

Успехи физиологических наук, 2021, T. 52, № 1, стр. 31-48
Основные биохимические аспекты патогенеза депрессии. Часть II
Е. Е. Дубинина a, *, Л. В. Щедрина a, **, Г. Э. Мазо a, ***
a ФГБУ Национальный медицинский исследовательский центр психиатрии и неврологии им. В.М. Бехтерева
Минздрава России
192019 Санкт-Петербург, Россия
* E-mail: eedubinina@rambler.ru
** E-mail: petrored@bekhterev.ru
*** E-mail: galina-mazo@yandex.ru
Поступила в редакцию 27.06.2020
После доработки 20.08.2020
Принята к публикации 10.09.2020
Аннотация
Данная статья посвящена изучению патофизиологических основ депрессивного состояния, отражающих значимость третичного звена передачи сигнала в патогенезе депрессии. Представлены обобщенные литературные данные, рассматривающие особенности метаболизма триптофана, нарушение метаболизма серотонина и норадреналина, дисфункции серотонинергической, норадренергической и дофаминоергической нейротрансмиссии при депрессии. Проанализирована роль нейротрофинов в качестве первичных мессенджеров в регуляции функциональной активности клеток. Нарушение функции нейротрофинов приводит к сдвигу между процессами пролиферации и апоптозом в сторону последнего, что является причиной снижения нейрогенеза и развития нейродегенерации при патологических состояниях, в частности, депрессивном расстройстве.
ВВЕДЕНИЕ
Симптоматика любого заболевания обусловлена характером изменений на уровне третичного звена передачи сигнала в клетке, связанного с влиянием первичных и вторичных мессенджеров. В первой части обзора была подробно освещена роль окислительного стресса (ОС), иммуновоспалительной и гормональной систем при депрессии, которые, выполняя функцию первичных и вторичных мессенджеров, вызывают нарушения третичного звена передачи сигнала в клетке [8] . Это, в свою очередь, приводит к специфическим для депрессии нарушениям метаболизма серотонина и норадреналина, дисфункции серотонинергической, норадренергической и дофаминоергической нейротрансмиссии. Параллельно нарушается сбалансированное в норме соотношение процессов пролиферации и апоптоза в клетке, сопряженное с нарушением процессов нейрогенеза и развитием нейродегенерации. Мы предполагаем, что в целом патогенез депрессивного расстройства следует рассматривать в свете нарушений механизма адаптации на уровне первичных, вторичных и третичных звеньев передачи информации. Вычленение из этого комплекса отдельных его компонентов и рассмотрение их в качестве возможных причин депрессии фактически не полностью отражают патогенез данного заболевания. Целью второй части нашего обзора является попытка представить существующие на эту тему теории, связанные фактически только с нарушением третичного звена передачи сигнала в клетке в свете его тесной взаимосвязи с первичными и вторичными мессенджерами.
ВОЗМОЖНЫЕ МЕХАНИЗМЫ НАРУШЕНИЯ МЕТАБОЛИЗМА БИОГЕННЫХ АМИНОВ (СЕРОТОНИН, НОРАДРЕНАЛИН) ПРИ ДЕПРЕССИВНОМ СОСТОЯНИИ
Основные гипотезы формирования депрессии базируются на роли биогенных аминов и нарушении их метаболизма [75]. Однако их значимость в патогенезе заболевания носит вторичный характер и их следует рассматривать в тесной связи с ОС и состоянием иммунно-воспалительной и гормональной систем.
При депрессии наблюдается дисфункция серотонинергической, норадренергической и дофаминоергической нейротрансмиссии [67, 102]. Развитие депрессивных состояний сопряжено с подавлением моноаминоергических медиаторных систем, в частности, серотонинергических и катехоламиноергических, снижением уровня моноаминовых нейротрансмитеров серотонина и норадреналина, нарушением структуры рецепторов [19, 81, 83, 93]. Многочисленные клинические и экспериментальные данные свидетельствуют о подавлении серотонинергической трансмиссии при депрессивных состояниях. Это может быть связано с нарушением метаболизма аминокислоты триптофана, которая является источником серотонина.
Известно, что распад триптофана у млекопитающих в основном осуществляется двумя путями. Основной – кинурениновый путь начинается с разрыва индольного кольца триптофана и образованием N-формилкинуренина. Эта реакция катализируется триптофан-2,3-диоксигеназой (ТДО) или индоламино-2,3-диоксигеназой (ИДО), отличающихся только разной локализацией в тканях и обладающих высокой индуцибельностью [54]. ТДО относится к геминовым ферментам, с которыми связано участие супероксидного анион-радикала – начального звена генерации активных форм кислорода (АФК). Активность этого фермента регулируется глюкокортикоидами, провоспалительными цитокинами на уровне синтеза специфической м-РНК, которая участвует в синтезе апофермента диоксигеназы.
Кинурениновый путь метаболизма триптофана в основном происходит в печени. В мозговой ткани в этот процесс включаются астроциты и микроглия, в которых распад кинуренина имеет свои особенности. Помимо кинуренина, синтезирующегося в мозге, наблюдается его активное поступление из печени, так как он легко проникает через гематоэнцефалический барьер [58, 60].
Другой путь метаболизма триптофана связан с синтезом ряда биологических соединений и, в частности, серотонина. На первой стадии наблюдается гидроксилирование триптофана с образованием 5-гидрокси-L-триптофана с участием биоптериновой монооксигеназы, содержащей атом Fe2+. В качестве кофермента здесь выступает тетрагидробиоптерин. Следует отметить, что именно триптофан-5-монооксигеназа является лимитирующим звеном синтеза серотонина. Особенности метаболизма триптофана отражены на рис. 1.
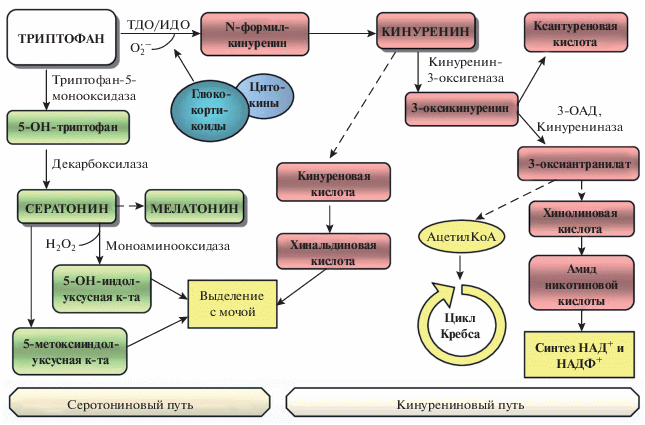
Анализируя возможные причины снижения уровня серотонина при депрессивных состояниях, можно выделить два направления. Во-первых, это снижение источника серотонина – триптофана, сдвиг его метаболизма в сторону синтеза кинуренина и интенсификация распада серотонина. Во-вторых, нарушение состояния рецепторного аппарата и переносчиков медиатора.
Триптофан в свободном состоянии и в составе белков подвергается активной атаке со стороны АФК, что приводит к снижению синтеза серотонина. Индольное кольцо триптофана является первичной мишенью для ОН•, что сопровождается в дальнейшем его гидроксилированием во 2-, 4-, 5-, 6-, и 7-ом положениях с последующим разрывом пятичленной структуры и образованием конечных продуктов нерадикальной природы [7, 21, 117].
В условиях ОС наблюдается активация ТДO, которая использует по ходу ферментативной реакции супероксидный анион-радикал в качестве косубстрата [72, 90]. Активация ТДО приводит к снижению уровня триптофана в плазме и, соответственно, снижению триптофана и серотонина в ткани мозга. Однако существует мнение, что снижение концентрации триптофана в плазме не следует рассматривать, как, своего рода, предрасположенность к развитию депрессии или появлению его определенных симптомов. Снижение уровня этой аминокислоты наблюдается и при других патологических состояниях [81].
При депрессии индукция ТДО приводит к снижению уровня триптофана в плазме и активации его катаболизма по кинурениновому пути, накоплению таких промежуточных продуктов, как кинуренин, хинолиновая, кинуреновая и ксантуреновая кислоты [90, 94].
Образующиеся продукты распада триптофана различным образом влияют на мозговую ткань. Так, кинуренин и хинолиновая кислота обладают анксиогенным и депрессогенным действием, нейротоксическим эффектом. Другие продукты метаболизма триптофана, такие как кинурениновая кислота, проявляют нейрозащитное действие [90].
Maes M. и соавторы считают, что развитие депрессивных симптомов больше связано с активацией ТДО и образованием промежуточных продуктов катаболизма триптофана, чем co снижением его уровня в плазме [94]. В экспериментальных исследованиях на животных при моделировании депрессии наблюдали экспрессию/повышение активности ТДО, концентрации кинуренина, гидроксикинуренина и хинолиновой кислоты в отдельных областях мозга (гиппокамп, гипоталамус, миндалина), с которыми связана регуляция настроения [41, 79]. Некоторые авторы отмечают связь между активацией ТДО или увеличением концентрации кинуренина, гидроксикинуренина и хинолиновой кислоты в сыворотке крови и степенью выраженности снижения настроения у депрессивных больных [55, 87, 99, 106]. Следует отметить, что несмотря на то, что активация ТДО сопряжена, в основном, с проявлением антиоксидантной активности, образующиеся в дальнейшем продукты метаболизма триптофана могут обладать анти- и прооксидантными свойствами [90].
При патологических условиях, в основном, проявляется действие прооксидантов. Так, с 5-оксиантраниловой кислотой, 3-гидроксикинуренином, 3-гидроксиантраниловой и хинолиновой кислотами связывают генерацию свободнорадикальных продуктов, интенсификацию перекисного окисления липидов (ПОЛ) и окислительное повреждение клеточных структур [94, 109, 134]. Некоторые продукты распада триптофана могут являться одной из причин нарушения энергетического метаболизма в митохондриях. В частности, кинуреновая кислота, 3-гидроксиантраниловая и/или 3-оксикинуренин cнижают синтез АТФ и дыхательный контроль в митохондриях. А это, в свою очередь, приводит к повышению образования АФК [26, 128].
Метаболизм триптофана связан с фолиевой кислотой (витамин В9), которая участвует в ферментативных реакциях в качестве кофермента в форме тетрагидрофолиевой кислоты (ТГФК, Н4-ФК). При депрессивном состоянии наблюдается снижение уровня фолиевой кислоты, что отражается на активности биоптериновой монооксигеназы [13, 14, 52]. Известно, что фолиевая кислота состоит из трех структурных единиц: остатка птеридина, парааминобензойной и глутаминовой кислот. Снижение уровня фолиевой кислоты при депрессивном состоянии, возможно, связано с нарушением синтеза птеридина из гуанозинтрифосфата.
Восстановленная фолиевая кислота – тетрагидрофолат функционирует в качестве кофермента ферментов, катализирующих реакции переноса одноуглеродных фрагментов. Реакции переноса метильных групп являются необходимым элементом в образовании и метаболизме некоторых нейротрансмитеров (адреналина, дофамина, норадреналина, серотонина) [107].
Таким образом, тетрагидрофолат выполняет роль своего рода регулятора синтеза нейротрансмитеров. С другой стороны, снижение концентрации фолиата при депрессивных состояниях приводит к повышению уровня гомоцистеина, который в норме образуется в процессе метаболизма метионина в результате гидролиза промежуточного продукта S-аденозилгомоцистеина. Гомоцистеин оказывает токсическое действие на обмен аминокислот и, соответственно, на синтез биогенных аминов. Накопление гомоцистеина сопряжено с усугублением ОС при депрессивных состояниях. Имеется ряд исследований, указывающих на связь между увеличением содержания гомоцистеина, нарушениями когнитивной функции и психическими расстройствами. Показано, что увеличение концентрации гомоцистеина в крови непосредственно коррелирует с когнитивными расстройствами у лиц пожилого возраста [16, 81, 108, 126].
В 2008 году Мaes M. и соавторы выдвинули гипотезу о связи ОС и интенсивного провоспалительного цитокининового ответа при депрессивном состоянии. Провоспалительные цитокины вызывают индукцию ТДО/ИДО, что приводит к активации метаболизма триптофана и накоплению его метаболитов, включая кинуренин, 3-гидроксикинуренин, кинурениновую, ксантуреновую, пиколиновую, хинолиновую кислоты и др. [91, 94]. Эти продукты обладают разным биологическим действием. Так, кинурениновая кислота на физиологическом уровне проявляет антиоксидантные и нейропротекторные свойства. С другой стороны, 3-гидроксикинуренин и хинолиновая кислота оказывают нейротоксический и прооксидантный эффект [54, 59, 98]. Следует отметить, что этот путь триптофана связан с увеличением продукции АФК, оказывающих токсическое действие на ткань мозга при депрессии [97, 142].
Переход метаболизма триптофана при депрессии на кинурениновый путь вследствие активации ТДО приводит к снижению уровня серотонина [57]. У депрессивных больных, совершающих суицидные попытки, наблюдается снижение уровня продукта метаболизма 5-гидроксииндолуксусной кислоты на фоне дефицита серотонина [15]. Это послужило основанием для гипотезы, согласно которой торможение метаболического оборота серотонина в некоторых отделах мозга, в частности, в стволовых структурах и префронтальной коре, является одним из нейробиологических механизмов формирования суицидального поведения. На сегодняшний день серотониновая система наиболее изучена с этих позиций и дефицит серотонинергической медиации является важным механизмом суицидального поведения. У жертв суицида и у лиц с высоким риском суицида, вероятнее всего, имеет место локальное снижение серотониновой медиации, сопровождающееся нарушением функции соответствующих постсинаптических рецепторов.
С кинуренином и его метаболитами связывают развитие определенной симптоматики депрессивных состояний (меланхолические симптомы, тревога и др.) [94, 95, 112]. Кинуреновая кислота, которую рассматривают в качестве эндогенного антагониста NMDA-рецепторов, предположительно снижает эксайтоксическое действие избытка глутамата на нейроны [125, 133]. Однако последующий распад кинуренина приводит к образованию 3-гидроксикинуренина, с которым связывают генерацию свободно-радикальных продуктов, а хинолиновую кислоту рассматривают в качестве агониста рецепторов глутамата. Имеются данные о роли повышения глутамат-рецепторной активности при депрессии. В первую очередь, с хинолиновой кислотой связано увеличение продукции АФК, нарушение функции митохондрий, агонистический эффект по отношению к NMDA-рецепторам. Это приводит к эксайтоксичности нейронов, ингибированию использования глутамата, деструкции постсинаптической системы, дегенерации и гибели нейронов, особенно в области гиппокампа, а также к снижению функционирования центральной холинергической системы [41, 136]. Все перечисленное дает основание рассматривать участие ТДО-опосредованного дисбаланса метаболизма кинуренинового пути в увеличении активности рецепторов глутамата при депрессии [110]. Подтверждением этого положения является снижение концентрации кинурениновой кислоты по сравнению с кинуренином у депрессивных больных [112].
Нарушения функционирования рецепторов серотонина вносят свой вклад в патофизиологию депрессии. Это обусловлено изменениями в 5-НТ (5-гидрокситриптофан, серотонин) опосредованной сигнальной системе на уровне вторичных мессенджеров, изменениями в транспортере серотонина (5-НТТ или SERT – serotonin transporters), 5-НТ2 или 5-HT1A рецепторах [95, 120, 121]. Именно нарушение работы этих рецепторов наиболее часто ассоциировано с депрессивным состоянием. Повреждения 5-НТ рецепторов при депрессии, возможно, связаны с окислительной деструкцией липидов и белков клеточных мембран.
Известно, что синтезированный серотонин накапливается в везикулах и выбрасывается в синаптическую щель при деполяризации 5-НТ-нейронов. Удаление медиаторов осуществляется за счет включения механизма обратного захвата с участием трансмебранного белка-транспортера SERT, который рассматривают как ключевой модулятор серотонинергической сигнализации в ЦНС [27, 62, 77]. SERT регулирует синаптическую активность 5-НТ в ЦНС, действие антидепрессантов в основном направлено на SERT [81]. Показано, что фактор некроза опухоли (ФНО-α) и интерлейкин- β (IL-β) вызывают быструю каталитическую активацию SERT, зависящую от р38 МАРК активации. Провоспалительные цитокины повышают уровень SERT-мРНК и, соответственно, содержание белка [83, 144]. Цитокин-модулированный путь активации SERT является своего рода сигналом к снижению синаптического 5-НТ уровня. При процессах воспаления выявлен дисбаланс в экспрессии рецепторов: увеличение экспрессии 5-НТ2А рецептора в среднем мозге и снижение 5-НТ1А в коре мозга взрослых мышей [77]. Изменение SERT уровня и его активности может быть ассоциировано с областью поведенческих и психологических расстройств [111, 138]. Полиморфизм гена, кодирующего транспортер в условиях стресса может являться фактором риска в развитии депресcии [71]. Ингибирование SERT селективными ингибиторами обратного захвата серотонина (SSRIs) приводит к повышению экстрацеллюларного 5-НТ и увеличению сигнала через постсинаптические 5-НТ рецепторы, также как и через пресинаптические рецепторы [24, 62]. Обнаружено, что стимуляция SERT активности in vitro и in vivo и регуляция генной экспрессии в астроцитах зависит от р38 МАРК активации. Это свидетельствует о роли р38 МАРК пути в цитокин-опосредованной регуляции SERT [103, 144, 145].
Большой вклад в развитие депрессии вносят изменения со стороны катехоламинергической системы. Эти изменения могут быть обусловлены нарушением метаболизма катехоламинов, состоянием функциональной активности их белков-переносчиков, а также состоянием рецепторной системы.
Так же, как и триптофан, фенилаланин и тирозин – предшественники норадреналина, относятся к ароматическим аминокислотам с развитой системой двойных связей и поэтому обладают высокой чувствительностью к окислению. Окисление тирозина приводит к генерации высокореактивного тирозильного радикала – перокси-тирозина, тирозин-нитросоединений. В условиях ОС наблюдается интенсивное образование 3,4-диоксифенилаланина (ДОФА), при котором молекула кислорода является главной детерминантой, участвующей в гидроксилировании тирозиновых остатков. Известно, что ДОФА подвергается дальнейшему окислению с образованием хинона и других циклических соединений [12].
При депрессии наблюдается снижение интенсивности норадренергической передачи. Возможно, это обусловлено нарушением АТФ-зависимого депонирования норадреналина и других катехоламинов, вследствие чего они спонтанно выбрасываются в синаптическую щель. Разрушение биогенных аминов осуществляется за счет реакции окислительного дезаминирования с участием моноаминоксидаз, что приводит к их быстрой инактивации в синаптической щели, не давая возможности связаться со своим рецептором.
Существует моноаминоксидазная теория депрессии, согласно которой длительное снижение уровня серотонина и катехоламина связывают с повышением активности неспецифической моноаминоксидазы (МАО-А) в различных отделах мозга и МАО-Б в тромбоцитах [2, 20].
Известно, что МАО-А регулирует уровень 3-х главных моноаминов: серотонина, норадреналина и дофамина. Процесс окислительного дезаминирования аминов, катализируемого МАО, связан с образованием перекиси водорода. У больных большой формой депрессии, которые никогда не получали антидепрессанты или не принимали их в течение 5 мес., с помощью позитронной эмиссионной томографии было выявлено значительное повышение плотности МАО-А, что является показателем повышения активности фермента в различных отделах мозговой ткани и сопряжено со снижением концентрации моноаминов [106].
Таким образом, состояние хронического ОС уже на начальных стадиях заболевания может являться причиной окислительной деструкции как самих нейротрансмитеров, так и их метаболитов, и дополнительной генерации АФК, вследствие нарушения катаболизма триптофановых рецепторов.
В литературе также прослеживается связь между дисфункцией серотонинергической, норадренергической и дофаминоэргической нейротрансмиссии и иммунологическим статусом организма при депрессивном состоянии. Считают, что ТДО-опосредованный дисбаланс метаболизма кинуренина вносит свой вклад в цитокин-опосредованную депрессию, так как ТДO является высоко индуцибельным по отношению к провоспалительным цитокинам [19, 73, 81, 83, 92].
Провоспалительные цитокины, такие как интерферон-γ, влияют не только на активность триптофан-5-монооксигеназы, но и на уровень тетрагидробиоптерина (ТГБП), который вовлекается в биосинтез нейротрансмиттера серотонина и катехоламинов. В условиях ОС интерферон-γ в макрофагах вызывает генерацию АФК, которые, вследствие реакции окисления, могут снижать уровень ТГБП. При хроническом воспалении, сопровождающемся интенсивной генерацией АФК, также наблюдается окисление ТГБП, приводя к снижению биосинтеза серотонина и катехоламинов. Эти факторы могут иметь непосредственное отношение к изменению нейротрансмиссии у больных депрессией [44, 83, 100].
Некоторые исследователи считают, что развитие депрессии в большей степени связано не с нарушением метаболизма серотонина и катехоламинов, а с ОС и эксайтоксичностью – процессом, ведущим к повреждению и гибели нервных клеток [91].
Исходя из парадигмы истощения серотонина и норадреналина, изучение роли моноаминов у здоровых людей и больных, при отсутствии антидепрессантов, позволило ряду исследователей прийти к заключению, что с позиции одних только биогенных аминов нельзя объяснить ни механизма действия антиоксидантов, ни патофизиологии самой депрессии. Ключевое место в патогенезе заболевания занимает ОС и связанная с ним активация клеточно-опосредованной иммунной системы и развитие процесса воспаления, которые приводят к нарушению других метаболических звеньев, в частности, нарушению метаболизма триптофана и синтеза биогенных аминов, что сопровождается дальнейшим усугублением симптомов депрессии [73, 81, 92].
ПРОЦЕССЫ НЕЙРОГЕНЕЗА И НЕЙРОДЕГЕНЕРАЦИИ ПРИ ДЕПРЕССИВНОМ СОСТОЯНИИ
В настоящее время большое внимание уделяется роли нейротрофинов, с которыми связаны процессы нейрогенеза [50, 53, 65]. Нейротрофины – это семейство низкомолекулярных, положительно заряженных белков, которые оказывают выраженное трофическое действие на основные процессы жизнедеятельности клеток нервной системы, начиная с дифференцировки и роста нейронов до синаптогенеза и синаптической пластичности [49]. Из головного мозга выделены и изучены следующие, близкие по своей химической структуре нейротрофины: фактор роста нервов (NGF), нейротрофический фактор головного мозга (BDNF), нейротрофины 3, 4/5 и 6 (NTs 3, 4/5, 6), глиальный нейротрофический фактор (GDNF). Действие последнего связано в основном с дофаминовыми нейронами. Однако GDNF является потенциальным промотором выживаемости нейронов ЦНС и периферической нервной системы. Он активно влияет на численность клеточной популяции чувствительных и вегетативных ганглиев, клеток Пуркинье, мозжечка, нейронов гиппокампа, в том числе норадренергических, серотонинергических и холинергических нейронов [22].
В многочисленных обзорных отечественных и зарубежных статьях детально освещены моменты, касающиеся структуры нейротрофинов, механизма их функционирования в нейронах [9, 48, 68, 86, 146].
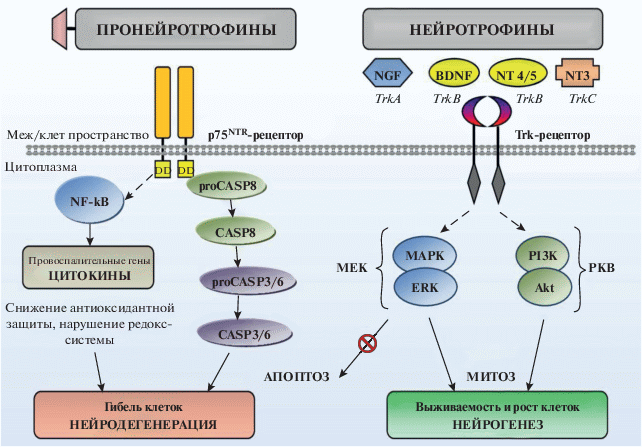
Нейротрофины участвуют в регуляции функциональной активности клеток, выступая в качестве первичных мессенджеров. Сигнальные пути, активированные нейротрофинами, координируют различные клеточные процессы, включающие клеточную выживаемость, рост аксонов и дендритов, синаптогенез и апоптоз [86, 146]. В зрелом мозге основная функциональная роль в контроле нейрогенеза принадлежит BDNF. Нормальное нейрон-глиальное взаимодействие обеспечивается за счет интегральной роли BDNF, участвующего в процессе нейрогенеза. BDNF структурно связан с фактором роста нервов и, соответственно, вовлечен в поддержание жизнедеятельности, пластичности нейронов, их рост и гибель [49, 135]. Каждый из нейротрофинов связывается со своим высокоэффективным Trk рецептором, относящимся к семейству протеинтирозинкиназных рецепторов (Trk A, B, C): NGF – c TrkA, BDNF и NT4/5 – c TrkB, NT3 – c TrkC. NT3 также может взаимодействовать c TrkА и TrkB.
Рецептор р75NTR, с которым могут взаимодействовать нейротрофины и пронейротрофины, не обладает такой избирательной способностью [68, 131]. Он является членом суперсемейства ФНО-α (фактор некроза опухолей), которые работают как рецепторы гибели клеток, запуская процесс апоптоза в различных популяциях нервных клеток [28, 80, 101, 115]. Рецептор р75NTR не обладает ферментативной активностью и состоит из 2-х доменов: экстрацеллюларного, с которым связываются пронейротрофины (нейротрофины), и цитоплазматического домена, который рассматривают как домен смерти (death down – DD). DD, также как Ras-рецептор и ФНО-α, стимулирует апоптоз через активацию каспаз и участвует в активации фактора транскрипции NF-κB (nuclear factor-κB) [30, 64].
Протеинтирозинкиназные рецепторы являются классическими мембран-связанными рецепторами с ферментативной активностью. Они представлены целым семейством (около 20 субъединиц), обладающих общей базисной структурой и состоящих из 2х доменов [30]. Один домен располагается экстрацеллюларно и участвует во взаимодействии с первичными мессенджерами. Второй домен располагается интрацеллюларно и представляет протеинкиназу, которая непосредственно участвует в трансдукции сигнала. Передача сигнала от рецептора на сигнальную систему обычно сопровождается включением разнообразных трансдукторов и усилителей с последующей мобилизацией различных вторичных мессенджеров. Лигандсвязывание первичных мессенджеров индуцирует димеризацию и фосфорилирование тирозиновых остатков цитоплазматического домена Trk с участием АТФ, что приводит к активации тирозинкиназы. Активированная тирозинкиназа обеспечивает передачу сигнала за счет фосфорилирования ряда белков.
Сигнал от первичных мессенджеров передается на систему вторичных мессенджеров, к которым относятся, в частности, АФК. Вторичные мессенджеры через различные эффекторы (факторы транскрипции, ионные каналы, метаболические пути и др.) осуществляют контроль над функциональной активностью клеток. Именно с нейротрофинами связана активация многочисленных сигнальных трансдукционных каскадов, определяющих рост и выживаемость клеток, апоптоз [30, 64]. Показано, что одной из функций нейротрофинов является контроль синаптической пластичности гиппокампа [74, 85, 86].
Лиганд-рецепторное взаимодействие нейтрофинов с Trk-рецепторами приводит к включению двух основных сигнальных путей, которые идентифицированы как наиболее значимые в процессе выживаемости клеток и являются фактически общими для большинства факторов роста. К ним, в частности, относятся семейства эпидермальных и тромбоцитарных факторов роста, семейство фактора роста фибробластов и др.
Первый путь связан с сигнальным каскадом фосфатидилинозитол 3-киназы PI3K (phosphatidyl inositol 3-kinase) – Akt (v-akt murinethymomaviral oncogene homologue), известный как протеинкиназа В (РКВ). Другой путь предусматривает участие митогенактивированной протеинкиназы МАРК – MEK (МАРК/ERK (extracellular signal-regulated kinase) kinase) сигнальный каскад [78, 141].
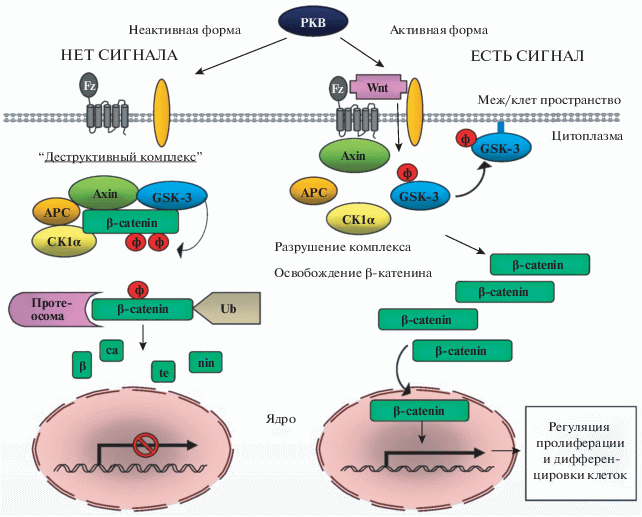
Действие PI3K сигнального каскада сопряжено с генерацией и метаболизмом фосфатидилинозитолов, регуляцией их активности. Вторичный липидный мессенджер PtdIns 3,4,5Р3 (phosphatidylinositol 3,4,5 trisphosphate) активирует фосфоинозитид-зависимую киназу (PDK1) с последующим фосфорилированием и активацией РКВ [31, 43, 47, 63].
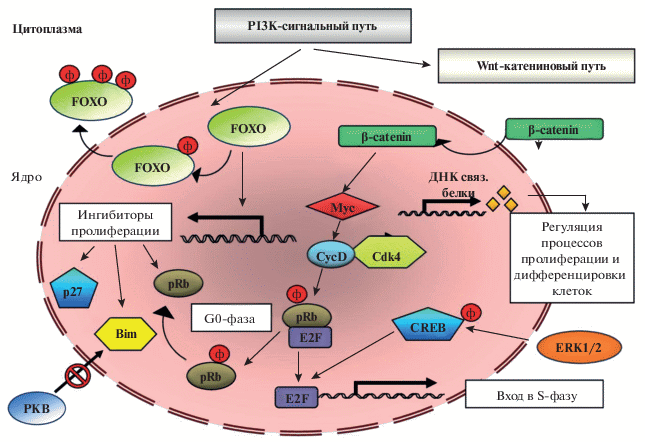
Активированная РКВ участвует во многих сигнальных системах, включая гликогенсинтазу киназу-3β (GSK-3β) и проапоптотический фактор Bad. Механизм действия GSK-3β связан с каноническим Wnt катениновым сигнальным путем. GSK-3β является частью мульти-белкового “деструктивного комплекса” (неактивного), фосфорилируя β-катенин. В результате действия активированной РКВ происходит фосфорилирование сериновых остатков GSK-3β в области N-конца, что является причиной ингибирования GSK-3β, разрушения комплекса и, как следствие, освобождение β-катенина, который начинает накапливаться в цитоплазме [40, 82, 114, 122]. Затем β-катенин перемещается в ядро, участвуя в качестве фактора транскрипции или активируя ряд других факторов транскрипции, контролируя, в частности, транскрипцию генов Wnt [34, 82, 118].
PI3K сигнальная система осуществляет регуляцию клеточного цикла за счет ингибирования транскрипционной активности семейства Fork head box O (FOXO1, FOXO3а, FOXO4). Кроме того, влияние PI3K на FOXO семейство приводит к модулированию апоптоза за счет изменения экспрессии проапоптотического фактора Bim. Удаление FOXO под действием PI3K из ядра приводит к снижению экспрессии Bim. Параллельно PKB инактивирует проапоптотический фактор Bad за счет фосфорилирования в области Ser-136 [36, 45, 46]. Таким образом, PI3K сигнальный каскад непосредственно влияет на выживаемость клеток, стимулируя клеточную пролиферацию и вызывая торможение процессов апоптоза.
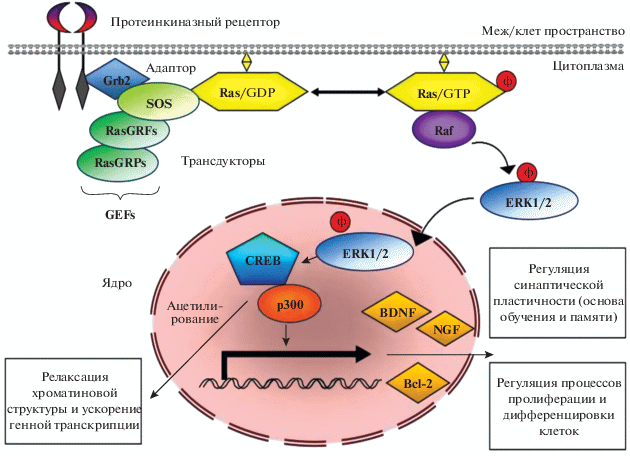
Второй сигнальный путь связан с экстрацеллюларной сигнал-регулирующей киназой ERK1/2 (extracellular-signal-regulated kinases). ERK путь является одним из главных в сигнал-регулируемом МАРК каскаде, выполняя множество важных сигнальных функций, в частности, контроль за клеточной пролиферацией и синаптической пластичностью, участие в процессах обучения и памяти.
Kлассическая рецепторно-опосредованная активация ERK1/2 сигнального пути происходит через цитоплазматические белки-адапторы (adaptor protein – Shc) и низкомолекулярные G-белки (Ras) мембран, которые активируют МАРК киназу киназ – Raf-1. Активность ЕRК1/2 связана с функционированием Ras (ГТФазы) – трансдуктора тирозинкиназных рецепторов. Активированная форма Ras затем взаимодействует с протеинкиназой Raf, которая инициирует фосфорилирование каскада ERK пути и его активацию [30, 31, 39]. Активированная фосфо-ЕRК1/2 покидает плазматическую мембрану и диффундирует из цитоплазмы в ядро, где она фосфорилирует и активирует ряд факторов транскрипции [31, 81].
ЕRK1/2 стимулирует активность или экспрессию антиапоптотических белков, включая BCL2 и фактор транскрипции CREB (cyclic AMP responsive element binding protein). ERK1/2 фосфорилирует CREB в области Ser-133. Фосфорилированный CREB связывается с транскрипционным ко-фактором р300, который является гистон-ацетилтрансферазой, ацетилирующей гистоны. Вследствие ацетилирования гистонов наблюдается релаксация хроматина в области промоторов, доступность ключевых элементов транскрипционного комплекса промотору, обеспечение стабилизации взаимодействий других факторов транскрипции с транскрипционным комплексом [33, 56, 69, 132].
Таким образом, в результате фосфорилирования CREB выступает в роли одного из ключевых регуляторов стимул-зависимой экспрессии генов. CREB фактически участвует в активации нейрональной генной транскрипции, в частности, в экспрессии BDNF. С СREB связывают процессы обучения и памяти [1, 11, 66, 123].
Таким образом, действие нейротрофинов, как регуляторов нейрогенеза, можно рассматривать как с позиций их влияния на процессы пролиферациии и, соответственно, на систему контроля регуляции клеточного цикла, так и с позиции поддержания соответствующего соотношения проапоптотических и антиапоптотических факторов. Для процесса нейрогенеза значимым является не только процесс пролиферации, но и степень отбора образующихся пронейронов, за счет апоптоза. Сигнальные пути нейротрофинов, сигнальная система клеточного цикла и апоптоз связаны между собой, участвуя в регуляции пролиферации клеток и их дальнейшем созревании [3, 4].
PI3K сигнальная система осуществляет регуляцию клеточной пролиферации разными путями, способствуя стабильности циклина D и экспрессии циклин D мРНК. Действие PI3K сигнальной системы может осуществляться через канонический Wnt-катениновый путь. Освобожденный β-катенин перемещается из цитоплазмы в ядро, где активирует фактор транскрипции Myc, увеличивает транскрипцию циклина D, с которым связан запуск всего клеточного цикла. Кроме того, Мус увеличивает экспрессию ингибитор-ДНК-связывающих белков, которые играют ключевую роль в активации пролиферации клеток [33].
Известно, что прохождение клеток через G1 фазу контролируется функциональным состоянием ретинобластомного белка (retinoblastoma protein – pRb), зависящим от степени его фосфорилирования. В фазе G0 pRb белок находится в связанном состоянии с членами семейства фактора транскрипции E2F и тем самым предотвращает E2F-зависимую транскрипцию [10, 42, 116, 140]. PI3K сигнальная система ответственна за ингибирование транскрипционной активности семейства FОХO, которое играет важную роль в регуляции клеточного цикла, участвуя в экспрессии ряда ингибиторов пролиферации, таких как ретинобластомный белок (Rb) и белок р27 – ингибитор циклин-зависимых киназ [32, 116]. Фактически действие PI3K обусловлено снятием состояния супрессии клеточного цикла G0, которая поддерживается за счет различных механизмов, в том числе, FOXO.
МАРкиназная сигнальная система также играет одну из ключевых ролей в клеточной пролиферации. Одной из главных мишений перемещенной в ядро МАРК1/2 является транскрипционный фактор CREB. В частности, CREB участвует в регуляции активности E2F, влияя, тем самым, на переход клеток из фазы G1 в фазу S [56].
Описанные нами сигнальные каскады, участвующие в поддерживании динамического равновесия между процессами пролиферации клеток и апоптозом, носят общий характер для всех ростовых факторов. Специфика их действия определяется функциональными особенностями тканей, зависящих от характера метаболических процессов, скоординированного и взаимосвязанного действия окислительно-восстановительной, иммуновоспалительной и эндокринной систем, осуществляющих регуляцию сигнальных каскадов клетки.
При депрессивном состоянии мы сталкиваемся с нейродегенеративными процессами, обусловленными преобладанием апоптоза и снижением пролиферации. Это может быть связано с состоянием самих рецепторов: их структурной дезорганизацией, вызванной ОС; нарушением лиганд-рецепторного взаимодействия в связи с изменениями синтеза и созревания нейротрофинов, что приводит к нарушению передачи сигналов на ключевые звенья третичного звена и на сами сигнальные системы клеток.
В физиологических условиях процесс регуляции амплитуды и кинетики сигнальной трансдукции, в частности, тирозинкиназных рецепторов связан со своевременным устранением активного лиганд-рецепторного комплекса с клеточной поверхности с последующей его деградацией. Это обеспечивается за счет включения рецепторной “down-regulation”. Это медленный процесс, направленный на устранение рецептора с поверхности клеток за счет эндоцитоза. В дальнейшем эндоцитозная везикула может подвергаться либо лизосомальной деградации, что приводит к протеолизу рецептора, либо за счет процесса деубиквитинизации, когда рецептор возвращается к плазматической мембране и снова участвует в клеточной сигнализации. В регуляции “down-regulation” тирозинкиназных рецепторов участвует адапторный белок Cdl, который связывается с активным рецептором и инициирует его деградацию через убиквитинизацию [30].
В условиях ОС АФК могут в качестве сигнальной молекулы действовать непосредственно через рецепторы, обладающие тирозинкиназной активностью. Показано, что инкубация клеток с Н2О2 приводит к быстрому фосфорилированию рецептора за счет активации тирозинкиназы без взаимодействия с факторами роста, что сопровождается включением сигнальных каскадов с участием, в частности, PKB и МАРкиназной системы. Это может приводить к нарушению передачи сигнала, развитию нейродегенерации и снижению нейрогенеза [51, 61].
При депрессии нарушается метаболизм самих лигандов-нейротрофинов, а именно их синтез, а также экспрессия самих рецепторов. В настоящее время снижение нейрогенеза при депрессии объясняют снижением уровня нейтрофинов и, в первую очередь, BDNF, что приводит к нарушению функции нейронов, уменьшению объема определенных клеточных структур, в частности, нарушению структуры дендридного дерева, уменьшение его длины и ветвления [2, 23, 96].
При депрессивных состояниях было зарегистрировано снижение уровня не только BDNF, но и рецептора TrkB в крови больных и в гиппокампе умерших больных [25, 70, 129 ] . Показано, что выраженная экспрессия BDNF в симпатических нейронах увеличивает плотность преганглионарных синапсов, а истощение BDNF сопряжено со снижением синаптической плотности и количества преганглиозных синаптических аксонов [37].
В ряде клинических исследований прослеживается ассоциативная связь между депрессивными нарушениями и снижением BDNF в сыворотке крови депрессивных больных. Проведение антидепрессантной терапии приводит к повышению BDNF. Анализ постсмертных тканей гиппокампа человека показал, что экспрессия BDNF была снижена у депрессивных суицидных пациентов и повышена у получавших антидепрессантную терапию. Наблюдается снижение сывороточного BDNF у больных не только при остром депрессивном состоянии, но и при предрасположенной чувствительности к депрессии [53, 129, 137 ] .
У больных с резистентной формой депрессии отмечается снижение объема серого вещества, особенно гиппокампа, которое коррелирует со снижением экспрессии BDNF. При эффективном антидепрессантном лечении изменение объема нервной ткани было менее выражено и параллельно отмечалось повышение BDNF [119, 130, 143].
Диагностическую значимость имеют не только абсолютные величины BDNF, но и соотношение между нейротрофинами и пронейротрофинами, зависящее от интенсивности их созревания при участии экстрацелюларной плазминпротеазной системы [38, 86]. Поддержание жизнеспособности нейронов зависит от сохранения оптимальной концентрации нейротрофинов и их соотношения с пронейротрофинами. Повышение уровня пронейротрофинов и образование лигандных комплексов пронейротрофин–р75NTR сопровождаются гибелью нейронов. Следует отметить, что интенсивность гибели нейронов зависит не только от активности экстрацелюларной плазминпротеазной системы, но и от степени ингибирования или экспрессии сортилина, который является частью рецептора р75NTR и специфически связывает продомен NGF [86, 115].
Таким образом, протеолитический процесс расщепления пронейротрофинов при образовании нативных нейротрофинов играет важную регулирующую роль в проявлении их биологической активности.
В последнее время появились исследования, в которых обращается внимание на полиморфизм BDNF при психических расстройствах. Полиморфизм гена BDNF и гена его высоко-аффинного рецептора TrkB2 обусловлен генетическими факторами в патофизиологии депрессивных расстройств. С полиморфизмом генов BDNF и TrkB2 рецептора связан риск развития устойчивости к антидепрессивной терапии. Однако вопросы полиморфизма и мутации гена при депрессии и связанные с ними селективная потеря определенных популяций нейронов и развитие устойчивости к антидепрессивной терапии у ряда больных, по мнению авторов, требуют дальнейшего изучения [49].
Прослеживается связь между депрессией, экспрессией BDNF и серотонинэргической системой мозга [18, 49]. Экстрацеллюларный серотонин может влиять на экспрессию BDNF, который, в свою очередь, регулирует уровень серотонина, влияя на активность транспортера [124]. Наличие обратной связи между серотонином и BDNF обеспечивает поддержание между ними определенного баланса, необходимого для сохранения нейрональной активности гиппокампа и других отделов мозга [62].
BDNF и серотонин способны регулировать синаптическую пластичность, нейрогенез и жизнеспособность нейронов головного мозга у взрослых [18]. Связь между BDNF и серотониновой системой мозга показана как в опытах in vivo, так и in vitrо. На культурах клеток крысиных эмбрионов при введении BDNF было выявлено увеличение количества 5НТ нейронов и рост аксонов. Параллельно отмечалось увеличение мРНК генов, которые кодируют рецепторы 5НТ1А и 5НТ1В и SERT [124]. Показано, что снижение уровня серотонина за счет торможения его синтеза и разрушения серотонергических рецепторов сопряжено со снижением процессов нейрогенеза [35]. У определенной линии мышей, предрасположенных к депрессивноподобному поведению, наблюдаются изменения со стороны серотонинергической системы мозга. Однократное введение BDNF приводило к повышению экспрессии гена триптофангидроксилазы – ключевого фермента синтеза серотонина и генов рецепторов 5НТ1А и 5НТ1В [18, 35, 113]. Более того, у линии мышей, обладающих агрессивностью, выявлены нарушения в серотонинергической системе, проявляющиеся в снижении метаболизма серотонина и нарушении плотности рецепторов. Параллельно у этих животных повышался уровень BDNF в гиппокампе, стриатуме и коре головного мозга [127, 139].
В опытах на взрослых животных показано, что разные нейротрофины оказывают различное влияние на рост и жизнедеятельность серотониновых нейронов. Наиболее выраженное влияние наблюдается со стороны BDNF, в меньшей степени – нейротрофина-3 и отсутствует у фактора роста нервов [104]. Таким образом, между нейротрофическими факторами и серотонинергической системой существует тесная связь, которая носит сложный функциональный характер в разных структурах мозга, зависит от влияния внешней и внутренней среды организма, генетической предрасположенности отдельных их компонентов [105].
В формирование нейробиологических механизмов при депрессивном состоянии вовлечена иммунная система и, в первую очередь, врожденный иммунитет, который не только влияет на метаболизм серотонина, мелатонина и норадреналина, но и на уровень нейротрофинов мозга. А это приводит к уменьшению объема гиппокампа, снижению его регуляторной роли в гипоталамо-гипофизарно-адреналовой системе и развитию депрессии [19, 29].
Maes М. в 2009 г. была сформулирована гипотеза патогенеза депрессии, основанная на связи процессов воспаления и нейродегенерации при депрессии. Авторы считают, что нейродегенерация и снижение нейрогенеза, наблюдаемого при депрессии, вызываются воспалением, их клеточно-опосредованной активацией [100]. Стресс-индуцированное депрессивно-подобное поведение сопровождалось повышением уровня интерлейкин-1β (IL-1β), ФНО-α, интерлейкин-6 (IL-6), NF-κB, активацией циклооксигеназы-2 (COX), перекисным окислением липидов (ПОЛ), снижением уровня BDNF и антиапоптотических факторов – Bcl-2 и BAG1, что приводит к нейродегенерации и снижению нейрогенеза, стимуляции апоптозa. Имеются доказательства, что IL-1β тормозит экспрессию ВDNF в гиппокампе [6, 29, 88]. Введение антидепрессантов приводит к снижению индукторов воспалительной реакции, стимуляции нейрональной дифференцировки, синаптической пластичности, аксональному росту и регенерации через экспрессию нейротрофинов, ослаблению апоптоза через активацию антиапоптотических факторов [49, 76, 100].
Ряд исследователей связывают уменьшение количества BDNF и нарушение функции нейронов с ингибирующим действием глюкокортикоидов [2, 6, 17]. Существует мнение, что клеточно-опосредованная иммунная активация и воспаление, и связанная с ними нейродегенерация при депрессии, опосредована увеличением глюкокортикоидов, способствующих продукции провоспалительных цитокинов, таких как IL-1β и IL-6 [89].
ЗАКЛЮЧЕНИЕ
При депрессивном состоянии нарушение функционирования рецепторного аппарата нейрона и деятельности нейротрансмитеров влияют на сигнальные системы, с которыми связано состояние отдельных компонентов клеточного цикла, регуляция активности ключевых ферментов синтеза соответствующих нейромедиаторов (биогенных аминов), экспрессия нейротрофинов. Все эти процессы развиваются на фоне окислительного стресса, нарушения в иммуновоспалительной и гормональной системах, приводящих к срыву адаптационной системы на уровне первичных и вторичных мессенджеров и третичного звена передачи сигнала в клетке.
Именно эти системы являются теми пусковыми факторами, которые на уровне основных сигнальных систем провоцируют перестройку метаболических процессов в сторону нейродегенерации. Все это свидетельствует о том, что депрессивное состояние следует рассматривать как полипатогенетическое, поскольку причиной развития депрессии может быть и нарушение метаболизма биогенных аминов, отдельных аминокислот, нейротрофинов, вызванного либо провоспалительными цитокинами, либо активными формами кислорода, либо гормонами. А это, в свою очередь, отражается на жизнедеятельности клеток, связанной с нарушением функционирования нейронов, апоптозом и развитием нейропластичной ригидности: атрофии и потери нейронов, нарушение синаптогенеза [5].
Возможно, развитие депрессии, ее латентное проявление на ранних стадиях опосредовано особенностями внутриутробного созревания плода, генетической предраспоженностью к возникновению болезни. Этому также способствуют стрессорные воздействия, неблагоприятные внешние и внутренние условия существования организма.
Список литературы
Бородинова А.А., Зюзина А.Б., Балабан П.М. Роль атипичных протеинкиназ в поддержании долговременной памяти и синаптической пластичности // Биохимия. 2017. № 2. С. 372.
Брусов О.С., Фактор М.И., Катасонов Ф.Б. Структурные и функциональные изменения в головном мозге при эмоциональных расстройствах: основы нейроциркуляторной и нейротрофической гипотезы депрессии // Журн. неврологии и психиатрии. 2012. Т. 7. С. 83.
Гомазков О.А. Нейрогенез как адаптивная функция мозга. Москва: Икар, 2013.
Гомазков О.А Нейрогенез как организующая функция взрослого мозга. Достаточно ли доказательств? // Успехи совр. биол. 2016. Т. 136. № 3. С. 227.
Гомазков О.А. Сигнальные молекулы как регуляторы нейрогенеза взрослого мозга // Нейрохимия. 2013. Т. 30. С. 1.
Григорьян Г.А., Дыгало Н.Н., Гехт А.Б. и др. Молекулярно-клеточные механизмы депрессии. Роль глюкокортикоидов, цитокинов, нейротрансмиттеров и трофических факторов в генезе депрессивных расстройств // Успехи физиологических наук. 2014. Т. 45. № 2. С. 3.
Дубинина E.E. Продукты метаболизма кислорода в функциональной активности клеток (жизнь и смерть, созидание и разрушение). Санкт-Петербург: Медицинская пресса, 2006.
Дубинина Е.Е., Щедрина Л.В., Мазо Г.Э. Основные биохимические аспекты патогенеза депрессии. Часть I //Успехи физиологических наук. 2018. № 1. С. 28.
Дубинина Е.Е., Щедрина Л.В., Мазо Г.Э. и др. Процессы нейрогенеза и нейродегенерации при депрессивных расстройствах // Психическое здоровье. 2016. Т. 122. № 7. С. 29.
Дубинина Е.Е., Щедрина Л.В., Незнанов Н.Г. и др. Окислительный стресс и его влияние на функциональную активность клеток при болезни Альцгеймера // Биомедицинская химия. 2015. № 1. С. 57.
Дубынина Е.В., Долотов О.В. Транскрипционный фактор CREB и процессы формирования памяти // Нейрохимия. 2009. Т. 26. № 3. С. 181.
Ещенко Н.Д. Биохимия психических и нервных болезней. Санкт-Петербург: СПб ГУ, 2004.
Жиляева Т.В., Касимова Л.Н. Нарушения одноуглеродного метаболизма. В кн. Депрессия и риск развития соматических заболеваний / Под ред. Н.Г. Незнанова, Г.Э. Мазо, А.О. Кибитова. Москва: Спец. изд. мед. кн., 2018.
иляева Т.В., Ларионова В.И., Мазо Г.Э. Птерины как потенциальные средства преодоления терапевтической резистентности при шизофрении // Современная терапия психических расстройств. 2018. №1. С. 2
Каплан Г., Сэдок Б. Клиническая психиатрия. Москва: Гэотар медицина, 1998.
Касьянов Е.Д., Мазо Г.Э. Клинический случай гипергомоцистеинемии и рекуррентного депрессивного расстройства // Фармакогенетика и фармакогеномика. 2018. № 2. С. 53.
Касьянов Е.Д., Мазо Г.Э. Функционирование гипоталамо-гипофизарно-надпочечниковой оси при депрессии: актуальное состояние проблемы // Психическое здоровье. 2017. № 8. С. 27.
Попова Н.К., Ильчибаева Т.В., Науменко В.С. Нейротрофические факторы (BDNF, GDNF) и серотонинергическая система мозга // Биохимия. 2017. Т. 82. № 3. С. 449.
Тиганов А.С., Копейко Г.И., Брусов О.С., Клюшник Т.П. Новое в исследовании патогенеза и терапии эндогенной депрессии // Журн. неврологии и психиатрии. 2012. Т. 11. С. 65.
Узбеков М.Г., Мисионжник Э.Ю. Неспецифический синдром эндогенной интоксикации как интегральный компонент патогенеза психических расстройств. Российский психиатрический журнал // 2000. Т. 4. С. 56.
Albani J.R. Motions of tryptophan residues in asialylated human alpha 1-acid glycoprotein // Biochim. Biophys. Acta. 1996. V. 1291. № 3. P. 215.
Allen S.J., Watson J.J., Shoemark D.K. et al. GDNF, NGF and BDNF as therapeutic options for neurodegeneration // Pharmacol. Ther. 2013. V. 138. № 2. P. 155.
Angelucci F., Brene S., Mathe A.A. BDNF in schizophrenia depression and corresponding animal models // Mol. Psychiatry. 2005. V. 10. № 4. P. 345.
Artigas F. Serotonin receptors involved in antidepressant effects // Pharmacol. Ther. 2013. V. 137. P. 119.
Autry A.E., Monteggia L.M. Brain-derived neurotrophic factor and neuropsychiatric disorders // Pharmacol. Res. 2012. V. 64. P. 238.
Baran H., Staniek K., Kepplinger B. et al. Kynurenines and the respiratory parameters on rat heart mitochondria // Life Sci. 2003. V. 72. № 10. P. 1103
Barnes N.M., Sharp T.A. A review of central 5-HT receptors and their junction // Neuropsychopharmacology. 1999. V. 38. P. 1083.
Barrett G.L. The p75 neurotrophin receptor and neuronal apoptosis // Prog. Neurobiol. 2000. V. 61. P. 205.
Barrientos R., Sprunger D.V., Campeau S. et al. Brain-derived neurotrophic factor mRNA downregulation induced by social isolation is blocked by intrahippocampal interleukin-1 receptor antagonist // Neuroscience. 2003. V. 121. P. 847.
Berridge M.J. Cell Signaling Biology // 2012. Introduction. Modul 1. https://doi.org/10.1042/csb0001001
Berridge M.J. Cell Signaling Biology // 2012. Cell signalling pathway. Modul 2. https://doi.org/10.1042/csb0001002
Berridge M.J. Cell Signaling Biology // 2012. Modul 4. Sensors and Effectors.; https://doi.org/10.1042/csb00010
Berridge M.J. Cell Signaling Biology // 2012. Modul 9. Cell Cycle and Proliferation. https://doi.org/10.1042/csb0001009
Beurel E., Grieco S.F., Jope R.S. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases // Pharmacol. Ther. 2015. V. 148. P. 114.
Brezun J.M., Daszuta A. Depletion in serotonin decreases neurogenesis in the dentate gyrus and the subventricular zone of adult rats // Neuroscience. 1999. V. 89. P. 999.
Brunet A., Bonni A., Zigmond M.J. et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor // Cell. 1999. V. 96. № 6. P. 857.
Causing C.G., Gloster A., Aloyz R. et al. Synaptic innervation density is regulated by neuron-derived BDNF // Neuron. 1997. V. 18. № 2. P. 257.
Chao M.V., Botwell M. Neurotrophins: to cleave or not to cleave // Neuron. 2002. V. 33. P. 9.
Chu C.T., Levinthal D.J., Kulich S.M. et al. Oxidative neuronal injury. The dark side of ERK1/2 // Eur. J. Biochem. 2004. V. 271. № 11. P. 2060.
Cole A., Frame S., Cohen P. Further evidence that the tyrosine phosphorylation of glycogen synthase kinase-3 (GSK3) in mammalian cells is an autophosphorylation event // Biochem J. 2004. V. 377. № 1. P. 249.
Connor T.G., Star N.O., Sullivan J.B., Harkin A. Introduction of indolamin 2,3-dioxygenase and kynurenine 3-monooxygenase in rat brain following a systemic inflammatory challenge: a role for IFN-gamma? // Neurosci. Lett. 2008. 441. V. 1. P. 29.
Currais A., Hortobagyi T., Soriano S. The neuronal cell cycle as a mechanism of pathogenesis in Alzheimer’s disease // Aging. 2009. V. 1. № 4. 363.
Czech M.P. PIP2 and PIP3 Complex Roles at the Cell Surface // Cell. 2000. V. 100. P. 603.
Dantzer R., o’Connor J.C., Lawson M.A., Kelley K.W. Inflammation-associated depression: From serotonin to kynurenine // Psychoneuroendocrinology. 2011. V. 36. P. 426.
Datta S.R. Akt phosphorylation of BAD couples survival to cell-mashinery // Cell. 1997. V. 91. P. 231.
Del Peso L., Gonzalez-Garcia M., Page C. et al. Interleukin-3-induced phosphorylation of BAD through the protein kinase // Science. 1997. V. 278. P. 687.
Di Paolo G., De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Review Article // Nature. 2006. V. 443. P. 651.
Duman R.S. Role of neurotrophic factors in the etiology and treatment of mood disorders // Neuromolecular Medicine. 2004. V. 5 P. 11.
Duman R.S., Monteggia L.M. A neurotrophic model for stress related mood disorders // Biol. Psychiatry. 2006. V. 59. P. 1116.
Dunham S., Deakin J.F., Miyajima F. et al. Expression of hippocampal brain-derived neurotrophic factor and its receptors in Stanley consortium brains // J. Psychiatric Research. 2009. V. 43. P. 1175.
Esposito F., Chirico G., Montesano Gesualdi N. et al. Protein kinase B activation by reactive oxygen species is independent of tyrosine kinase receptor phosphorylation and requires Src activity // J. Biol. Chem. 2003. V. 278. № 23. P. 20828.
Fava M., Mischoulon D. Folate in depression: efficacy, safety, differences in formulations, and clinical issues // J. Clin. Psychiatry. 2009. V. 70. № 5. P. 12.
Fernandes B.S., Gama C.S., Cereser K.M. et al. Brain-derived neurotrophic factor as a state-marker of mood episodes in bipolar disorders: a systematic review and meta-regression analysis // J. Psychiatric Research. 2011. V. 45. P. 995.
Filho A.J.M., Lima C.N.C., Vasconcelos S.N.N. et al. IDO chronic immune activation and tryptophan metabolic pathway: A potential pathophysiological link between depression and obesity // Prog. Neuropsychopharmacol. Biol. Psychiatry. 2018. V. 80. P. 234.
Gabbay V., Klein R.G., Katz Y. et al. The possible role of the kynurenine pathway in adolescent depression with melancholic features // J. Child. Psychol. Psychiatry. 2010. V. 51. № 8. P. 935.
Goodman R.H., Smolik S. CBP/p300 in cell growth, transformation, and development // Genes Dev. 2000. V. 14. №13. P. 1553.
Graeff R.G., Guimaraes P.S., De Andrade T.G., Deakin J.F. Role of SHT in stress, anxiety and depression // Pharm. Biochem. Behav. 1998. V. 54. P. 129.
Grant R.S., Naif Y., Espinola M. et al. IDO after INF-gamma activated astroglia: a role in improvin cell viability during oxidative stress // Redox Rep. 2000. V. 5. P. 101.
Guillemin G.J., Kerr S.J., Smythe G.A. et al. Kynurenine pathway metabolism in human astrocytes: a paradox for neuronal protection // Neurochem. 2001. V. 78. № 4. P. 842.
Guillemin G.J., Smythe G., Tokikawa O. et al. Expression of indoleamine 2, 3-dioxygerase and production of quinolinic acid by human microglia, astrocytes, and neurons // Glia. 2005. V. 49. P. 15.
Guyton K.Z., Liu Y., Gorospe M. et al. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury // J. Biol. Chem. 1996. V. 271. № 8. P. 4138.
Haase J., Brown E. Integrating the monoamine, neurotrophin and cytokine hypothesis of depression – A central role for the serotonin transporter? // Pharmacology and Therapeutics. 2015. V. 147. P. 1.
Hawkins P.T., Anderson K.E., Davidson K., Stephens L.R. Signalling through Class I PI3Ks in mammalia n cells // Biochem. Soc. Trans. 2006. V. 34. P. 647.
Heldin C.H. Protein tyrosine kinase receptor signaling overview / Ed. Bradshaw R.A., Dennis E.A. San Diego: Academic Press, 2003. P. 391.
Henn F., Vollmayer B., Sartorius A. Mechanism of depression: the role of neurogenesis // Drug Discov. Today Dis. Mech. 2004. V. 1. P. 407.
Herold S., Jagasia R., Merz K. et al. CREB signalling regulates early survival, neuronal gene expression and morphological development in adult subventricular zone neurogenesis // Mol. Cell Neurosci. 2011. V. 46. № 1. P. 79.
Hindmarch I. Beyong the monoamine hypothesis: mechanisms, molecules and methods // Eur. Psychiatry. 2002. V. 17. № 3. P. 294.
Huang E.J., Peichardt L.F. Neurotrophins: roles in neuronal development and function // Ann. Rev. Neurosci. 2001. V. 24. P. 677.
Ianari A., Gallo R., Palma M. et al. Specific role for p300/CREB-binding protein-associated factor activity in E2F1 stabilization in response to DNA damage // J. Biol. Chem. 2004. V. 279. № 29. P. 30830.
Karege F., Vaudan G., Schwald M. et al. Neurotrophin levels in postmortem brains of suicide and the effects of antemortem diagnosis and psichotropic drugs // Brain Res. Mol. Brain Res. 2005. V.136. P. 29.
Karg K., Burmeister M., Shedden K. et al. The serotonin transporter promoter variant (5-HTTLPR), stress and depression meta-analysis revisted: Evidence of genetic moderation // Arch. Gen. Psychiatry. 2011. V. 68. P. 444.
Kobayashi K., Hayashi K., Sono M. Effects of tryptophan and pH on the kinetics of superoxide radical binding to indoleamine2,3-dioxygenase studied by pulse radiolysis // J. Biol. Chem. 1989. V. 264. № 26. P. 15280.
Kobrosly R., van Wijngaarden E. Associations between immunologic, inflammatory, and oxidative stress markers with severity of depressive symptoms: An analysis of the 2005–2006 National Health and Nutrition Examination Survey // NeuroToxicology. 2010. V. 31. P. 126.
Korte M., Carroll P., Wolf E. et al. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor // Proc. Natl Acad. Sci. 1995. V. 92. P. 8856.
Krishnan V., Nestler E.J. The molecular neurobiology of depression // Nature. 2008. V. 455. P. 894.
Kubera M., Obuchowicz E., Goehler L. et al. In animal models, psychosocial stress-induced (neuro)inflammation, apoptosis and reduced neurogenesis are associated to the onset of depression. Prog. Neuropsychopharmacol // Biol. Psychiatry. 2011. V. 35. P. 744.
Kulikov A.V., Naumenko V.S., Tsybko A.S. et al. The role of glicoprotein gp 130 in serotonin mediator system in mouse brain // Mol. Biol. 2010. V. 44. № 4. P. 801.
Kuruvilla R.,Ye H., Ginty D.D. Spatially and functionally district roles of the PI3-K effector pathway during NGF signaling in sympathetic neurons // Neuron. 2000. V. 27. P. 499.
Laugeray A., Launay J.M., Callebert J. et al. Peripheral and cerebral metabolic abnormalities of the tryptophan-kynurenine pathway in a murine model of major depression // Behav Brain Res. 2010. V. 210. № 1. P. 84.
Lee F.S., Kim A.N., Khursigara G. et al. The uniqueness of being a neurotrophin receptor // Curr. Opin. Neurobiol. 2001. V. 11. P. 281.
Leonard B., Maes M. Mechanistic explanations how cell-mediated immune activation, inflammation and oxidative and nitrosative stress pathways and their sequels and concomitans play a role in the pathophysiology of unipolar depression // Neurosci. Biobehav. Rev. 2012. V. 36. P. 764.
Lie D.C., Colamarino S.A., Song H.J. et al. Wnt signaling regulates adult hippocampal neurogenesis // Nature. 2005. V. 437. № 7063. P. 1370.
Loftis J.M., Huckans M., Morasco B.J. Neuroimmune mechanisms of cytokine-induced depression: current theories and novel treatment strategies // Neurobiology of Disease. 2010. V. 37. P. 519.
Logan C.Y., Nusse R. The Wnt signaling pathway in development and disease // An. Review of Cell and Developmental Biology. 2004. V. 20. P. 781
Lu B. BDNF and activity-dependent synaptic modulation // Learn Mem. 2003. V. 10. № 2. P. 86.
Lu B., Pang P.T., Woo N.H. The yin and yang of neurotrophin action // Neurosci. 2005. V. 6. P. 603.
Mackay G.M., Forrest C.M., Christofides J. et al. Kynurenine metabolites and inflammation markers in depressed patients treated with fluoxetine or counselling // Clin. Exp. Pharmacol. Physiol. 2009. V. 36. № 4. P. 425.
Maes M. Depression is an inflammatory disease, but cell-mediated immune activation is the key component of depression // Prog. Neuropsychopharmacol. Biol. Psychiatry. 2011. V. 35. № 3. P. 664.
Maes M. Evidence for an immune response in major depression: a review and hypothesis // Progress in Neuro-Psychopharmacology & Biological Psychiatry. 1995. V. 19. P. 11.
Maes M. Mechanistic explanations how cell-mediated immune activation, inflammation and oxidative and nitrosative stress pathways and their sequels and concomitans play a role in the pathophysiology of unipolar depression // Neuroscience and Biobehavioral Reviews. 2012. V. 36. P. 764.
Maes M. The cytokine hypothesis of depression: inflammation, oxidative & nitrosative stress and leaky gut as new targets for adjunctive treatments in depression // Neuroendocrinol. Lett. 2008. V. 29. № 3. P. 287.
Maes M., Galecki P., Chang Y.S., Berk M. A review on the oxidative and nitrosative stress [O&NS] pathways in major depression and their possible contribution to the neurodegenerative processes in that illness // Prog. Neuropsychopharmacol. Biol. Psychiatry. 2011. V. 35. № 3. P. 676.
Maes M., Galecki P., Verkerk R., Rief W. Somatization, but not depression, is characterized by disorders in the tryptophan catabolite (TRYCAT) pathway, indicating increased indoleamine 2,3-dioxygenase and lowered kynurenine aminotransferase activity // Neuro. Endocrinol. Lett. 2011. V. 32. № 3. P. 264.
Maes M., Leonard B. E., Myint A. M., Kubera M., Verkerk R. The new 5-HT hypothesis of depression: cell-mediated immune activation induces indoleamine 2,3-dioxygenase, wich leads to lower plasma tryptophan and an increased synthesis of detrimental tryptophan catabolites (TRYCATs), both of which contribute to the onset of depression // Prog. Neuro. Psychopharmacol. Biol. Psychiatry. 2011. V. 35. № 3. P. 702.
Maes M., Meltzer H.V. The serotonin hypothesis of major depression. Selected chapters on mood disorders / Eds. Bloom F., Kupfer D. Psychopharmacology. The fourth generation of progress. USA: Raven. Press, 1995.
Maes M., Michaylova L., Kubera M. et al. Coenzyme Q10 deficiency in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) is related to fatique, autonomic and neurocognitive symptoms and is another risk factor explaning the early mortality in (ME/CFS) due to cardiovascular disorder // Neuro. Endocrinol. Lett. 2009. V. 30. № 4. P. 470.
Maes M., Michaylova L., Leunis J.C. Increased serum IgM antibodies directed against phosphatidyl inositol in chronic fatigue syndrome (CFS) and major depression: evidence that an IgM-mediated immune response against Pi is one factor underpinning the comorbidity between both CFS and depression // Neuro. Endocrinol. Lett. 2007. V. 28. № 6. P. 861.
Maes M., Mihaylova I., Ruyter M.D. et al. The immune effects of TRYCATs (tryptophan catabolites along the IDO pathway): relevance for depression – and other conditions characterized by tryptophan depletion induced by inflammation // Neuro. Endocrinol. Lett. 2007. V. 28. № 6. P. 826.
Maes M., Verkerk R., Bonaccorso S. et al. Depressive and anxiety symptoms in the early puerperium are related to increased degradation of tryptophan into kynurenine, a phenomenon which is related to immune activation // Life Sciences. 2002. V. 71. № 16. P. 1837.
Maes M., Yirmyia R., Noraberg J. et al. The inflammatory and neurodegenerative [I&ND] hypothesis of depression: leads for future research and new drug developments in depression // Metab. Brain. Dis. 2009. V. 24. P. 27.
Majdan M., Miller F. Neuronal life and death decisions: functional antagonism between the Trk and p75 neurotrophin receptors // Int. J. Dev. Neurosci. 1999. V. 17. P. 153.
Malhi G.S., Parker G.B., Greenwood J. Structural and functional models of depression: from sub-types to substrates // Acta Psychiatr. Scan. 2005. V. 111. P. 94.
Malynn S., Campos-Torres A., Moynagh P., Haase J. The pro-inflammatory cytokine TNF-alfa regulates the activity and expression of the serotonin transporter (SERT) in astrocytes // Neurochem. Res. 2013. V. 38. P. 694.
Mamounas L.A., Blue M.E., Siuciak J.A., Altar C.A. Brain-derived neurotrophic factor promotes the survival and sprouting of serotonergic axons in rat brain // J. Neurosci. 1995. V. 15. P. 7929.
Mattson M.P., Maudsley S., Martin B. BDNF and 5-HT: a dynamic duo in age-related neuronal plasticity and neurodegenerative disorders // Trends Neurosci. 2004. V. 10. P. 589.
Meier T.B., Drevets W.C., Wurfel B.E. et al. Relationship between neurotoxic kynurenine metabolites and reductions in right medial prefrontal cortical thickness in major depressive disorder // Brain Behav. Immun. 2016. V. 53. P. 39.
Miller A.L. The methylation, neurotransmitter, and antioxidant connections between folate and depression // Altern. Med. Rev. 2008. V. 13. № 3. P. 216.
Moustafa A.A., Doaa H.H., Abeer M.E. et al. Homocysteine levels in schizophrenia and affective disorders-focus on cognition // Front. Behav. Neurosci. 2014. V. 8. P 343.
Muller N., Schwarz M. J. COX-2 inhibition in schizophrenia and major depression// Curr. Pharm. Des. 2008. Vol. 14. P. 1452.
Muller N., Schwarz M.J. The immune-mediated alteration of serotonin and glutamate: towards an integrated view of depression // Mol. Psychiatry. 2007. V. 12. P. 988.
Murphy D.L., Fox M.A., Timpano K.R. et al. How the serotonin story is being rewritten by new gene-based discoveries principally related to SLC6A4, the serotonin transporter gene. Which functions to influence all cellilar serotonin systems // Neuropharmacology. 2008. V. 55. P. 932.
Myint A.M., Kim Y.K., Verkerk R. et al. Kynurenine pathway in major depression: evidence of impaired neuroprotection // J. Affect. Disord. 2007. V. 98. № 1–2. P. 143.
Naumenko V.S., Kondaurova E.M., Popova N.K. Effect of brain-derived neurotrophic factor on behavior and key members of the brain serotonin system in genetically predisposed to behavioral disorders mouse strainsn // Neuroscience. 2012. V. 214. P. 59.
Nelson W.J., Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways // Science. 2004. V. 303. № 5663. P. 1483.
Nykjser A., Lee R., Teng K. et al. Sortilin is essential for proNGF-induced neuronal cell death // Nature. 2004. V. 427. P. 843.
Ogawa O., Lee H.G., Zhu X. et al. Increased p27, an essential component of cell cycle control, in Alzheimer’s disease // Aging Cell. 2003. V. 2. № 2. P. 105.
Okajima T., Kawata Y., Hamaguchi K. Chemical modification of tryptophan residues and stability changes in proteins // Biochemistry. 1990. V. 29. № 39. P. 9168.
Ozlem K.O., Doberauer K.C.,Vadodaria K.C. et al. Prospero-related homeobox 1 gene (Prox1) is regulated by canonical Wnt signaling and has stage-specific role in adult hippocampal neurogenesis // Proc. Natl. Acad. Sci. U S A. 2011. V. 108. № 14. P. 5807.
Piccinni A. Del Debbio A., Medda P. et al. Plasma Brain-Derived Neurotrophic Factor in treatment-resistant depressed patients receiving electroconvulsive therapy // Eur. Neuropsychopharmacol. 2009. V. 19. P. 349.
Plein H., Berk M. Changes in the platelet intracellular calcium response to serotonin in patients with major depression treated with electroconvulsive therapy: state or trait marker status // Int. Clin. Psychopharmacol. 2000. V. 15 № 2. P. 93.
Plein H., Berk M., Eppel S., Butkow N. Augmented platelet calcium uptake in response to serotonin stimulation in patients with major depression measured using Mn2+ influx and 45Ca2+ uptake // Life Sci. 2000. V. 66. № 5. P. 423.
Polter A.M., Yang S., Jope R.S. Functional significance of glycogen synthase kinase-3 regulation by serotonin // Cellular Signalling. 2012. V. 24. P. 265.
Ruan L., Wui-Man Lau B., Wang J. et al. Neurogenesis in neurological and psychiatric diseases and brain injury: From bench to bedside // Progress in Neurobiology. 2014. V. 115. P. 116.
Rumajogee P., Madeira A., Verge D. et al. Up-regulation of the neuronal serotoninergic phenotype in vitro: BDNF and cAMP share Trk B-dependent mechanisms // J. Neurochem. V. 83. P. 1525.
Sapko M.T., Guidetti P., Yu P. et al. Endogenous kynurenate controls the vulnerability of striatal neurons to quinolinate: implications for Huntington’s disease // Exp. Neurol. 2006. V. 197. № 1. P. 31.
Schafer J.H., Glass T.A., Bolla K.I. et al. Homocysteine and cognitive function in a population–based study of older adults // J. Am. Geriatr. Soc. 2005. V. 53. P. 381
Schiller L., Donix M., Jähkel M., Oehler J. Serotonin 1A and 2A receptor densities, neurochemical and behavioural characteristics in two closely related mice strains after long-term isolation // Prog. Neuropsychopharmacol. Biol. Psychiatry. 2006. V. 30. P. 492.
Schuck P.F., Tonin A., da Costa Ferreira G. et al. Kynurenines impair energy metabolism in rat
Sen S., Duman R., Sanacora G. Serum brain-derived neurotrophic factor, depression, and antidepressant medications: Meta-analyses and implications // Biol. Psychiatry. 2008. V. 64. P. 527.
Shah P.J., Glabus M.F., Goodwin G.M., Ebmeier K.P. Chronic, treatment-resistant depression and right fronto-striatal atrophy // Br. J. Psychiatry. 2002. V. 180. P. 434.
Sofroniew M.V., Howe C.L., Mobley W.C. Nerve growth factor signaling, neuroprotection, and neural repair // Ann. Rev. Neurosci. 2001. V. 24. P. 1217.
Stauffer D., Chang B., Huang J. et al. p300/CREB-binding protein interacts with ATR and is required for the DNA replication checkpoint // J. Biol. Chem. 2007. V. 282. № 13. P. 9678.
Swartz K.J., During M.J., Freese A., Beal M.F. Cerebral synthesis and release of kynurenic acid: an endogenous antagonist of excitatory amino acid receptors // J. Neurosci. 1990. V. 10. № 9. P. 2965.
Talowska M., Orzechowska A., Szemraj J., Kuan-Pin S., Maes M., Galecki P. Manganese superoxide dismutase gene expression and cognitive functions in recurrent depressive disorder // Neuropsychology. 2014. V. 70. № 1. P. 23.
Tapia-Aruncibia L., Rage F., Givalous L., Aruncibia S. Physiology of BDNF: focus on hypothalamic function // Front. Neuroendocrinol. 2004. V. 25. P. 77.
Tavares R.G., Tasca C.I., Santos C.E. et al. Quilolinic acid stimulates synaptosomal glutamate release and inhibits glutamate uptake into astrocytes // Neurochem. Int. 2002. V. 40. P. 621.
Terracciano A., Lobina M., Piras M.G., Mulas A., Cannas A. et al. Neuroticism, depressive symptoms, and serum BDNF // Psychosom. Med. 2011. V. 73. № 8. P. 638.
Torres G.E., Gainetdinov R.R., Caron M.G. Plasma membrane monoamine transporters: Structure, regulation and function // Nat. Rev. Neurosci. 2003. V. 4. P. 13.
Undine E., Lang L., Günther K. et al. Higher BDNF concentrations in the hippocampus and cortex of an aggressive mouse strain // Behav. Brain Res. 2009. V. 197. P. 246.
Vincent I., Pae C.I., Hallows J.L. The cell cycle and human neurodegenerative disease // Prog. Cell Cycle. 2003. V. 5. P. 31.
Watson F.L. Neurotrophins use the Erk5 pathway to mediate a retrograde survival response // Nature Neurosci. 2001. V. 4. P. 981.
Wichers M.C., Maes M. The role of indoleamine 2,3-dioxygenase (IDO) in the pathophysiology of interferon-alpha-induced depression // Psychiatry Neurosci. 2004. V. 29. № 1. P. 11.
Xingbing Huang, Xiong Huang et al. Association of serum BDNF levels with psychotic symptomin chronic patients with treatment-resistant depression in a Chinese Han population // Psychiatry Res. 2017. V. 257. P. 279.
Zhu C.B., Blakely R.D., Hewlett W.A. The proinflammatory cytokines interleukin-1beta and tumor necrosis factor-alpha activate serotonin transportes // Neuropsychopharmacology. 2006. V. 31. № 10. P. 2121.
Zhu C.B., Lindler K.M., Owens A.W. et al. Interleukin-1 receptor activation by systemic lipopolysaccharide induces behavioral despair linked to MAPK regulation of CNS serotonin transporters // Neuropsychopharmacology. 2010. V. 35. P. 2510.
Zweifel L.S., Kuruvilla R., Ginty D.D. Functions and mechanisms of retrograde neurotrophin signaling // Neurosci. 2005. V. 6. P. 615.
Дополнительные материалы отсутствуют.
Инструменты
Успехи физиологических наук