

Успехи современной биологии, 2021, T. 141, № 2, стр. 107-117
Ассоциация между эволюционной консервативностью длинных некодирующих РНК и присутствием CPG-островов
И. А. Сидоренко 1, И. Б. Рогозин 1, 2, В. Н. Бабенко 1, 3, *
1 Институт цитологии и генетики СО РАН
Новосибирск, Россия
2 Национальные институты здоровья, Роквилл Пайк
Бесезда, США
3 Новосибирский государственный университет
Новосибирск, Россия
* E-mail: bob@bionet.nsc.ru
Поступила в редакцию 02.10.2020
После доработки 02.10.2020
Принята к публикации 02.10.2020
Аннотация
Гены длинных некодирующих РНК (lncRNA) в большом количестве представлены у млекопитающих, но их функции остаются в значительной степени неизвестными. Одним из возможных способов их изучения является использование крупномасштабных сравнений различных характеристик lncRNA с характеристиками кодирующих белок генов, для которых имеется множество функциональной информации. Характерной особенностью белок-кодирующих генов у млекопитающих является высокая эволюционная консервативность последовательностей экзонов. Хотя консервативность ДНК-последовательностей экзонов lncRNA не столь выражена, как у белок-кодирующих генов, но, тем не менее, она существенно выше, чем у интронов генов lncRNA. Мы оценили консервативность указанных характеристик с помощью множественного выравнивания четырех млекопитающих – человека, шимпанзе, мыши и крысы. Была проведена разметка экзон-интронной структуры каждого гена некодирующей lncRNA, а также их промоторной части (300 п.н.), и вычислена консервативность по позициям полных выравниваний. Исследование связи консервативности генов lncRNA и наличием/отсутствием CpG-островов (CGI) выявило более высокую консервативность генов lncRNA, которые локализованы рядом с CGI. Эта тенденция может быть причиной выявленной ранее ассоциации между консервативностью и уровнем экспрессии lncRNA. Важной задачей также являлась классификация выборок по типу lncRNA (сенс-, антисенс-, межгенных РНК, и псевдогенных локусов). Выяснилось, что lncRNA, находящиеся в белок-кодирующем локусе в прямой цепи (сенс-lncRNA), обладают наибольшим количеством промоторных CGI. На втором месте по встречаемости CGI были псевдогены, и на третьем месте – антисенс-lncRNA. Межгенные lncRNA обладали наименьшей долей CGI. Это позволило сделать вывод, что CGI-острова более присущи промоторам кодирующих генов, нежели некодирующих. Оценка консервативности CGI-островов путем сравнения геномов человек–мышь по наличию ортологичных CGI выявила степень их консервативности около 45%. Оценка консервативности еще раз подтвердила наличие ген-специфичных сигналов в некодирующих РНК. Расширение репертуара ген-специфичных сигналов для lncRNA с помощью CGI было сделано впервые, насколько известно авторам.
ВВЕДЕНИЕ
В последнее время растет внимание к длинным некодирующим РНК (lncRNA) – сравнительно недавним объектам изучения в области геномики. В то время, как роль многих некодирующих молекул РНК (рибосомных, транспортных (tRNA), малых ядрышковых (snoRNA), микро- (miRNA) и piwi) уже определена, функции lncRNA остаются в значительной степени неопределенными (Сидоренко и др., 2019; Goodrich, Kugel, 2006; Mercer et al., 2009; Ng et al., 2013; Kapusta, Feschotte, 2014).
Существует предположение, что основная масса lncRNA выступает побочным продуктом фоновой транскрипции (van Bakel, Hughes, 2009; Robinson, 2010). Это основано на их невысоком уровне экспрессии и малой эволюционной консервативности при сравнении с белок-кодирующими генами и такими малыми РНК, как miRNA и snoRNA (Marques, Ponting, 2009). В противовес этой точке зрения заметим, что часть lncRNA соответствуют эволюционно консервативным областям (Siepel et al., 2005), и существенная доля lncRNA выявляет меньшее по сравнению с остальными (некодирующими) районами число инсерций/делеций, указывая на присутствие селективного отбора (Ponjavic et al., 2007; Guttman et al., 2009; Managadze et al., 2011; Guttman, Rinn, 2012; Guttman et al., 2013; Kannan et al., 2015). При невысокой экспрессии lncRNA (Bertone et al., 2004; Amaral et al., 2013) секвенирование транскриптомов ряда видов привело к массовому обнаружению новых транскрибирующихся последовательностей. К примеру, лишь только проект FANTOM каталогизировал больше 30 000 предполагаемых длинных некодирующих транскриптов в тканях мыши методом клонирования полноразмерной комплементарной ДНК (кДНК) (Liu et al., 2006).
ДНК-последовательности большинства lncRNA гораздо менее консервативны ДНК-последовательностей белок-кодирующих генов. Тем не менее, уровень их ортологичности высок: от 60 до 70% генов lncRNA считаются ортологичными у человека и мыши (Managadze et al., 2013). lncRNA показывают своеобразную субклеточную локализацию и часто экспрессируются тканеспецифично. Они являются процессированными (полиаденилированными и сплайсированными) транскриптами; это позволяет предположить их функциональность (Сидоренко и др., 2019; Kapusta, Feschotte, 2014; Vance, Ponting, 2014). Другой возможный фактор функциональности lncRNA – наличие регуляторных сигналов сплайсинга, как, собственно, и самого сплайсинга (Chodroff et al., 2010; Schüler et al., 2014). Экспериментальное исследование lncRNA с уже определенной клеточной функцией выявило, что они функционируют в процессированной форме (Kapusta, Feschotte, 2014; Vance, Ponting, 2014). Сравнение около 3000 генов lncRNA мыши с ортологами крысы и человека выявило сохранение экзон-интронной структуры: 65%; и 40% |GT-AG|-сайтов сплайсинга lncRNA мыши консервативны у крысы и человека, что позволяет рассматривать сплайсинг как неотъемлемое свойство lncRNA (Ponjavic et al., 2007).
В составе класса lncRNA есть также многочисленные длинные межгенные некодирующие РНК (lincRNA), то есть молекулы РНК длиной больше 200 п.н., которые расположены за пределами белок-кодирующих локусов. Одной из наиболее изученных lincRNA считается Xist, которая принимает участие в инактивации Х-хромосомы самок плацентарных млекопитающих (Brockdorff et al., 1992; Chang et al., 2006). В частности, сообщалось, что ген Xist эволюционно произошел от псевдогена Lnx3, появившегося у ранних плацентарных. Вслед за тем последовала интеграция мобильных элементов (Duret et al., 2006; Elisaphenko et al., 2008), которые также наблюдаются в ряде других lincRNA (Kapusta et al., 2013).
Для геномов позвоночных характерно наличие большого количества CpG-островов (CGI), характеризующихся увеличенной долей CG-динуклеотидов (Gardiner-Garden, Frommer, 1987). Формальное определение CGI подразумевает три основных характеристики: процентная доля cg-динуклеотидов больше 60%; отношение cg_obs/ cg_exp > 0.6; длина острова >300 п.н. Упомянутое отношение видоспецифично, в частности у мыши плотность cg-динуклеотидов в CGI меньше, чем у человека (Illingworth, Bird, 2009). В геноме человека их количество составляет примерно 26 тыс. (www.genome.ucsc.edu). Медиана распределения длины CGI около 1 кб. CGI находится в промоторах 50–70% генов. Эти промоторы именуют CpG-промоторами (Babenko et al., 1999; Deaton, Bird, 2011). В генах гомеобоксов (Hox, Pax) и еще приблизительно в 5% других CpG-островов промоторов, CGI метилируются на определенной стадии или в определенной ткани (<6% островов). В силу кооперативности, статус метилирования островов имеет U-образное распределение по их численности против доли метилирования, с подавляющим (> 0%) левым плечом (гипометилирование; Zeng et al., 2014). Остальные 30–40% островов, не расположенные в промоторах генов, присутствуют во внутригенных районах (20%) и в межгенных областях (12–15%) (Deaton, Bird, 2011). Также, в силу кооперативности метилирования динуклеотидов CGI (Haerter et al., 2014), существуют способы интерполяции значений статуса метилирования подмножества нескольких CpG-динуклеотидов острова на все его CpG-динуклеотиды с учетом плотности CpG и длины острова (Zou et al., 2018).
Целью проведенной работы была оценка доли промоторов генов lncRNA, содержащих CpG-острова, для сравнения с тем же для кодирующих генов. Для этого в предоставленной работе мы поставили задачу проклассифицировать lncRNA по типу промоторов (CGI-содержащие, CGI-несодержащие (^CGI)), а также оценили консервативность промоторных районов, используя геномные выравнивания lncRNA четырех видов: человек–орангутанг–мышь–крыса. Мы также оценили уровень ассоциации консервативности длинных некодирующих генов и наличием/отсутствием CpG-островов.
МАТЕРИАЛЫ И МЕТОДЫ
Для нашего исследования были выбраны четыре вида млекопитающих: человек, шимпанзе, мышь и крыса. Множественные выравнивания геномов этих четырех видов были взяты из базы данных выравниваний UCSC (genome.ucsc.edu). В этой же базе данных были выбраны белок-некодирующие гены человека (lncRNA) длиною более 200 п.н. и состоящие из двух и более экзонов, как было предложено ранее (Cabili et al., 2011). Результирующая выборка lncRNA содержала 7941 транскриптов 5826 генов (табл. 1). В случае альтернативных изоформ соответствующий им ген в выборке был представлен максимальной по длине изоформой (число изоформ было указано, но специфицирована только одна). Геномные последовательности транскриптов lncRNA человека из результирующей выборки были пересечены с CpG-островами (CpG Island, CGI) по локализации. Затем эти районы были наложены на трек множественного выравнивания lncRNA. Анализируемый район включал в себя транскрипт, а также 300 п.н. промоторной области и 300 п.о. фланкирующего района. Использовался вариант множественного выравнивания с геномом человека (версия hg19) в качестве начального генома для построения выравнивания. В итоге было получено две выборки: lncRNA с CGI в гене и/или промоторе (CGI-выборка) и остальные lncRNA (^CGI-выборка). Статистика приведена в табл. 1.
Таблица 1.
Распределение числа элементов в выборке lncRNA человека, попавших в анализ
| Транскриптов в выборке | 7941 |
| Транскриптов, содержащих CGI | 4567 |
| Средний размер транскриптов | 44 086 |
| Минимальный размер транскриптов | 548 |
| Максимальный размер транскриптов | 1 698 418 |
| Среднее количество экзонов | 6.1 |
| Среднее количество интронов | 5.1 |
| Число генов (CGI, ^CGI) | 3091, 2735 |
| Число генов с >1 изоформ (CGI, ^CGI) | 758, 223 |
| Число уникальных CGI (без учета дуплицированных генов и изоформ) | 2969 |
Мы оценили число случаев, когда альтернативные lncRNA транскрипты начинались с альтернативного старта транскрипции. В случае CGI-содержащих промоторов было 26 генов, содержащих два альтернативных CGI-промотора на ген (более двух не наблюдалось). Таким образом, поскольку, в отличие от матричной РНК (mRNA), их было менее 1% от всех случаев, мы предпочли использовать гены, представленные максимальным транскриптом в части задач, связанной с оценкой консервативности, и ряда других.
Полученные на основе множественного выравнивания геномов четырех видов позиции использовались для оценки консервативности генов lncRNA. Пропуски исключались из рассмотрения, рассматривалась позиция с остальными нуклеотидами. Исключались также те участки генов lncRNA (интроны, экзоны, промоторы, 3'-районы), где количество информативных позиций было меньше 70. Эволюционная консервативность считалась как [1 – (количество замен)/(общее количество позиций)]. В качестве меры консервативности в каждой позиции мы использовали число замен, полученное по методу максимальной парсимонии, а именно: при наличии одинаковой замены в паре грызунов или приматов, считалось, что произошла одна общая предковая замена в соответствующей паре, а не в обоих видах.
Для анализа типов CGI с учетом вероятности и особенностей их метилирования было взято разбиение статуса метилирования на 5 классов из работы (Zeng et al., 2014). После оценки распределения 5 классов CGI, в силу малочисленности дифференциально метилированных CGI-классов в нашей выборке (Zeng et al., 2014), мы исследовали только два класса CpG-островов: гипометилированные (промоторные) CGI, и негипометилированные (все остальные; табл. 2). Для CGI-выборок человека мы провели поиск ортологичных CGI в мыши c помощью инструмента LiftOver (UCSC Genome Browser; http://genome.ucsc.edu/cgi-bin/hgLiftOver/).
РЕЗУЛЬТАТЫ
Анализ состава CGI/^CGI-выборок
Путем сравнения имен некодирующих генных локусов lncRNA с кодирующими мы выяснили, что из 3091 (различных) генов lncRNA, содержащих в основном промоторный CGI, по крайней мере 1402 белок-кодирующих генных локуса содержат некодирующую изоформу из нашей выборки (2395 транскриптов) в той же цепи, то есть перекрывающиеся с кодирующей частью (предполагающего NMD (nonsense-mediated mRNA decay, нонсенс-опосредованный распад мРНК) – деградацию; Lareau et al., 2007). Порядка 800 генов являются аннотироваными антисенс-РНК. Остальные lncRNA (порядка 1000) попадают в межгенные районы или находятся в локусе белок-кодирующих генов. В табл. 3 приведены cоставы CGI/^CGI-выборок по распределению в генах, кодирующих белки и длинные некодирующие РНК. Заметно, что в выборке ^CGI число случаев lncRNA в белок-кодирующем локусе в 4 раза меньше, чем в CGI-содержащих генах.
Анализ альтернативного сплайсинга и экспрессии генов lncRNA
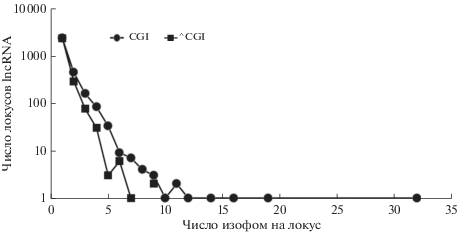
Существенно меньшее число белок-кодирующих локусов в ^CGI-выборке (344) может приводить к тому (табл. 3, выделенные значения: 1402/344 = 4 раза), что число некодирующих изоформ на кодирующий локус значительно меньше в ^CGI-выборке по сравнению с CGI-выборкой (рис. 1), что может свидетельствовать о большой интенсивности альтернативного сплайсинга lncRNA именно в кодирующих локусах. Кроме этого, подтверждается более интенсивная экспрессия lncRNA генов, содержащих CGI в промоторе.
Рис. 1.
Распределение числа транскриптов по локусам в CGI/^CGI-выборках lncRNA. Первые 2 точки (1 изоформа, 2 изоформы) охватывают 70% генов каждой выборки.
Мы использовали базу данных GTEX (ENCODE Consortium, 2017) для исследования экспрессии транскриптов lncRNA в выборке. Как правило, высокоэкспрессирующиеся гены lncRNA являются функциональными (Niazi, Valadkhan, 2012). Мы оценили распределение величины экспрессии в транскриптах, представленных на рис. 2. Из 6718 транскриптов (1103 транскриптов не было найдено в базе данных GTEX), 3782 транскрипта вошли в состав CGI-выборки и 2930 транскриптов вошли в состав ^CGI-выборки. В качестве оценки экспрессии генов нами были взяты максимальные значения экспрессии (transcripts per million, TPM) транскрипта среди 53 тканей в базе данных GTEX, результаты представлены на рис. 2. Для 67% CGI- и 79% ^CGI-выборок максимальный уровень экспрессии был менее 5 TPM (первые измерения на шкале TPM на рис. 2).
Рис. 2.
Распределение числа транскриптов в зависимости от максимальной величины экспрессии транскрипта по базе данных GTEX в CGI/^CGI-выборках.
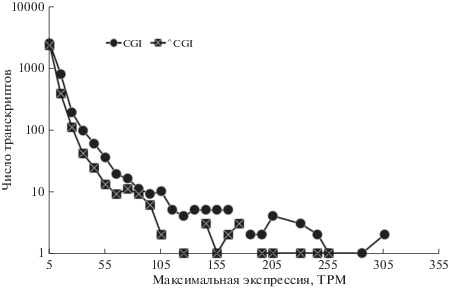
Таким образом, можно оценить, что в общей выборке около 71% транскриптов имеют экспрессию ниже 5 TPM при максимальном значении в какой-либо ткани (либеральный критерий). Это также соответствует приведенным данным в литературе по lncRNA (Niazi, Valadkhan, 2012). Необходимо отметить сходство распределения числа транскриптов по локусам в CGI/^CGI-выборках (рис. 1) и распределения числа транскриптов в зависимости от максимальной величины экспрессии транскрипта (рис. 2). Возможно, именно уровень экспрессии является основным фактором, определяющим повышенное число альтернативных изофорим в CGI-выборке.
В качестве дополнительного анализа, мы оценили группу lncRNA, у которых наблюдалась тканеспецифическая экспрессия. Она идентифицировалась по эмпирическому порогу: stdev/avg > 1.3, которые считались по 53 тканям в базе данных GTEX (описание процедуры приведено на Доп. рис. 1 в дополнительных материалах (доступно только в электронной версии журнала)). Мы смогли найти 117 lncRNA в GTEX по экспрессии в 53 тканях, приведенных в табл. 4, удовлетворяющих этому критерию. Стоит заметить, что lncRNA из этой группы эспрессируются крайне тканеспецифично, и, при этом, с высоким уровнем экспрессии (см. Доп. мат., Приложения 1, 2 ). Наибольшее число тканеспецифичных генов наблюдались в семенниках (53 транксрипта), а также в поджелудочной железе (5 транскриптов: см. Приложения). Высокий уровень тканеспецифической экспрессии генов lncRNA отмечался ранее (Kapusta, Feschotte, 2014; Vance, Ponting, 2014; Postnikova et al., 2019). Наличие большого количества альтернативных изоформ lncRNA, которые частично перекрываются с белок-кодирующими экзонами (сенс-lncRNA), может быть связана с регуляцией экспрессии белок-кодирующих генов (Kapusta, Feschotte, 2014; Vance, Ponting, 2014, Postnikova et al., 2019).
Распределение CGI по статусу метилирования
Используя данные (Zeng et al., 2014; табл. 2), мы произвели идентификацию статуса метилирования CGI, попавших в нашу выборку. Результаты приведены в табл. 5.
Таблица 5.
Число перекрывания CGI с генными сегментами в CGI-выборке. I – гипометилированные (промоторные) CGI, ^I – вариабельные и гиперметилированные CGI
| Категория | I | ^I | N/A | Всего |
|---|---|---|---|---|
| 3'-фланкирующие районы | 44 | 21 | 7 | 72 |
| Экзоны | 1768 | 188 | 274 | 2230 |
| Интроны | 1556 | 305 | 260 | 2121 |
| Промоторы | 1717 | 104 | 250 | 2071 |
| Всего | 5085 | 618 | 791 | 6494 |
В табл. 5 указано число генов, перекрывающихся с CGI разными генными компартментами. Из нее можно сделать вывод, что наибольшее число метилированных островов (32%) находятся в 3'-районах, наименьшее (5.6%) – в промоторах, 16% CGI метилированы в интронах, 9% – в экзонах. Это в целом соотносится с теми же показателями в кодирующих генах (в промоторах кодирующих генов 3.6% метилированных CGI, в 3'-районах – 61%, в экзонах – 17%, и в интронах – 14%).
Перекрывание CGI c lncRNA по сегментам гена
Поскольку число физических (различных) CGIs в исследуемой выборке всего 2969 (табл. 1), а число попаданий в экзоны, интроны и промоторы ненамного меньше этого числа (1556–1768), то ожидаемая частота попадания (перекрывания) одного CGI трех (начальных) сегментов гена – около 60% (табл. 5). Только небольшое число CGI находится в 3'-районе (табл. 5, рис. 3).
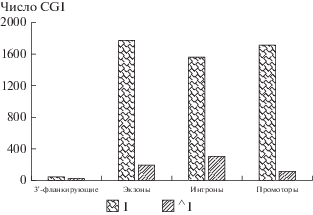
Мы оценили распределение одновременного перекрывания CGI с 1–4 сегментами гена: 3'-фланкирующие районы, экзоны, интроны, промоторы, приведенные в табл. 6. Большинство промоторных CGI одновременно перекрывается с экзоном и интроном. Учитывая, что ген в нашей выборке содержит по крайней мере 2 и более экзона (среднее – 6; табл. 1), среднее число перекрытий сегментов – 2.4 на ген lncRNA (как можно вычислить из табл. 6).
Оценка промоторного потенциала по базе данных GeneHancer
Мы оценили качество и количество промоторных областей lncRNA двух выборок. Для этого мы использовали данные промоторного потенциала, вычисленные в работе (Fishilevich et al., 2017) по семи источникам данных, включая хроматиновые метки, Eqtl-данные, данные конформационного анализа хроматина и позиции относительно гена среди 38 тканей. Мы выяснили, что 2677 генов из 3091 (табл. 1) CGI-выборки найдена в базе данных GeneHancer, для выборки ^CHI – 1926 генов из 2973. В зависимости от активности и подтверждения промоторного статуса по семи типам данных, а также его присутствия в различных типах клеток, каждый промотор оценивался по шкале от 1 до 1000 (z-score; Haeussler et al., 2019). Путем пересечения таблицы промоторов GeneHancer с CpG-островами CGI-выборки мы установили, что 2455 генов из 2677 доступных имеют CGI в промоторном сегменте, аннотированном в GeneHancer. Для выборки ^CGI 1118 генов из 1926 доступных имеют аннотированный в GeneHancer промотор в пределах –300…+300 п.н.
При оценке промоторного потенциала каждой из выборок мы наблюдали следующую картину: в выборке ^CGI не было выраженного пика, в медиане величина была равна 120 (из 1000 максимальных баллов) (рис. 4), в то время как в выборке CGI наблюдался пик при значении промоторного потенциала 500 (рис. 5).
Рис. 4.
Распределение промоторного потенциала (GeneHancer Database) в ^CGI-выборке. Мода (пик) промоторного потенциала – порядка 130, 1118 генов.
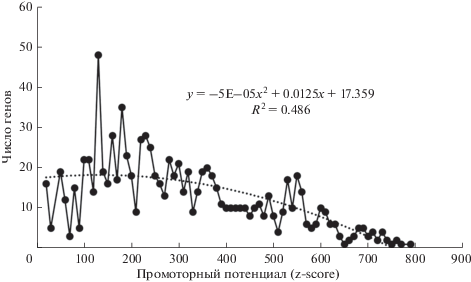
Рис. 5.
Распределение промоторного потенциала (GeneHancer Database) в CGI-выборке. Пик промоторного потенциала – 550, 2455 генов.
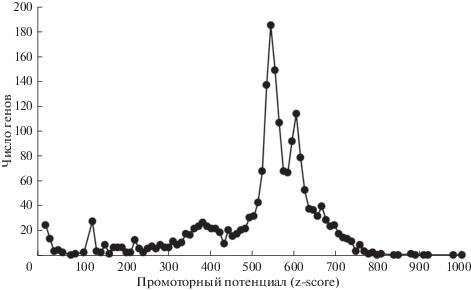
Здесь же мы оценили число генов lncRNA, имеющих 2 альтернативных CGI-промотора, их оказалось 26 (в основном, антисенс-РНК): MEF2C-AS1, HOXB-AS3, ZNF718, LOC441242, MAP3K14-AS1, LEF1-AS1, TRAM2-AS1, LOC100507557, MSTO2P, CBR3-AS1, EGFL7, LINC01159, MAGI2-AS3, LIPE-AS1, KCTD2, LOC101929340, EPS15L1, THTPA, LOC100506125, DHRS4L1, MIR124-2HG, PGAP2, ID2-AS1, CRIP2, CYB561D2, HOXA-AS3.
Анализ консервативности lncRNA в выборках CGI и ^CGI
Эволюционная консервативность является важным индикатором функциональных районов генома (Koonin, Rogozin, 2003). Важным фактором, который может существенно влиять на консервативность lncRNA является перекрывание с белок-кодирующими мРНК, для которых повышенная консервативность является общепризнанным фактом (Koonin, Rogozin, 2003). Для этого выборка lncRNA была разделена на две подвыборки: lncRNA, которые перекрываются с мРНК (рис. 3) и не перекрываются с мРНК (рис. 4).
Из рис. 6 и 7 можно видеть, что фактор перекрывания c мРНК не сильно влияет на распределение консервативности. Напротив, наличие CGI-острова в промоторе lncRNA существенно увеличивает величину консервативности, особенно в экзонах (рис. 6г и 7г). На основе распределений графиков консервативности (рис. 6 и 7) мы оценили достоверность отличия распределений консервативности в зависимости от наличия или отсутствия CpG-островов тестом Манна–Уитни и получили значения вероятности (P-value), приведенные в табл. 7. Видно, что вышеупомянутые свойства увеличения консервативности при наличии CGI в экзонах имеют статистически значимую поддержку вне зависимости от перекрывания с мРНК (табл. 7). Интересно, что наблюдаемая тенденция была найдена для интронов (рис. 6 и 7). Возможной причиной повышенной консервативности интронов (не столь четко выраженной по сравнению с экзонами) может быть наличие в их составе многочисленных неаннотированных альтернативных экзонов, что согласуется с повышенной частотой альтернативных изоформ генов lncRNA, ассоциированных с CpG-островами (рис. 2).
Рис. 6.
Распределение уровня консервативности в промоторах (а), интронах (б), экзонах (в) и 3'-фланкирующих районах (г), содержаших (светло-серый цвет) и не содержаших CGI (темно-серый цвет) при условии перекрывания lncRNA и мРНК.
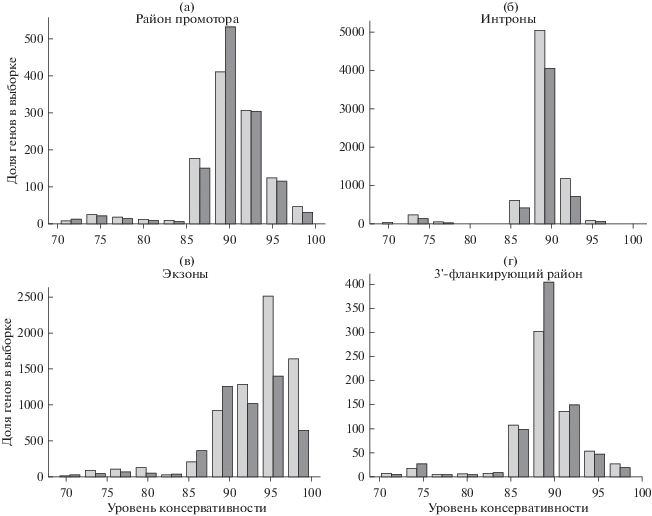
Рис. 7.
Распределение уровня консервативности в промоторах (а), интронах (б), экзонах (в) и 3'-фланкирующих районах (г), содержаших (светло-серый цвет) и не содержаших CGI (темно-серый цвет) при условии неперекрывания lncRNA и мРНК.
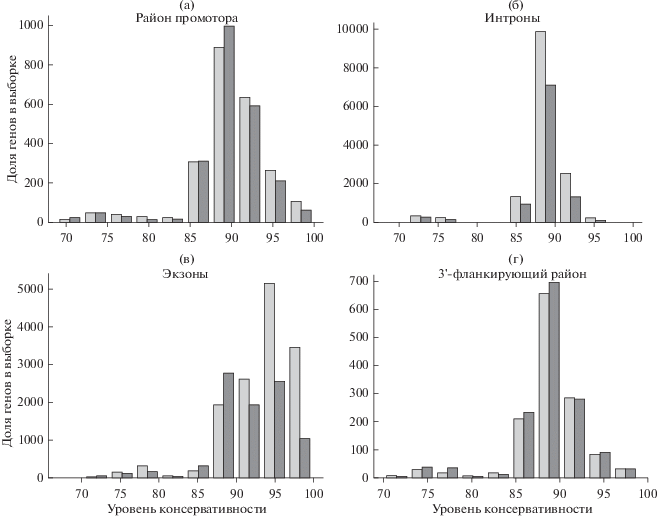
Таблица 7.
Таблица вероятностей (P-values) сравнения распределений категорий lncRNA c наличием CGI и без CGI (рис. 6 и 7)
| Категория | P-values | |||
|---|---|---|---|---|
| промоторы | интроны | экзоны | 3'-конец | |
| Выборка с перекрыванием с мРНК | 0.11 | 0.017 | 0.0001 | 0.21 |
| Выборка без перекрывания с мРНК | 0.005 | 0.001 | 0.0001 | 0.051 |
ОБСУЖДЕНИЕ
Эволюционная консервативность показывает функциональную значимость районов геномной ДНК. Классическим примером является критерий отбора в белок-кодирующих генах. Критерий определяется отношением несинонимических (Ka) к синонимическим (Ks) заменам. Предполагается, что положительный отбор наблюдается при Ka/Ks > 1, в то время как отрицательный отбор может наблюдаться при Ks/Ka > 1 (Hurst, 2002). При рассмотрении генов lncRNA скорость замен экзонов (Ke) можно считать аналогичной Ka, а интронов (Ki), соответственно, – Ks (Louie et al., 2003; Hoffman, Birney, 2007; Resch et al., 2007). Селективный отбор в экзонах lncRNA потенциально может быть определен при условии Ke/Ki < 1.
Ранее было показано (Managadze et al., 2011), что скорость эволюции экзонов lncRNA мыши и человека значительно ниже скорости замещения интронов (Ke/Ki < 1). Результаты исследования говорят о том, что селективный отбор действует на экзоны генов lncRNA и согласуются с более ранними наблюдениями (Ponjavic et al., 2007; Guttman et al., 2009). Также было показано, что распределение скоростей замен значительно шире для выборки экзонов lncRNA мыши и человека, чем для выборки интронов. Это указывает на меняющуюся интенсивность селективного отбора на гены lncRNA. Наши результаты в целом подтверждают эти наблюдения (рис. 6 и 7).
В работе (Managadze et al., 2013) также были оценены уровни экспрессии. Исследователи выявили высокую корреляцию между уровнями экспрессии lncRNA. В экзонах lncRNA мыши и человека наблюдалась статистически значимая отрицательная корреляция между скоростью эволюции последовательности и ее уровнем экспрессии. Коэффициенты корреляции в основном находились в диапазоне 0.1–0.16. Напротив, для интронов аналогичные коэффициенты корреляции были очень низкими и статистически не значимыми. Таким образом, авторами была показана отрицательная корреляция между скоростью эволюции и уровнем экспрессии lncRNA (Managadze et al., 2013).
В данной работе нами исследовалась связь консервативности генов lncRNA и наличием/отсутствием CpG-островов (CGI). Была показана более высокая консервативность генов lncRNA, которые локализованы рядом с CGI. Этот результат может объяснить выявленную ранее отрицательную корреляцию между скоростью эволюции и уровнем экспрессии lncRNA (Managadze et al., 2013). Наличие или отсутствие CGI может быть причиной наблюдаемой корреляции. Однако мы не можем исключить, что более высокая консервативность генов lncRNA является следствием отрицательной корреляции между скоростью эволюции и уровнем экспрессии lncRNA. В любом случае, наличие значимых отличий между классами генов lncRNA указывает на то, что значительная доля этих генов является функционально важной и становится приоритетным кандидатом при поиске генов lncRNA, которые регулируют различные процессы в клетках млекопитающих.
Список литературы
Сидоренко И.А., Рогозин И.Б. Бабенко В.Н. Эволюция экзонов и экзон-интронной структуры генов длинных межгенных некодирующих РНК у плацентарных млекопитающих // Успехи соврем. биол. 2019. Т. 139. №3. С. 221–234.
Amaral P.P., Dinger M.E., Mattick J.S. Non-coding RNAs in homeostasis, disease and stress responses: an evolutionary perspective // Brief. Func. Genom. 2013. V. 12. P. 254–278.
Babenko V.N., Kosarev P.S., Vishnevsky O.V. Investigating extended regulatory regions of genomic DNA sequences // Bioinformatics. 1999. V. 15 (7–8). P. 644–653.
Bertone P., Stolc V., Royce T.E. et al. Global identification of human transcribed sequences with genome tiling arrays // Science. 2004. V. 306. P. 2242–2246.
Brockdorff N., Ashworth A., Kay G.F. et al. The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus // Cell. 1992. V. 71. P. 515–526.
Cabili M.N., Trapnell C., Goff L. et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses // Gen. Dev. 2011. V. 25. P. 1915–1927.
Chang S.C., Tucker T., Thorogood, N.P. et al. Mechanisms of X-chromosome inactivation // Front. Biosci. 2006. V. 11. P. 852–866.
Chodroff R.A., Goodstadt L., Sirey T.M. et al. Long noncoding RNA genes: conservation of sequence and brain expression among diverse amniotes // Gen. Biol. 2010. V. 11. P. R72.
Deaton A. M., Bird A. CpG islands and the regulation of transcription // Genes Dev. 2011. V. 25. P. 1010–1022.
Duret L., Chureau C., Samain S. et al. The Xist RNA gene evolved in eutherians by pseudogenization of a protein-coding gene // Science. 2006. V. 312. P. 1653–1655.
Elisaphenko E.A., Kolesnikov N.N., Shevchenko A.I. et al. A dual origin of the Xist gene from a protein-coding gene and a set of transposable elements // PLoS One. 2008. V. 3. P. e2521.
Fishilevich S., Nudel R., Rappaport N. et al. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards // Database (Oxford). 2017. https://doi.org/10.1093/database/bax028
Gardiner-Garden M., Frommer M. CpG islands in vertebrate genomes // J. Mol. Biol. 1987. V. 196 (2). P. 261–282.
Goodrich J.A., Kugel J.F. Non-coding-RNA regulators of RNA polymerase II transcription // Nat. Rev. Mol. Cell. Biol. 2006. V. 7. P. 612–616.
Guttman M., Rinn J.L. Modular regulatory principles of large non-coding RNAs // Nature. 2012. V. 482. P. 339–346.
Guttman M., Amit I., Garber M. et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals // Nature. 2009. V. 458. P. 223–227.
Guttman M., Russell P., Ingolia N.T. et al. Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins // Cell. 2013. V. 154. P. 240–251.
Haerter J. O., Lövkvist C., Dodd G. et al. Collaboration between CpG sites is needed for stable somatic inheritance of DNA methylation states // Nucl. Acids Res. 2014. V. 42. P. 2235–2244.
Haeussler M., Zweig A.S., Tyner C. et al. The UCSC genome browser database: 2019 update // Nucl. Acids Res. 2019. V. 47 (1). P. D853–D858.
Hoffman M.M., Birney E. Estimating the neutral rate of nucleotide substitution using introns // Mol. Biol. Evol. 2007. V. 24. P. 522–531.
Hurst L.D. The Ka/Ks ratio: diagnosing the form of sequence evolution // Trends Genet. 2002. V. 18. P. 486.
Illingworth R.S., Bird A.P. CpG islands – 'a rough guide' // FEBS Lett. 2009. V. 583 (11). P. 1713–1720.
Kannan S., Chernikova D., Rogozin I.B. et al. Transposable element insertions in long intergenic non-coding RNA genes // Front. Bioeng. Biotechnol. 2015. V. 3. P. 71.
Kapusta A., Feschotte C. Volatile evolution of long noncoding RNA repertoires: mechanisms and biological implications // Trends Genet. 2014. V. 30. P. 439–452.
Kapusta A., Kronenberg Z., Lynch V.J. et al. Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs // PLoS Genet. 2013. V. 9. P. e1003470.
Koonin E.V., Rogozin I.B. Getting positive about selection // Gen. Biol. 2003. V. 4 (8). P. 331.
Lareau L.F., Brooks A.N., Soergel D.A. et al. The coupling of alternative splicing and nonsense-mediated mRNA decay // Adv. Exp. Med. Biol. 2007. V. 623. P. 190–211.
Liu J., Gough J., Rost B. Distinguishing protein-coding from non-coding RNAs through support vector machines // PLoS Genet. 2006. V. 2. P. e29.
Louie E., Ott J., Majewski J. Nucleotide frequency variation across human genes // Gen. Res. 2003. V. 13. P. 2594–2601.
Managadze D., Rogozin I.B., Chernikova D. et al. Negative correlation between expression level and evolutionary rate of long intergenic noncoding RNAs // Gen. Biol. Evol. 2011. V. 3. P. 1390–1404.
Managadze D., Lobkovsky A.E., Wolf Y.I. et al. The vast, conserved mammalian lincRNome // PLoS Comput. Biol. 2013. V. 9. P. e1002917.
Marques A.C., Ponting C.P. Catalogues of mammalian long noncoding RNAs: modest conservation and in-completeness // Gen. Biol. 2009. V. 10. P. R124.
Mercer T.R., Dinger M.E., Mattick J.S. Long non-coding RNAs: insights into functions // Nat. Rev. Genet. 2009. V. 10. P. 155–159.
Ng S.Y., Lin L., Soh B.S., Stanton L.W. Long non-coding RNAs in development and disease of the central nervous system // Trends Genet. 2013. V. 29. P. 461–468.
Niazi F., Valadkhan S. Computational analysis of functional long noncoding RNAs reveals lack of peptide-coding capacity and parallels with 3' UTRs // RNA. 2012. V. 18 (4). P. 825–843.
Ponjavic J., Ponting C.P., Lunter G. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs // Gen. Res. 2007. V. 17. P. 556–565.
Postnikova O.A., Rogozin I.B., Samuel W. et al. Volatile evolution of long non-coding RNA repertoire in retinal pigment epithelium: insights from comparison of bovine and human RNA expression profiles // Genes (Basel). 2019. V. 3. P. 205.
Resch A.M., Carmel L., Marino-Ramírez L. et al. Widespread positive selection in synonymous sites of mammalian genes // Mol. Biol. Evol. 2007. V. 24. P. 1821–1831.
Robinson R. Dark matter transcripts: sound and fury signifying nothing? // PLoS Biol. 2010. V. 8. P. e1000370.
Schüler A., Ghanbarian. A.T., Hurst L.D. Purifying selection on splice-related motifs, not expression level nor RNA folding explains nearly all constraint on human lincRNAs // Mol. Biol. Evol. 2014. V. 31. P. 3164–3183.
Siepel A., Bejerano G., Pedersen J.S. et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes // Gen. Res. 2005. V. 15. P. 1034–1050.
van Bakel H., Hughes T.R. Establishing legitimacy and function in the new transcriptome // Brief. Funct. Gen. Proteom. 2009. V. 8. P. 424–436.
Vance K.W., Ponting C.P. Transcriptional regulatory functions of nuclear long noncoding RNAs // Trends in Genetics. 2014. V. 30. P. 348–355.
Zeng J., Nagrajan H.K., Yi S.V. Fundamental diversity of human CpG islands at multiple biological levels // Epigenetics. 2014. V. 9. P. 483–491.
Zou L. S., Erdos M. R., Taylor D.L. et al. BoostMe accurately predicts DNA methylation values in whole-genome bisulfite sequencing of multiple human tissues // BMC Genom. 2018. V. 19. P. 390.
Дополнительные материалы
- скачать ESM.docx
- Рис. 1. «Калькулятор» вычисления числа тканей, в которых ген экспрессируется, от отношения stddev/avg (для таблиц внизу) для 53 величин экспрессии тканей в GTEX.
Приложение 1. Таблица экспрессии в CGI выборке тканеспецифичных генов (65 входов), за исключением семенников (приложение 3)
Приложение 2. Таблица экспрессии тканеспецифичных генов в ^CGI выборке (44 входа), за исключением семенников (приложение 3).
Приложение 3. Семенник - специфические некодирующие lncRNA из CGI (выделено желтым) и ^CGI выборок.
Инструменты
Успехи современной биологии