8–18. https://doi.org/10.1016/j.jpsychires.2020.04.005
Lum JS, Millard SJ, Huang XF, Ooi L, Newell KA (2018) A postmortem analysis of NMDA ionotropic and group 1 metabotropic glutamate receptors in the nucleusaccumbens in schizophrenia. J Psychiatry Neurosci 43: 102–110. https://doi.org/10.1503/jpn.170077
Fukuyama K, Kato R, Murata M, Shiroyama T, Okada M (2019) Clozapine Normalizes a Glutamatergic Transmission Abnormality Induced by an Impaired NMDA Receptor in the Thalamocortical Pathway via the Activation of a Group III Metabotropic Glutamate Receptor. Biomolecules 9: 234. https://doi.org/10.3390/biom9060234
Limongi R, Jeon P, Théberge J, Palaniyappan L (2021) Counteracting Effects of Glutathione on the Glutamate-Driven Excitation/Inhibition Imbalance in First-Episode Schizophrenia: A 7T MRS and Dynamic Causal Modeling Study. Antioxidants (Basel) 10: 75. https://doi.org/10.3390/antiox10010075
Wang G, Weber-Fahr W, Frischknecht U, Hermann D, Kiefer F, Ende G, Sack M (2021) Cortical Glutamate and GABA Changes During Early Abstinence in Alcohol Dependence and Their Associations With Benzodiazepine Medication. Front Psychiatry 12: 656468. https://doi.org/10.3389/fpsyt.2021.656468
Laniepce A, Cabé N, André C, Bertran F, Boudehent C, Lahbairi N, Maillard A, Mary A, Segobin S, Vabret F, Rauchs G, Pitel AL (2020) The effect of alcohol withdrawal syndrome severity on sleep, brain and cognition. Brain Commun 2:fcaa123. https://doi.org/10.1093/braincomms/fcaa123
Prendergast MA, Mulholland PJ (2012) Glucocorticoid and polyamine interactions in the plasticity of glutamatergic synapses that contribute to ethanol-associated dependence and neuronal injury. Addict Biol 17: 209–223. https://doi.org/10.1111/j.1369-1600.2011.00375.x
Laukkanen V, Kärkkäinen O, Kautiainen H, Tiihonen J, Storvik M (2019) Increased [3H]quisqualic acid binding density in the dorsal striatum and anterior insula of alcoholics: A post-mortem whole-hemisphere autoradiography study. Psychiatry Res Neuroimaging 287: 63–69. https://doi.org/10.1016/j.pscychresns.2019.04.002
Hadad NA, Schwendt M, Knackstedt LA (2020) Hypothalamic-pituitary-adrenal axis activity in post-traumatic stress disorder and cocaine use disorder. Stress. 23(6): 638–650. https://doi.org/10.1080/10253890.2020.1803824
Montemitro C, Angebrandt A, Wang TY, Pettorruso M, Abulseoud OA (2021) Mechanistic insights into the efficacy of memantine in treating certain drug addictions. Prog Neuropsychopharmacol Biol Psychiatry 111: 110409. https://doi.org/10.1016/j.pnpbp.2021.110409
Дополнительные материалы отсутствуют.
Инструменты
Российский физиологический журнал им. И.М. Сеченова