

Успехи физиологических наук, 2021, T. 52, № 3, стр. 24-40
Пуринергическая регуляция нейровоспаления при черепно-мозговой травме
Н. Б. Серебряная 1, *, Е. Е. Фомичева 1, П. П. Якуцени 2
1 ФГБНУ Институт экспериментальной медицины Минобрнауки РФ
Санкт-Петербург, Россия
2 ФГАОУ Санкт-Петербургский политехнический университет Петра Великого Минобрнауки РФ
Санкт-Петербург, Россия
* E-mail: nbvma@mail.ru
Поступила в редакцию 11.01.2021
После доработки 13.01.2021
Принята к публикации 20.01.2021
Аннотация
Пуринергическая система определяется как универсальная регуляторная система, позволяющая калибровать и синхронизировать индивидуальные клеточные ответы, чтобы они соответствовали интересам целого организма. Важнейшие пуриновые медиаторы АТФ и аденозин обеспечивают позитивную и негативную модуляцию сигналов в центральной и периферической нервной системе, иммунной системе и других системах организма. Синтез и высвобождение этих медиаторов, скорость их ферментативного метаболизма, экспрессия пуринергических рецепторов существенно влияют на течение нормальных физиологических и патологических процессов, в том числе и посттравматических. В обзоре рассмотрены основные компонент пуринергической системы, влияющие на развитие нейровоспаления после черепно-мозговой травмы и возможности корректирующих воздействий.
ВВЕДЕНИЕ
Черепно-мозговая травма (ЧМТ) − разнообразная группа травматических повреждений, которая представляет серьезную социальную проблему, актуальную для людей разного возраста и социального статуса [133]. ЧМТ возникает в результате физического воздействия, которое нередко приводит к потере сознания и далее к развитию осложнений и заболеваний, различных по тяжести, патогенезу и клиническому исходу.
Прямые первичные повреждения нервной ткани могут вызвать фокальное внутричерепное кровоизлияние, эпидуральные и субдуральные гематомы, субарахноидальные и внутрижелудочковые кровоизлияния, очаговые ушибы и отек мозга (рис. 1) [96]. При гистологических исследованиях отмечают гибель клеток в мозговых оболочках и паренхиме мозга, в венозных синусах, растяжение и разрыв аксонов, диффузное повреждение подкорковых нейронов и разрывы на стыках между белым и серым веществом [130]. В случае легкого повреждения ЧМТ может завершаться благоприятно, а при тяжелом течении может стать как причиной смерти, в том числе от внутрибольничных инфекций [89], так и последующей хронической патологии, включая хроническую травматическую энцефалопатию, эпилепсию, болезнь Альцгеймера, болезнь Паркинсона и ряда других заболеваний [44, 106]. Считается, что связанный с ЧМТ неврологический дефицит является долгосрочным почти у 50% людей, перенесших даже легкую ЧМТ [122]. Первичное повреждение определяет развитие клеточных и молекулярных каскадов, ведущих к развитию вторичного воспаления и дополнительной гибели клеток.
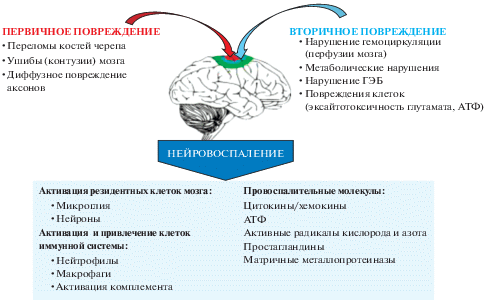
Вторичное воспаление в случае его гиперактивного, несбалансированного течения препятствует восстановлению поврежденной ткани, поддерживает нарушение физиологических функций и, в конечном счете, определяет степень нейродегенерации, ведущей к развитию неврологических заболеваний и поведенческих нарушений [1, 170]. Развитие и поддержание вторичного воспаления связывают с такими патогенными факторами, как эксайтотоксичность, вызванная избытком нейромедиатора глутамата и АТФ [59], образование свободных радикалов, вызывающее повреждение белков и фосфолипидных мембран нервных клеток [8], и воспалительный ответ, связанный как с местной, так и системной иммунной активации [153]. Считается, что первичная гибель клеток после ЧМТ неизбежна, но вторичное повреждение поддается терапевтическому вмешательству путем воздействия на перечисленные процессы. Однако достаточно эффективные терапевтические подходы, способные минимизировать последствия полученных повреждений, пока не реализованы [54]. Сложность лечения ЧМТ связана с неоднородностью задействованных патогенетических процессов, их комбинаций и наложениями.
РАЗВИТИЕ ТКАНЕВОГО ВОСПАЛЕНИЯ ПРИ ЧМТ
Нейровоспаление является ключевой реакцией ЦНС на травму [142]. Резидентные в ЦНС и периферические иммунные клетки (микроглия, тучные клетки, астроциты, моноциты и макрофаги, нейтрофилы, Т-лимфоциты) быстро реагируют на ЧМТ и участвуют в процессе развивающегося воспаления и последующего восстановления [49]. Ранним событием при нейротравме является активация тучных клеток менингеальной оболочки, которая приводит к нарушению гематоэнцефалического барьера (ГЭБ) [14, 103]. Признаки дисфункции ГЭБ определялась уже через 3 минуты после сотрясения мозга [55]. В местах сосудистого повреждения активируются система комплемента, тромбоциты и нейтрофилы, которые затем проникают к мозговым оболочкам и в периваскулярные пространства. При этом формируются микротромбы, усиливаются гемоциркуляторные нарушения [43, 97], что ведет к гипопефузии и отеку головного мозга [66, 138].
Поврежденные клетки, в свою очередь, высвобождают различные молекулы, обладающие характеристиками сигналов опасности – аларминов или DAMP – среди которых белки теплового шока, белок 1 группы высокой подвижности (HMGB1), белки S-100, аденозинтрифосфат (АТФ), микрокристаллы мочевой кислоты, ДНК или РНК, интерлейкин (IL)1α и другие молекулы [27, 144]. При распознавании DAMP активируются резидентные миелоидные клетки врожденного иммунитета ЦНС – микроглия. Распознавание через Toll-подобные рецепторы (TLR) и нуклеотид-связывающие рецепторы, подобные домену олигомеризации (NLR) и запускает стерильную иммунную реакцию, предназначенную для восстановления гомеостаза тканей [3, 111]. На клетках микроглии присутствуют различные TLR (TLR1, TLR2, TLR3, TLR4, TLR5, TLR7, TLR8, TLR9) и корецептор CD14 [29], причем их экспрессия при воспалении увеличивается [137]. Наиболее высокая экспрессия TLR наблюдается в областях мозга, прилегающих к мозговым оболочкам и околожелудочковым органам [2, 39]. Интересно, что в условиях церебральной ишемии активация микроглии через TLR3 (лигандами которого определены двуцепочная РНК и полиинозин-полицитидиловая кислота) способствует выживанию нейронов [98], а стимуляция микроглии агонистом TLR4, например, эндотоксином, блокирует пролиферацию предшественников олигодендроцитов и стимулирует их апоптоз из-за высвобождения большого количества TNFα, препятствуя процессу ремиелинизации [114].
Регулятором нейровоспалительного ответа, возникающего после ЧМТ, также является микроглия [57]. К факторам микроглиального происхождения относят провоспалительные цитокины, включая TNF-α, IL-1α, IL-1β, IL-6, IFNγ и хемокины (CCL2/MCP-1, хемоаттрактантный белок-1 моноцитов и CCL3/MIP1-α, воспалительный белок макрофагов 1-альфа), действующие как хемоаттрактанты для макрофагов и моноцитов [111, 149]. Опасность избыточного воспалительного ответа в ЦНС связана с тем, что повышенные концентрации TNFα, IL-6 и оксида азота (NO) вызывают повреждение нейронов [3]. Однако активированная микроглия высвобождает также нейротрофические факторы, включая фактор роста нервов (NGF), нейротрофины (NT)-4/5, трансформирующий фактор роста β1 (TGF-β1), нейротрофический фактор глиального происхождения (GDNF), фактор роста фибробластов 2 (FGF2), IL-10 и IL-3, которые также поддерживают выживание нейронов [131]. В итоге, суммарную провоспалительную секреторную функцию микроглии определяет тяжесть повреждения и наличие/выраженность различных сигналов (хемокинов, АТФ и т.д.).
ПУРИНЕРГИЧЕСКАЯ СИСТЕМА
В начале 1960 года Джеффри Бернсток начал исследования ДНК как нейромедиаторной молекулы и к 1972 г. он создал концепцию пуринергической иннервации [32], показав, что АТФ действует как медиатор. В центральной нервной системе АТФ, высвобождаемый из синаптических окончаний, вызывает быстрые возбуждающие постсинаптические токи, а в периферической нервной системе АТФ был определен как участник передачи сигналов в сенсорных и вегетативных ганглиях. Метаболизация АТФ приводит к формированию аденозина, который оказывает ингибирующую тоническую модуляцию при синаптической передаче сигналов в ЦНС [36].
Теория Бернстока не была сразу принята научным сообществом, которое было настроено воспринимать АТФ только как переносчик химической энергии между макромолекулами, что показал Фриц Липман в 1941 г. [115]. Однако с годами труды Джеффри Бернсток и его многочисленных коллег и учеников по всему миру доказали, что АТФ и его метаболит аденозин являются важными внеклеточными сигнальными молекулами, задействованными в регуляции множества процессов. Первоначально были определены краткосрочные эффекты пуринергической активации, такие как нейротрансмиссия, нейромодуляция, секреция, хемоаттрактация и индукция острого воспаления. Позднее были выявлены и долгосрочные эффекты АТФ, его способность контролировать пролиферацию клеток, их дифференцировку, подвижность, гибель в процессе развития, регенерацию, заживление ран, развитие опухолей и при клеточном старении [32]. И АТФ, и аденозин были определены как особенно важные сигнальные молекулы при патологических состояниях, например, при ишемии, когда их внеклеточная концентрация резко возрастает [140].
В последние годы совокупную систему пуринергической регуляции обозначают как “пурином” [60]. Пурином считается универсальной системой, позволяющей так калибровать и синхронизировать индивидуальные клеточные ответы, чтобы они соответствовали интересам целого организма. Передача пуринергических сигналов обеспечивается следующими основными элементами:
(i) высвобождением АТФ (и ГТФ) в перицеллюлярное пространство;
(ii) пуринергическими рецепторами (Р2), которые распознают высвобожденный АТФ/ГТФ и его метаболиты и проводят сигналы внутрь клеток, регулируя их функции;
(iii) прекращением передачи пуринергических сигналов, что осуществляется путем быстрого ферментативного расщепления АТФ до аденозина;
(iv) в свою очередь, аденозин осуществляет регуляторную функцию, связываясь с пуринергическими рецепторами (Р1), в тех случаях, когда внеклеточная концентрация аденозина начинает превышать внутриклеточную, он захватываются и направляются внутрь клетки белками-транспортерами.
В физиологических условиях АТФ присутствует в основном внутри клетки и высвобождается во внеклеточное пространство в низких (микромолярных) концентрациях [33], создавая “пуринергический ореол” в их ближайшем окружении [164]. Такой “ореол” АТФ представляет собой сигнал низкой интенсивности, адресованный соседним клеткам, и отражает состояние клеток, испускающих АТФ [164]. Интактные клетки могут высвобождать АТФ при экзоцитозе везикул или через проницаемые для АТФ мембранные каналы, такие как коннексиновые полуканалы, паннексиновые каналы, модулятор гомеостаза кальция 1, максианионные каналы и анионные каналы с регулируемым объемом (рис. 2) [113, 162]. В состоянии покоя или в физиологических условиях клетки выделяют низкие уровни АТФ.
Рис. 2.
Основные компоненты пуринергической системы. Молекулы АТФ высвобождаются из клетки через паннексиновые (Panx1) и другие типы каналов, связываются с Р2-рецепторами как на клетке, из которой они вышли, так и на соседних клетках. Пуриновые рецепторы для нуклеотидов Р2R определяют как ионотропные P2XR (P2X1–7) и метаботропные P2Y (P2YR1,2,4,6,11–14). Нуклеотиды подвергаются гидролизу мембранными и растворимыми эктонуклеотидазами (ENTPD1, CD39) и экто-5'-нуклеотидазой (5'-нтд, CD73), проходя путь от АТФ, АДФ и АМФ до аденозина, который, в свою очередь, активирует пуриновые рецепторы первого типа (P1R). Лигандом всех P2XR на микроглии является АТФ, а для P2YR – лигандами могут быть АTФ (P2Y2 и P2Y11), АДФ (P2Y1). Активация рецепторов P2X приводит к поступлению в клетку ионов Na+, K+ и Ca2+, а в случае рецептора P2X7, при действии высоких концентрация АТФ формируется большая пора, проницаемая для крупных молекул. Активация Р2YR по-разному действуют на аденилатциклазу: рецептор P2Y11 стимулирует этот фермент, а рецепторы P2Y12,13 – ингибируют. Аденилатциклазы, связанные с плазматической мембраной, синтезируют цАМФ, который является аллостерическим эффектором протеинкиназы A и ионных каналов. Активация рецепторов P2Y1,2,4,6 и P2Y11, связанных с альфа-субъединицей G-белка (Gq), приводит к стимуляции фосфолипазы C, которая инициирует выработку инозитолтрифосфата (IP3) и диацилглицерола (DAG) и высвобождение кальция из клеточных депо и последующей активацией протеинкиназы С . Аденозин может действовать на четыре типа Р1-рецепторов (A1, A2A, A2B и A3), которые объединены с G-белками, как ингибирующими (Gi), так и стимулирующими (Gs) аденилатциклазу. Способность подавлять активность аденилатциклазы определена для рецепторов A1 и A3, тогда как A2A и A2B стимулируют этот фермент. Действие аденозина прекращается после его ферментативной деградации аденозиндезаминазой (ADA) до инозина или трансформацией аденозинкиназой (ADK) в АМФ. Перераспределение аденозина и других нуклеозидов между клетками и внеклеточной средой осуществляется специфическими белками, транспортерами нуклеозидов, которые облегчают их перемещение через плазматические мембраны и мембраны некоторых органелл. Эти белки-переносчики определяют как “концентрирующие транспортеры нуклеозидов” (CNT) или как “уравновешивающие транспортеры нуклеозидов” (ENT).
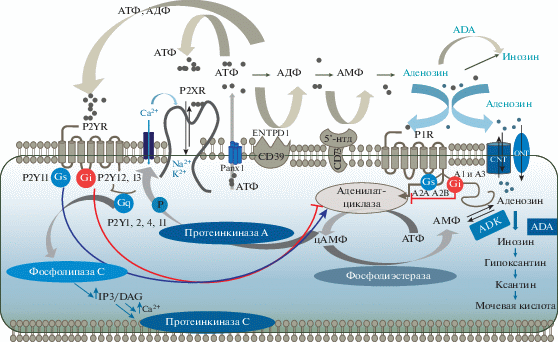
При клеточном стрессе и/или повреждении происходит быстрое пассивное (связанное с повреждением мембраны) высвобождении АТФ во внеклеточную среду, и АТФ достигает высоких (миллимолярных) концентраций, которые запускают воспаление [33], в том числе и активацию микроглии [4].
После выхода во внеклеточное пространство нуклеотиды, включая АТФ и АДФ связываются со специфическими P2 рецепторами, которые подразделяются на ионотропные P2XR (P2X1–7) и метаботропные P2Y (P2YR1, 2, 4, 6, 11–14) [9]. Значительно слабее с некоторыми P2YR могут взаимодействовать и пиримидины УТФ, УДФ и УДФ-глюкоза. Р2ХR имеют тримерную топологию с двумя трансмембранными доменами и управляют в первую очередь потоками Na+, K+, Ca2+ и иногда Cl– [139]. Активация рецепторов P2Y1,2,4,6 и P2Y11, связанных с альфа-субъединицей G-белка (Gq), приводит к стимуляции фосфолипазы C, которая инициирует выработку инозитолтрифосфата и диацилглицерола [72]. Инозитолтрисфосфат увеличивает внутриклеточные уровни Ca2+, а диацилглицерол стимулирует протеинкиназу C. Кроме того, рецепторы Р2Y по-разному действуют на аденилатциклазу: рецептор P2Y11 ее стимулирует, а рецепторы P2Y12,13 − ингибируют [72].
Пуринергические рецепторы сильно различаются по кинетике десенсибилизации и сродству к индивидуальным лигандам. Внеклеточные концентрации лигандов зависят от активности эктоферментов, как экспрессируемых на поверхности клетки, так и растворимых [174]. На поверхности клеток различных типов имеются три семейства ферментов, гидролизующих АТФ: экто-нуклеотидпирофосфатазы/фосфодиэстеразы (E-NPP), щелочные фосфатазы (AP) и экто-нуклеозидтрифосфатдифосфогидролазы (E-NTPD). Дальнейшая метаболизация осуществляется экто-5'-нуклеотидазой, которая отщепляет фосфатную группу от АМФ с образованием аденозина, и аденилаткиназой (AK), которая катализирует обратимую реакцию между АМФ и АТФ с образованием двух молекул АДФ [174].
Ось эктоферментов на клеточной поверхности, состоящая из фосфатазы ENTPD1 и экто-5'-нуклеотидазы (CD39−CD73), является основным механизмом деградации внеклеточного АТФ, АДФ и АМФ до аденозина [12]. Кроме того, выделяют ось ферментов гидролаза CD38−CD203a (экто-нуклеотидпирофосфатаза/фосфодиэстераза-3), которая действует на поверхности клетки независимо или совместно с путем CD39−CD73, и также способствует метаболизму внеклеточных пуринов [15]. В частности, CD38 катализирует синтез циклической АДФ-рибозы из никотинамидадениндинуклеотида (НАД+) и опосредует гидролиз циклической АДФ-рибозы до линейной формы АДФ-рибозы на поверхности клетки [85]. Пирофосфатаза/фосфодиэстераза CD203a способна гидролизировать НАД+, АДФ-рибозу и АТФ с образованием АМФ, который далее может расщепляться до аденозина с помощью CD73 [92, 143]. Ферменты, синтезирующие и катаболизирующие производные пуринов, вовлечены в точную регулировку (“колибрование”) состава, величин и продолжительности существования “пуринергического ореола” [10, 12, 92].
Пуринергическая регуляция поддерживает гомеостаз и регулирует важнейшие системы органов, в том числе сердечно-сосудистую, выделительную (почки), желудочно-кишечную, центральную нервную и иммунную системы [11, 18, 30, 35]. Большинство подтипов пуринергических рецепторов широко представлены на различных клетках организма и высоко экспрессируются в центральной и периферической нервной системе. В физиологических условиях они обычно вовлечены в двунаправленную нейронно-глиальную коммуникацию, оказывая долгосрочное влияние на пролиферацию, дифференцировку и миграцию нейронов (в том числе рост и направленное движение аксонов) и глиальных клеток [52]. В пресинаптических окончаниях и конусах роста аксонов располагаются рецепторы P2X7, которые модулируют действие других нейромедиаторов, влияя на активность нейронов, и могут координировать ответы микроглии, нейронов и астроглии [70, 158].
При нарушениях гомеостаза пурины накапливаются во внеклеточном пространстве в ответ на повреждение, травмы и изменения во внеклеточной среде (например, гипоксия, ацидоз, нарушения ионного баланса и изменения уровня гормонов и нейротрансмиттеров) и передача пуринергических сигналов в этих условиях связана с регуляцией функции органа или ткани.
АТФ КАК РЕГУЛЯТОР НЕЙРОВОСПАЛЕНИЯ ПРИ ЧМТ
АТФ, высвобождаемый клетками с нарушенной плазматической мембраной, действует как провоспалительный DAMP [34], при связывании с P2XR он может усиливать воспаление и проявлять повреждающие свойства [73].
После ЧМТ высвобождение АТФ из поврежденных астроцитов в течение нескольких минут активирует микроглию, которая при этом меняет свои морфологические характеристики [94]. Показано, что в состоянии покоя микроглия имеет очень разветвленную форму и непрерывно сканирует определенную территорию паренхимы множеством мелких подвижных отростков, регистрируя тканевые аномалии. При активации отростки клеток микроглии направляются к месту повреждения со средней скоростью выдвижения и втягивания 1.47 мкм в минуту (в диапазоне от 0.4 и 3.8 мкм в минуту соответственно) [135]. Затем отростки начинают втягиваться, тело клетки увеличивается в размерах и приобретает сферическую (амебоидную) форму [93]. Наконец, измененная клетка микроглии начинает мигрировать к месту повреждения со скоростью 1–2 мкм в час [135]. АТФ определен как хемоаттрактант для микроглии, а его высокие концентрации во внеклеточном пространстве связаны с плохими исходами ЧМТ [136]. Таким образом, микроглия не только очищает поврежденный мозг от клеточного дебриса, но и помогает поддерживать целостность глиального барьера, блокируя проникновение крупных молекул и дебриса через пограничную глиальную мембрану в паренхиму мозга, закрывая бреши, возникающие на месте погибших или поврежденных астроцитов [44].
Исследование функциональных характеристик активированной микроглии показало, что она может принимать “классический” активированный фенотип M1 (CD11b+, CD16+) [83, 120], при такой активации она способна фагоцитировать патогенные бактерии, выделять микровезикулы [171] и высвобождать провоспалительные цитокины (IL-1β, TNF-α, IFNγ, IL-12), хемокины, протеазы, активные формы кислорода/азота, а также АТФ и глутамат, используя экзоцитоз везикул (рис. 3) [105].
Рис. 3.
Пуринергический контроль активности микроглии в участках повреждения мозга. Распространением отростков микроглии в участке паренхимы мозга управляют рецепторы P2Y12. Втягивание отростков определяется повышением концентрации АТФ/АДФ и активацией P2Y12R и P2Y13R у человека и P2Y12R и A2AR (у грызунов). Миграционная активность микроглии по градиенту пуринов контролируется рецепторами P2Y12 и P2X4. Приобретая амебоидную морфологию, клетки микроглии приступает к фагоцитозу и пиноцитозу, которые индуцируется активацией P2Y6R и P2Y4R. Активированная микроглии может принимать классический активированный фенотип M1, в этом состоянии она способна к фагоцитозу и киллингу патогенных бактерий, продукции воспалительных медиаторов, реактивных радикалов кислорода и азота. Фенотип “альтернативной активации” M2 связан с фагоцитозом клеточного дебриса и продукцией многочисленных факторов роста, что характерно для поддержки репаративных процессов. Для фенотипа M2 выделяют так называемые смешанные фенотипы (подтипы a, b, c). Формирование фенотипов М1 и М2 связано с активацией пуринергических рецепторов, роль в этом процессе некоторых пуринорецепторов уже определена.
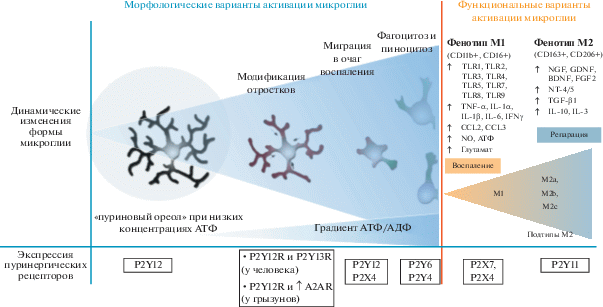
Кроме того, был выявлен также фенотип “альтернативной активации” микроглии M2 (CD163+, CD206+), для которого характерно вовлечение в фагоцитоз клеточного дебриса и высвобождение многочисленных защитных факторов (IL-4, IL-13, фактор роста нервов (NGF), фактор роста фибробластов (FGF)) [71]. Считается, что микроглия в состоянии M1 участвует в воспалительных и иммунных защитных реакциях ЦНС, а в состоянии M2 – в репаративных реакциях. Позднее микроглию M2 (как и макрофаги) разделили на три подтипа (M2a, M2b, M2c) [90], а также выявили существование клеток смешанных фенотипов M1/M2 [38]. В настоящее время представление о дихотомии M1/M2 признано упрощенным. Показано, что не существует определенного дискретного числа реактивных фенотипов микроглии, а имеется разнообразие состояний, которые формируются сигналами окружающей среды, что позволяет клеткам микроглии выполнять определенные функции в различных физиологических и патологических состояниях [90]. Также показано, что активация микроглии может быть частично обратимой, что также зависит от контекста, стимулов и условий, в которых эти стимулы появляются [129]. Показано, что блокирование рецепторов P2X4 и P2X7 отменяет активацию микроглии по типу М1 [77]. Недавно показано, что в макрофагах, поляризующихся как М2, повышается уровень экспрессии P2Y11, однако тождественность этого процесса для микроглии еще предстоит подтвердить [80].
Хотя АТФ связывает все рецепторы семейства P2X [76], участие в регуляции процессов после ЧМТ определено только для P2X7R [108]. Для P2X4R показано, что он опосредует с увеличение секреции нейротрофического фактора головного мозга (BDNF) в микроглии спинного мозга [110, 123].
Р2Х7R экспрессируется преимущественно в микроглии коры головного мозга [106] и значительно отличается от других Р2ХR по строению (может собираться как гомотример, так и гетеродимер) [150] и тем, что для активации ему необходима такая высокая (миллимолярная) концентрация АТФ, какая присутствует в участках воспаления и повреждения мозга через 6–12 ч после ЧМТ [116]. P2X7R регулирует активацию инфламмасом, индукцию медиаторов воспаления и выработку свободных радикалов [17, 78, 102]. Через P2X7R АТФ также стимулирует секрецию провоспалительных цитокинов микроглией [7, 155, 156], и секрецию IL-1β нейтрофилами (опосредованную инфламмасомой NLRP3) [101], а также усиливает образование внеклеточных ловушек нейтрофилов (NET) [157]. Помимо немедленных эффектов активации, P2X7R вызывает длительные молекулярные изменения, такие как пролиферация и апоптоз [121]. Однако длительное (30 мин) воздействие миллимолярных концентраций АТФ снижает фагоцитарную активность микроглии через P2X7R, что опосредуется Ca2+-независимым механизмом [68].
P2X7R активирует несколько внутриклеточных сигнальных каскадов, таких как пути кальмодулина, митоген-активируемой протеинкиназы и фосфолипазы D. При низких концентрациях АТФ (микромолярный диапазон) активация P2X7R открывает катионный канал, как и другие рецепторы P2X. Однако в присутствии высоких концентраций АТФ (0.05–1 мМ) он открывает путь, позволяющий проходить более крупным органическим катионам и анионам [6].
Р2Х7 обладает разной способностью к созданию большой поры в культурах астроцитов коры и гипоталамуса. Несмотря на то, что функциональный Р2Х7 рецептор экспрессирован в обоих перечисленных регионах мозга, только в коре он способен индуцировать открытие большой поры, что ведет к повышенной экспрессии IL-1β. Возможно, это приводит к тому, что кора больших полушарий больше подвержена травматическим воздействиям [108]. Р2Х7 рассматривают как основную потенциальную мишень при создании лекарственных средств, предотвращающих развитие вторичной травмы [36].
Из всего семейства рецепторов P2Y АТФ связывают только P2Y2 и P2Y11 [169], однако их роль в регуляции микроглии мало изучена. Показано, что стимуляция АДФ-связывающих рецепторов P2Y1 уменьшает отек, ослабляет реактивный астроглиоз и усиливает неврологическую функцию после ЧМТ [40, 161].
Регуляция функций микроглии в условиях нейровоспаления сопровождается изменением экспрессии пуринергических рецепторов (рис. 3), что было первоначально исследовано у грызунов. Показано, что на раннем этапе активации стимуляция рецепторов P2Y12 запускает процесс удлинения и расширения отростков микроглии грызунов по направлению к месту повреждения [50]. Расширением отростков микроглии совместно управляют рецепторы P2Y12 [88, 154] и аденозиновый рецептор семейства Р1 A3R [110]. Впоследствии этот процесс останавливается из-за снижения экспрессии P2Y12R и повышения экспрессии A2AR. Появляющаяся после этих изменений миграционная активность микроглии контролируется рецепторами P2Y12 и P2X4. После втягивания микроглиальных отростков и приобретения амебоидной морфологии, клетки приступают к фагоцитозу и пиноцитозу, которые индуцируется активацией P2Y6R и P2Y4R соответственно. При фармакологическом блокировании рецепторов P2Y6 после ЧМТ увеличивается проницаемость пограничной глиальной мембраны и усиливается гибель клеток мозга [147].
Недавно было выполнено исследование подвижности микроглии, управляемой пуриновыми молекулами, у человека (на образцах ткани мозга, полученной от больных мезиальной височной эпилепсии или в перитуморальной ткани коры мозга). В культуре микроглии человека АДФ в разных концентрациях запускал противоположные типы движения: низкие дозы приводили к удлинению отростков, а более высокие – к втягиванию отростков и сморщиванию мембраны. Блокирование этих эффектов достигалось при совместном применении антагонистов рецепторов P2Y1 и P2Y13. Эти наблюдения позволили авторам заключить, что индуцированная пуринами подвижность микроглии человека сходна с таковой у микроглии грызунов в том, что удлинение ее отростков инициирует P2Y12R, но отличается тем, что втягивание отростков запускается совместной активацией рецепторов P2Y1/P2Y13R, а не через аденозиновый рецептор A2A, который на микроглии человека экспрессирован слабо [125].
Суммируя последствия АТФ-регуляции в условиях ЧМТ, отмечают, что при легкой форме повреждения АТФ вызывает острую воспалительную реакцию, имеющую нейропротекторную направленность, однако при длительной активации микроглия становится инициатором вторичных повреждений, связных АТФ-зависимой активацией инфламмасом, что усиливает активацию клеток врожденного иммунитета и увеличивает размер травматического повреждения при ЧМТ [51, 147].
АДЕНОЗИН КАК РЕГУЛЯТОР НЕЙРОВОСПАЛЕНИЯ
В стерильных условиях воспалительные и иммунные реакции обычно быстро ослабевают, поскольку АТФ теряет свои фосфатные группы и трансформируются в АДФ/АМФ и аденозин при участии ряда ферментов, а именно эктонуклеозидтрифосфатдифосфогидролазы 1 (Е-NTPDasa1, CD39) и экто-5'-нуклеотидазы(СD73), представленных как на клетках ЦНС, так и иммунных клетках [24, 146]. На астроцитах и микроглии экспрессируется, по крайней мере, по одному из этих эктоферментов, что позволяет им совместно контролировать АТФ-опосредованное нейровоспаление [28, 134]. Последовательная доступность пуриновых регуляторов от АТФ до аденозина обеспечивает условия для активации микроглии, повышения уровня внеклеточного аденозина и изменение экспрессии пуринергических рецепторов.
Аденозин может продуцироваться внутриклеточно и напрямую транспортироваться во внеклеточное пространство, но при патологических состояниях он в основном образуется внеклеточно при ферментативном вычленении фосфатных групп из АТФ/АДФ ферментными каскадами CD39–CD73 и/или CD38–CD203a [15, 128].
Свободный аденозин в головном мозге обычно находится в наномолярном диапазоне [112]. Однако его уровни значительно повышаются при состояниях, связанных с повышенной метаболической потребностью, гипоксией, воспалением и повреждением тканей. В частности, повышенные уровни внеклеточного аденозина наблюдались при таких патологических состояниях, как боль [58], эпилепсия [79, 63 ], ишемия [74, 141], злокачественные новообразования [124] и воспаление [13].
Аденозин определяют как регуляторный аутакоид, обладающий множеством эффектов. Аденозин может действовать на четыре типа пуринергических Р1-рецепторов (A1, A2A, A2B и A3), которые объединены с ингибиторными (Gi) или стимулирующими (Gs) G-белками [25]. Рецепторы A2, повышающие уровни цАМФ, разделены на две группы: A2A с высоким сродством и рецепторы А2B с низким сродством к лиганду [141]. Ингибиторные свойства показаны для рецепторов A1 и A3, которые подавляют активность аденилатциклазы, тогда как A2A и A2B стимулируют этот фермент.
Рецепторы A1 и A2A дополняют друг друга в процессах ингибиции высвобождения возбуждающих аминокислот и поддержания постсинаптической гиперполяризации [48], участвуют в формировании кальциевой волны в астроцитарном синцитиуме [82]. Астроциты высвобождают аденозин к эндотелиальным клеткам, вызывая вазодилатацию за счет активации на них рецептора A2A, который усиливает местную гемоциркуляцию и обеспечивает дополнительную метаболическую поддержку, необходимую во время интенсивной синаптической стимуляции [87]. Рецепторы A2B и A3 низкоаффинные, чувствительны к микромолярным концентрациям аденозина [62]. Они широко экспрессируются на астроцитах, хотя и на низких уровнях [56]. Рецепторы A2B активируется в условиях ишемии [173] и быстро запускают выработку IL-6, что поддерживает воспалительную реакцию после травмы [165].
Действие аденозина, в свою очередь, прекращается после его деградации ферментом аденозиндезаминазой (ADA) до инозина [65] или трансформацией в АМФ аденозинкиназой (ADK) [127]. Внеклеточный аденозин быстро поглощается клетками окружающей ткани с помощью транспортеров нуклеозидов, уравновешивающих его вне- и внутриклеточные концентрации (ENT1–4) [152].
Помимо своей роли в качестве сигнальной молекулы, аденозин является посредником в метаболическом пути, который включает азотистое основание аденин, нуклеотид АТФ и вторичный мессенджер циклический аденозинмонофосфат (цАМФ) [118]. Модуляция уровней цАМФ активирует или ингибирует большое количество путей передачи сигналов в зависимости от типа вовлеченных клеток.
Хотя иногда сам аденозин способствует поддержанию различных патологических состояний (например, при опухолевом росте) [124], его обычно считают защитным и гомеостатическим медиатором в условиях повреждения тканей и стрессовых состояний [26]. В физиологических и стрессовых условиях внеклеточные концентрации аденозина поддерживаются на низком уровне в результате его быстрого метаболизма и поглощения [168].
АДЕНОЗИН ПРИ ЧМТ
Через 1 ч после ЧМТ у человека и животных повышается уровень аденозина в спинномозговой жидкости и интерстициальном пространстве [19, 42]. Самые высокие уровни аденозина обычно наблюдаются в ранний период после ЧМТ, хотя у детей было отмечено и более позднее повышение (через 72 ч) что, как считают, отражает прогрессирование вторичной травмы [145].
При экспериментальной черепно-мозговой травме генетическая или фармакологическая блокада A1R подавляет морфологическую активацию микроглии, вероятно, подавляя АТФ-индуцированное проникновение Са2+ в клетки [84, 117]. Аналогичным образом и передача аденозиновых сигналов через A3R способствует смягчению вторичного повреждения и уменьшению площади ишемии в окружающей мозговой ткани, поддерживая защитные эффекты и сохранение когнитивной функции после ЧМТ [69]. Хотя конститутивная экспрессия A1AR и A2AR в головном мозге выше, чем A3AR [25, 64], однако при быстром повышении внеклеточного уровня аденозина вскоре после ЧМТ задействованы, вероятно, бывают все три перечисленных типа рецепторов. Привлечение всех этих рецепторов обеспечивает контроль над нейровоспалением, развивающимся после первичного повреждения.
Регуляция внеклеточного аденозина в значительной степени управляется внутриклеточной аденозинкиназой (ADK), которая превращает внутриклеточный аденозин в аденозинмонофосфат, что приводит к втягиванию внеклеточного аденозина в клетку по градиенту его концентрации [23]. Повышенная активность аденозинкиназы, наблюдаемая при патологических состояниях, истощает внутриклеточный аденозин [23]. После ЧМТ повышенная экспрессия ADK в астроцитах связана с гибелью. нейронов и астроглиозом, а подавление ADK в астроцитах снижает их провоспалительный фенотип [99]. Кроме того, у мышей со сверхэкспрессией ADK после повреждения коры при ЧМТ была нарушена пролиферация нервных стволовых клеток, а при фармакологическом ингибировании ADK пролиферация нервных стволовых клеток после ЧМТ усиливалась [75]. На уровень внеклеточного аденозина влияет и превращение внеклеточного АТФ в аденозин эктонуклеотидазами [12, 21].
Однако высокие уровни аденозина, связанные с его быстрым высвобождением, оказываются краткосрочными, поскольку после ЧМТ церебральный кровоток не обеспечивает оптимального потребления кислорода в месте повреждения, что приводит к нарушению метаболизма аденозина [118]. На экспериментальных моделях показано, что после ЧМТ одновременно с повышением высвобождения аденозина нарушается гидролиз АМФ экто-5'-нуклеотидазой (CD73) в областях коры [21, 132] и в гиппокампе [21].
При низкой внеклеточной концентрации обеспечивается связывание аденозина только с A1AR и A2AR, но при такой концентрации не обеспечивается необходимая экспрессии и связывания A3AR, что создает условия для поддержания нейровоспаления.
При расщеплении аденозина ферментом ADA образуется инозин, который имеет иммуномодулирующее, кардиопротекторное и цитопротекторные действие [86, 159, 166]. Эндогенные аденозин и инозин обладают цитопротекторной активностью при различных формах ишемии, в том числе при ЧМТ [167]. Инозин оказывает сильное защитное действие [61, 119] благодаря своим антиоксидантным и противовоспалительным эффектам [148, 163]. Действуя как антиоксидант, инозин снижает уровни реактивных радикалов кислорода и азота, увеличивает активность ферментов каталазы (CAT), глютатионпероксидазы (GPx) и супероксиддисмутазы в структурах мозга [148] и повышает уровни восстановленного глутатиона. Инозин действует и как поглотитель свободных радикалов [81], способный снизить выработку перекиси водорода и гидроксильных радикалов и предотвратить окислительное повреждение ДНК. Другие возможные механизмы, вовлеченные в антиоксидантный эффект инозина, могут быть связаны с его дальнейшим расщеплением до мочевой кислоты.
ПЕРСПЕКТИВЫ КЛИНИЧЕСКОГО ИСПОЛЬЗОВАНИЯ СЕЛЕКТИВНЫХ РЕГУЛЯТОРОВ ПУРИНОВОГО МЕТАБОЛИЗМА
Поскольку пуринергические сигналы вовлечены в регуляцию широкого спектра физиологических и патофизиологических процессов, протекающих в ЦНС, иммунной и других системах, имеются теоретические предпосылки для исследования влияния нуклеотидных и нуклеозидных препаратов на функции различных органов и систем организма в норме и при патологии [30, 35, 36]. Известно, что широкий ряд противовоспалительных средств, таких как циклоспорин, салицилаты (аспирин), метотрексат, сульфасалазин и его аналоги, а также тофацитиниб (ингибитор JAK–STAT пути), оказывают свое противовоспалительное действие (в том числе) путем увеличения внеклеточного уровня аденозина [37, 46, 47, 109].
В последние годы, преимущественно благодаря использованию методов рентгеноструктурного анализа рецепторных белков и позитронно-эмиссионной томографии различных отделов мозга, удалось существенно расширить представления о физиологической роли пуринергических рецепторов ЦНС и лучше изучить молекулярные механизмы связывания их селективных агонистов и антагонистов. Так, в частности, недавно была пересмотрена специфичность некоторых соединений, считавшихся селективными для A1AR или A3AR [95]. Показано, многие из разработанных лигандов P2R пока еще не обладают биодоступностью к целевым тканям-мишеням, в частности, забарьерным структурам ЦНС, или являются гидролитически нестабильными [3, 95]. Значительную проблему для исследований и разработок представляют и межвидовые различия пуринопецепторов, что сказывается на снижении степени селективности используемых лигандов в опытах на различных видах модельных животных и при попытке применения препаратов, изученных на животных, в клинических экспериментах у пациентов.
Эксперименты показали, что при ЧМТ использование антагонистов метаботропных рецепторов P2Y2, P2Y4, P2Y6 и P2Y12 снижает повреждение нейронов [172]. Однако наиболее популярной мишенью в исследованиях пуринергической регуляции при ЧМТ стал рецептор P2X7. По результатам оценки поведенческих тестов показано, что полная генетическая или фармакологическая блокада P2X7 уменьшает нейровоспаление и улучшает исходы в моделях ЧМТ с контролируемым кортикальным повреждением у мышей [108]. Использование фармакологических антагонистов P2X7R значительно снижало количество регистрируемых микровезикул в поврежденных и прилегающих областях мозга, в спинномозговой жидкости, и улучшало исходы после ЧМТ, в том числе и связанные с поведением травмированных животных [116]. Аналогичное защитное действие блокирования P2X7 было показано и при травме спинного мозга (уменьшение площади повреждения, потери нейронов, ослабление местной воспалительной реакции при улучшении исходов) [41]. Поскольку в экспериментах на клеточных культурах и животных моделях фармакологические антагонисты Р2Х7R способствовали выживаемость нейронов и нормализации поведения животных после экспериментальной ЧМТ, это дает основание для предположений о возможности их использование в качестве препаратов для лечения вторичного повреждения [53].
Одна из трудностей, препятствующих получению препаратов для фармакологического воздействия на пуринергическую систему, связана с широким распространением в организме рецепторов P1 и P2, их участием в регуляции различных физиологических функций, что увеличивает вероятность проявления побочных эффектов. Оказалось, хотя многие лиганды, действующие на пуринергическую систему, являются достаточно селективными и демонстрируют обнадеживающие положительные эффекты в доклинических исследованиях, их эффективность в клиническом эксперименте оказалась невысокой. Антагонисты рецептора Р2Х7 являются ярким примером этого. Когда новые и безопасные антагонисты рецепторов Р2Х7, которые были эффективны в экспериментальных моделях воспаления (рассеянный склероз, хроническое воспалительное заболевание кишечника и ревматоидный артрит), были испытаны в клинике, результаты оказались неудовлетворительными [67, 104, 160].
Анализируя причину клинических неудач, ссылаются на наличие индивидуальных особенностей рецептора P2X7, влияющих на его функцию (однонуклеотидный полиморфизм) [7]. Также оказалось, что сборка и функционирование рецептора P2X7 зависит от липидного состава клеточной мембраны, в которой этот рецептор собирается [100]. Следовательно, на один и тот же фармакологический агент клетки в разном функциональном состоянии (что отражается на составе липидов их мембран) будут отвечать по-разному. Очевидно, что это создает дополнительные трудности при прогнозировании организменных эффектов препаратов, специфичных для P2X7.
Характеризуя имеющиеся лиганды различных подтипов пуринергических рецепторов, представленных в нервной системе, Jacobson и Müller отмечают, что пока имеющиеся агенты пригодны только как фармакологические инструменты для определения физиологических функций различных подтипов пуринорецепторов [95].
Кроме химических лигандов пуринергических рецепторов, разрабатываются также новые фармакологические агенты, способные модулировать активность ферментов, участников метаболизма пуринов [9]. Однако пока нет данных о получении селективных ингибиторов эктонуклеозидтрифосфатдифосфогидролаз, способных проникать через ГЭБ [16]. Мишенями при разработке препаратов, направленных на повышение уровня аденозина, могут быть ингибиторы ферментов ADA и ADK [151] Однако препаратов, готовых к клиническому использованию в этой группе ферментных ингибиторов также пока нет.
ЗАКЛЮЧЕНИЕ
В физиологических условиях в ткани мозга человека и экспериментальных животных пуриновые нуклеотиды и нуклеозиды, в частности АТФ и аденозин, модулируют сигналы, обеспечивающие связь между нейронами, а также между нейронами и клетками микроглии, синхронизируя совместную деятельность этих структур. Черепно-мозговая травма приводит к развитию несбалансированного воспаления и нарушению пуринергической регуляции как в структурах мозга, так и в иммунной системе. При этом действие внеклеточного АТФ на клетки иммунной системы приводит к их стимуляции и запуску защитных реакций организма [30]. Информация, переносимая АТФ и аденозином в ЦНС влияет на изменения сна, памяти, обучаемости, двигательной активности и восстановление тканевых повреждений после ЧМТ [32, 36]. Эти события определяют информативность исследования уровней АТФ и аденозина в ЦСЖ и/или активности ферментов, контролирующих их метаболизм, а их результаты позволяют получать важную в патогенетическом и прогностическом плане информацию о течении посттравматического процесса.
Основные усилия фармакологов в области биомедицинских исследований ЧМТ пока направлены на разработку и испытание селективных агонистов/антагонистов, блокаторов ключевых структур пуринергической системы (рецепторы, ферменты), способных изменять течение воспалительных процессов в ЦНС. Однако, нам представляется, что на современном этапе центральное внимание исследователей должны привлечь, если и не системные, то хотя бы комплексные аспекты многообразных изменений пуринергической регуляции, рассматриваемые с учетом характера патологии, локализации повреждения, фактора времени и предшествующего состояние тканей и организма.
Пока многолетнее и многостороннее изучение пуринергической регуляции привело к появлению и клиническому внедрению только одного антагониста пуринергического рецептора Р2Y12R, препарата Clopidogrel, способного ингибировать активацию тромбоцитов. Причем относительная эффективность результативность этого ингибитора связана с тем, что в условиях целого организма рецепторы Р2Y12 представлены только на тромбоцитах и микроглии. Учитывая отсутствие столь избирательно локализации для других перинергических рецепторов и ряд вышеперечисленных физиологических, биохимических и генетических особенностей пуринергической регуляции нейровоспаления, представляется, что использование селективных антагонистов или агонистов пуриновых рецепторов еще долго будет оставаться исследовательским инструментом, а для клинической практики потребуются препараты другого типа, способные оказывать комплексное корректирующее воздействие в патофизиологических условиях дефицита или избытка пуринергических медиаторов при остром, затяжном или хроническом воспалении различных структур ЦНС, любой нейротравме, в частности, ЧМТ.
Список литературы
Шевченко К.В., Четвертных В.А., Кравцов Ю.И. Иммунопатологические изменения при тяжелой черепно-мозговой травме // Иммунология. 2009. Т. 30. № 3. С. 180. https://www.medlit.ru/journal/403
Янкелевич И.А., Шустов М.В., Мартышкина Ю.С., Филатенкова Т.А. Стресс-индуцированное повышение экспрессии генов TLR2, TLR3 и TLR4 в клетках гипоталамуса // Медицинский академический журн. 2020. Т. 20. № 2. С. 11. https://doi.org/10.17816/MAJ33432
Abe N., Nishihara T., Yorozuya T., Tanaka J. Microglia and Macrophages in the Pathological Central and Peripheral Nervous Systems // Cells. 2020. V. 9. P. 2132. https://doi.org/10.3390/cells9092132
Abiega O., Beccari S., Diaz-Aparicio I., Nadjar A. et al. Neuronal Hyperactivity Disturbs ATP Microgradients, Impairs Microglial Motility, and Reduces Phagocytic Receptor Expression Triggering Apoptosis/Microglial Phagocytosis Uncoupling // PLoS Biol. 2016. 14:e1002466. https://doi.org/10.1371/journal.pbio.1002554
Able S.L., Fish R.L., Bye H. et al. Receptor localization, native tissue binding and ex vivo occupancy for centrally penetrant P2X7 antagonists in the rat // Br. J. Pharmacol. 2011. V. 162. P. 405. .https://doi.org/10.1111/j.1476-5381.2010.01025.x
Alves L., de Melo Reis R., de Souza C. et al. The P2X7 receptor: shifting from a low- to a high-conductance channel – an enigmatic phenomenon? // Biochim. Biophys. Acta. 2014. V. 1838. P. 2578. https://doi.org/10.1016/j.bbamem.2014.05.015
Andrejew R., Oliveira-Giacomelli Á., Ribeiro D.E. et al. The P2X7 Receptor: Central Hub of Brain Diseases // Front. Mol. Neurosci. 2020. V. 31. P. 124. https://doi.org/10.3389/fnmol.2020.00124
Anthonymuthu T.S., Kenny E.M., Bayır H. Therapies targeting lipid peroxidation in traumatic brain injury // Brain Res. 2016. V. 1640 (Pt A). P. 57. https://doi.org/10.1016/j.brainres.2016.02.006
Antonioli L., Blandizzi C., Pacher P., Haskó G. The Purinergic System as a Pharmacological Target for the Treatment of Immune-Mediated Inflammatory Diseases // Pharmacol. Rev. 2019. V. 71. P. 345. https://doi.org/10.1124/pr.117.014878
Antonioli L., Colucci R., La Motta C. et al. Adenosine deaminase in the modulation of immune system and its potential as a novel target for treatment of inflammatory disorders // Curr. Drug Targets. 2012. V. 13. P. 842. https://doi.org/10.2174/138945012800564095
Antonioli L., Fornai M., Colucci R. et al. Control of enteric neuromuscular functions by purinergic A(3) receptors in normal rat distal colon and experimental bowel inflammation // Br. J. Pharmacol. 2010. V. 161. P. 856. https://doi.org/10.1111/j.1476-5381.2010.00917.x
Antonioli L., Pacher P., Vizi E.S., Haskó G. CD39 and CD73 in immunity and inflammation // Trends Mol. Med. 2013. V. 19. P. 355. https://doi.org/10.1016/j.molmed.2013.03.005
Antonioli L., Csóka B., Fornai M. et al. Adenosine and inflammation: What’s new on the horizon? // Drug Discov. Today. 2014. V. 19. P. 1051. https://doi.org/10.1016/j.drudis.2014.02.010
Arac A., Grimbaldeston M.A., Galli S.J., Bliss T.M., Steinberg G.K. Meningeal mast cells as key effectors of stroke pathology // Front. Cell Neurosci. 2019. V. 13 P. 126. https://doi.org/10.3389/fncel.2019.00126
Bahri R., Bollinger A., Bollinger T., Orinska Z., Bulfone-Paus S. Ectonucleotidase CD38 demarcates regulatory, memory-like CD8+ T cells with IFN-γ-mediated suppressor activities // PLoS One. 2012. V. 7. e45234. https://doi.org/10.1371/journal.pone.0045234
Baqi Y., Rashed M., Schäkel L. et al. Development of Anthraquinone Derivatives as Ectonucleoside Triphosphate Diphosphohydrolase (NTPDase) Inhibitors With Selectivity for NTPDase2 and NTPDase3 // Front. Pharmacol. 2020. V. 11. P. 1282. https://doi.org/10.3389/fphar.2020.01282
Bartlett R., Yerbury J.J., Sluyter R. P2X7 receptor activation induces reactive oxygen species formation and cell death in murine EOC13 microglia // Mediators Inflamm. 2013. V. 5. P. 271813. https://doi.org/10.1155/2013/271813
Bele T., Fabbretti E. P2X receptors, sensory neurons and pain // Curr. Med. Chem. 2015. V. 22. P. 845. https://doi.org/10.2174/0929867321666141011195351
Bell M.J., Kochanek P.M., Carcillo J.A. et al. Interstitial adenosine, inosine, and hypoxanthine are increased after experimental traumatic brain injury in the rat // J. Neurotrauma. 1998. V. 15. P. 163–70. 10.1089/neu.1998.15.163 PMID: 9528916
Bjelobaba I., Parabucki A., Lavrnja I. et al. Dynamic changes in the expression pattern of ecto-5'-nucleotidase in the rat model of cortical stab injury // J. Neurosci. Res. 2011. V. 89. P. 862. https://doi.org/10.1002/jnr.22599
Bjelobaba I., Stojiljkovic M., Lavrnja I. et al. Regional changes in ectonucleotidase activity after cortical stab injury in rat // Gen. Physiol. Biophys. 2009. V. 28 Spec No. P. 62.
Block M.L., Zecca L., Hong J.S. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms // Nat. Rev. Neurosci. 2007 Jan; 8(1): 57–69. https://doi.org/10.1038/nrn2038]
Boison D. Adenosine kinase: exploitation for therapeutic gain // Pharmacol. Rev. 2013. V. 65. P. 906. https://doi.org/10.1124/pr.112.006361
Bono M.R., Fernández D., Flores-Santibáñez F., Rosemblatt M., Sauma D. CD73 and CD39 ectonucleotidases in T cell differentiation: Beyond immunosuppression // FEBS Letters. V. 2015. V. 589. P. 3454. https://doi.org/10.1016/j.febslet.2015.07.027
Borea P.A., Gessi S., Merighi S., Vincenzi F., Varani K. Pharmacology of Adenosine Receptors: The State of the Art // Physiol. Rev. 2018. V. 98. P. 1591. https://doi.org/10.1152/physrev.00049.2017
Borea P.A., Gessi S., Merighi S., Varani K. Adenosine as a Multi-Signalling Guardian Angel in Human Diseases: When, Where and How Does it Exert its Protective Effects? // Trends Pharmacol. Sci. 2016. V. 37. P. 419. https://doi.org/10.1016/j.tips.2016.02.006
Braun M., Vaibhav K., Saad N.M. et al. White matter damage after traumatic brain injury: A role for damage associated molecular patterns // Biochim. Biophys. Acta Mol. Basis Dis. 2017. V. 1863(10 Pt B). P. 2614–2626. https://doi.org/10.1016/j.bbadis.2017.05.020
Braun N., Sévigny J., Robson S.C. et al. Assignment of ecto-nucleoside triphosphate diphosphohydrolase-1/cd39 expression to microglia and vasculature of the brain // Eur. J. Neurosci. 2000. V.12. P. 4357. PMID: 11122346
Bsibsi M., Ravid R., Gveric D., van Noort J.M. Broad expression of Toll-like receptors in the human central nervous system // J. Neuropathol. Exp. Neurol. 2002. V. 61. P. 1013. https://doi.org/10.1093/jnen/61.11.1013
Burnstock G., Boeynaems J.M. Purinergic signalling and immune cells // Purinergic Signal. 2014. V. 10. V. 529. https://doi.org/10.1007/s11302-014-9427-2
Burnstock G., Dumsday B., Smythe A. Atropine resistant excitation of the urinary bladder: the possibility of transmission via nerves releasing a purine nucleotide // Br. J. Pharmacol. 1972. V. 44. P. 451.
Burnstock G., Knight G.E. Cellular distribution and functions of P2 receptor subtypes in different systems // Int. Rev. Cytol. 2004; V. 240. P. 31. https://doi.org/10.1016/S0074-7696(04)40002-3 PMID: 15548415
Burnstock G. Physiopathological roles of P2X receptors in the central nervous system // Curr. Med. Chem. 2015. V. 22. P. 819–844. https://doi.org/10.2174/0929867321666140706130415
Burnstock G. Purine and pyrimidine receptors // Cell. Mol. Life Sci. 2007. V. 64. P. 1471. https://doi.org/10.1007/s00018-007-6497-0
Burnstock G. Purinergic Signaling in the Cardiovascular System // Circ. Res. 2017. V. 120. P. 207. https://doi.org/10.1161/CIRCRESAHA.116.309726
Burnstock G. Purinergic Signalling and Neurological Diseases: An Update // CNS Neurol. Disord. Drug Targets. 2017. V. 16. P. 257. https://doi.org/10.2174/1871527315666160922104848
Capecchi P.L., Rechichi S., Lazzerini P.E. et al. Cyclosporin and tacrolimus increase plasma levels of adenosine in kidney transplanted patients // Transpl. Int. 2005. V. 18. P. 289–95. https://doi.org/10.1111/j.1432-2277.2004.00036.x
Casella G., Garzetti L., Gatta A.T. et al. IL4 induces IL6-producing M2 macrophages associated to inhibition of neuroinflammation in vitro and in vivo // J. Neuroinflammation. 2016. V. 13. V. 139. https://doi.org/10.1186/s12974-016-0596-5
Chakravarty S., Herkenham M. Toll-like receptor 4 on nonhematopoietic cells sustains CNS inflammation during endotoxemia, independent of systemic cytokines // J. Neurosci. 2005. V. 25. P. 1788. https://doi.org/10.1523/JNEUROSCI.4268-04.2005
Choo A.M., Miller W.J., Chen Y.C. et al. Antagonism of purinergic signalling improves recovery from traumatic brain injury // Brain. 2013. V. 136(Pt 1). P. 65. https://doi.org/10.1093/brain/aws286
Cisneros-Mejorado A.J., Pérez-Samartín A., Domercq M. et al. P2X7 Receptors as a Therapeutic Target in Cerebrovascular Diseases // Front. Mol. Neurosci. 2020. V. 18. P. 92. https://doi.org/10.3389/fnmol.2020.00092
Clark R.S., Carcillo J.A., Kochanek P.M. et al. Cerebrospinal fluid adenosine concentration and uncoupling of cerebral blood flow and oxidative metabolism after severe head injury in humans // Neurosurgery. 1997 V. 41. P. 1284. https://doi.org/10.1097/00006123-199712000-00010
Cohen M.J., Brohi K., Ganter M.T., Manley G.T, Mackersie R.C, Pittet J.F. Early coagulopathy after traumatic brain injury: the role of hypoperfusion and the protein C pathway // J. Trauma. 2007. V. 63. P. 1254. https://doi.org/10.1097/TA.0b013e318156ee4c
Corps K.N., Roth T.L, McGavern D.B. Inflammation and neuroprotection in traumatic brain injury // JAMA Neurol. 2015. V. 72. P. 355. https://doi.org/10.1001/jamaneurol.2014.3558
Crain J.M., Nikodemova M., Watters J.J. Expressionof P2 nucleotide receptors varies with age and sex in murine brain microglia // J. Neuro. Inflammation. 2009; 6: 24. https://doi.org/10.1186/1742-2094-6-24
Cronstein B.N., Montesinos M.C, Weissmann G. Salicylates and sulfasalazine, but not glucocorticoids, inhibit leukocyte accumulation by an adenosine-dependent mechanism that is independent of inhibition of prostaglandin synthesis and p105 of NFkappaB // Proc. Natl. Acad. Sci. U S A. 1999. V. 96. P. 6377. https://doi.org/10.1073/pnas.96.11.6377
Cronstein B.N. Going with the flow: methotrexate, adenosine, and blood flow // Ann. Rheum. Dis. 2006. V. 65. P. 421. PMID: .https://doi.org/10.1136/ard.2005.04960116531550
Cunha R.A. Different cellular sources and different roles of adenosine: A1 receptor-mediated inhibition through astrocytic-driven volume transmission and synapse-restricted A2A receptor-mediated facilitation of plasticity // Neurochem. Int. 2008. V. 52. P. 65–72. https://doi.org/10.1016/j.neuint.2007.06.026
Das M., Mohapatra S., Mohapatra S.S. New perspectives on central and peripheral immune responses to acute traumatic brain injury // J. Neuroinflammation. 2012. V. 9. P. 236. https://doi.org/10.1186/1742-2094-9-236
Davalos D., Grutzendler J., Yang G. et al. ATP mediates rapid microglial response to local brain injury in vivo // Nat. Neurosci. 2005. V. 8. P. 752. https://doi.org/10.1038/nn1472
de Rivero Vaccari J.P., Lotocki G., Alonso O.F. et al. Therapeutic neutralization of the NLRP1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injury // J. Cereb. Blood Flow. Metab. 2009. V. 29. P. 1251. https://doi.org/10.1038/jcbfm.2009.46
Del Puerto A., Wandosell F., Garrido J.J. Neuronal and glial purinergic receptors functions in neuron development and brain disease // Front. Cell Neurosci. 2013. V. 7. P. 197. https://doi.org/10.3389/fncel.2013.0019
Deussing J.M., Arzt E. P2X7 receptor: a potential therapeutic target for depression? // Trends Mol Med. 2018. V. 24. P. 736. https://doi.org/10.1016/j.molmed.2018.07.005
Diaz-Arrastia R., Kochanek P.M., Bergold P. et al. Pharmacotherapy of traumatic brain injury: state of the science and the road forward: report of the Department of Defense Neurotrauma Pharmacology Workgroup // J. Neurotrauma. 2014. V. 31. P. 135. https://doi.org/10.1089/neu.2013.3019
Dinet V., Petry K.G., Badaut J. Brain-immune interactions and neuroinflammation after traumatic brain injury // Front. Neurosci. 2019. V. 13. P. 1178. https://doi.org/10.3389/fnins.2019.01178
Dixon A.K., Gubitz A.K., Sirinathsinghji D.J., Richardson P.J., Freeman T.C. Tissue distribution of adenosine receptor mRNAs in the rat // Br. J. Pharmacol. 1996. V. 118. P. 1461. https://doi.org/10.1111/j.1476-5381.1996.tb15561.x
Donat C.K., Scott G., Gentleman S.M., Sastre M. Microglial activation in traumatic brain injury // Front Aging Neurosci. 2017. V. 9. P. 208. https://doi.org/10.3389/fnagi.2017.00208
Donnelly-Roberts D., McGaraughty S., Shieh C.C., Honore P., Jarvis M.F. Painful purinergic receptors // J. Pharmacol. Exp. Ther. 2008. V. 324. P. 409. https://doi.org/10.1124/jpet.106.105890
Dorsett C.R., McGuire J.L., Niedzielko T.L. et al. Traumatic Brain Injury Induces Alterations in Cortical Glutamate Uptake without a Reduction in Glutamate Transporter-1 Protein Expression // J. Neurotrauma. 2017. V. 34. P. 220. https://doi.org/10.1089/neu.2015.4372
Dos Santos-Rodrigues A., Grane-Boladeras N., Bicket A., Coe I.R. Nucleoside transporters in the purinome // Neurochem. Int. 2014. V. 73. P. 229. https://doi.org/10.1016/j.neuint.2014.03.014
Doyle C., Cristofaro V., Sullivan M.P., Adam R.M. Inosine – a Multifunctional Treatment for Complications of Neurologic Injury // Cell. Physiol. Biochem. 2018. V. 49. P. 2293. https://doi.org/10.1159/000493831
Dunwiddie T.V., Masino S.A. The role and regulation of adenosine in the central nervous system. Annu // Rev. Neurosci. 2001. V. 24. P. 31. https://doi.org/10.1146/annurev.neuro.24.1.31
During M.J., Spencer D.D. Adenosine: A potential mediator of seizure arrest and postictal refractoriness // Ann. Neurol. 1992. V. 32. P. 618. https://doi.org/10.1002/ana.410320504
Effendi W.I., Nagano T., Kobayashi K., Nishimura Y. Focusing on Adenosine Receptors as a Potential Targeted Therapy in Human Diseases // Cells. 2020. V. 9. P. 785. https://doi.org/10.3390/cells9030785
Eltzschig H.K., Faigle M., Knapp S. et al. Endothelial catabolism of extracellular adenosine during hypoxia: The role of surface adenosine deaminase and CD26 // Blood. 2006. V. 108. P. 1602. https://doi.org/10.1182/blood-2006-02-001016
Emerich D.F., Dean R.L 3rd, Bartus R.T. The role of leukocytes following cerebral ischemia: pathogenic variable or bystander reaction to emerging infarct? // Exp. Neurol. 2002. V. 173. P. 168–81. https://doi.org/10.1006/exnr.2001.7835
Eser A., Colombel J.F., Rutgeerts P. et al. Safety and Efficacy of an Oral Inhibitor of the Purinergic Receptor P2X7 in Adult Patients with Moderately to Severely Active Crohn’s Disease: A Randomized Placebo-controlled, Double-blind, Phase IIa Study // Inflamm. Bowel Dis. 2015. V. 21. P. 2247. https://doi.org/10.1097/MIB.0000000000000514
Fang K.M., Yang C.S., Sun S.H., Tzeng S.F. Microglial phagocytosis attenuated by short-term exposure to exogenous ATP through P2X receptor action // J. Neurochem. 2009. V. 111. P. 1225-37. https://doi.org/10.1111/j.1471-4159.2009.06409.x
Farr S.A., Cuzzocrea S., Esposito E. et al. Adenosine A3 receptor as a novel therapeutic target to reduce secondary events and improve neurocognitive functions following traumatic brain injury // J. Neuroinflammation. 2020. V. 17. P. 339. https://doi.org/10.1186/s12974-020-02009-7
Fields R.D., Stevens B. ATP: an extracellular signaling molecule between neurons and glia // Trends Neurosci. 2000. V. 23. P. 625. https://doi.org/10.1016/s0166-2236(00)01674-x
Franco R., Fernandez-Suarez D. Alternatively activated microglia and macrophages in the central nervous system // Prog. Neurobiol. 2015. V. 131. P. 65. https://doi.org/10.1016/j.pneurobio.2015.05.003
Franke H., Krügel U., Illes P. P2 receptors and neuronal injury // Pflugers Arch. 2006. V. 452. P. 622. https://doi.org/10.1007/s00424-006-0071-8
Franke H., Schepper C., Illes P., Krugel U. Involvement of P2X and P2Y receptors in microglial activation in vivo // Purinergic Signal. 2007. V. 3. P. 435. https://doi.org/10.1007/s11302-007-9082-y
Ganesana M., Venton B.J. Early changes in transient adenosine during cerebral ischemia and reperfusion injury // PLoS ONE. 2018. 13:e0196932. https://doi.org/10.1371/journal.pone.0196932
Gebril H.M., Rose R.M., Gesese R. et al. Adenosine kinase inhibition promotes proliferation of neural stem cells after traumatic brain injury // Brain Commun. 2020. 2. fcaa017. https://doi.org/10.1093/braincomms/fcaa017
Gever J.R., Cockayne D.A., Dillon M.P., Burnstock G., Ford A.P. Pharmacology of P2X channels // Pflugers Arch. 2006. V. 452. P. 513–37. https://doi.org/10.1007/s00424-006-0070-9
Gilbert D.F., Stebbing M.J., Kuenzel K. et al. Store-Operated Ca2+ Entry (SOCE) and Purinergic Receptor-Mediated Ca2+ Homeostasis in Murine bv2 Microglia Cells: Early Cellular Responses to ATP-Mediated Microglia Activation // Front. Mol. Neurosci. 2016. V. 28. P. 111. https://doi.org/10.3389/fnmol.2016.00111
Giuliani A.L., Sarti A.C., Falzoni S., Di Virgilio F. The P2X7 Receptor-Interleukin-1 Liaison // Front. Pharmacol. 2017. V. 8. P. 123. ,https://doi.org/10.3389/fphar.2017.00123
Gompel J.J.V., Bower M.R., Worrell G.A. et al. Increased cortical extracellular adenosine correlates with seizure termination // Epilepsia. 2014. V. 55. P. 233. https://doi.org/10.1111/epi.12511
Gruenbacher G., Gander H., Rahm A. et al. The Human G Protein-Coupled ATP Receptor P2Y11 Is Associated With IL-10 Driven Macrophage Differentiation // Front. Immunoly. 2019. V. 10. P. 1870. https://doi.org/10.3389/fimmu.2019.018700
Gudkov S.V., Shtarkman I.N., Smirnova V.S., Chernikov A.V., Bruskov V.I. Guanosine and inosine as natural antioxidants and radioprotectors for mice exposed to lethal doses of gamma-radiation // Dokl. Biochem. Biophys. 2006. V. 407. P. 47. https://doi.org/10.1134/s1607672906020013
Halassa M.M., Fellin T., Haydon P.G. The tripartite synapse: roles for gliotransmission in health and disease // Trends Mol. Med. 2007. V. 13. P. 54. https://doi.org/10.1016/j.molmed.2006.12.005
Hanisch U.K., Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain // Nat. Neurosci. 2007. V. 10. P. 1387. https://doi.org/10.1038/nn1997
Haselkorn M.L., Shellington D.K., Jackson E.K. et al. Adenosine A1 receptor activation as a brake on the microglial response after experimental traumatic brain injury in mice // J. Neurotrauma. 2010. V. 27. P. 901. https://doi.org/10.1089/neu.2009.1075
Haskó G., Antonioli L., Cronstein B.N. Adenosine metabolism, immunity and joint health // Biochem. Pharmacol. 2018. V. 151. P. 307. https://doi.org/10.1016/j.bcp.2018.02.002
Haskó G., Sitkovsky M.V., Szabó C. Immunomodulatory and neuroprotective effects of inosine // Trends Pharmacol. Sci. 2004. V. 25. P. 152. https://doi.org/10.1016/j.tips.2004.01.006
Haydon P.G., Carmignoto G. Astrocyte control of synaptic transmission and neurovascular coupling // Physiol. Rev. 2006. V. 86. P. 1009. https://doi.org/10.1152/physrev.00049.2005
Haynes S.E., Hollopeter G., Yang G. et al. The P2Y12 receptor regulates microglial activation by extracellular nucleotides // Nat. Neurosci. 2006. V. 12. P. 1512. https://doi.org/10.1038/nn1805
Hazeldine J., Lord J.M., Belli A. Traumatic brain injury and peripheral immune suppression: primer and prospectus // Front. Neurol. 2015. V. 6. P. 235. https://doi.org/10.3389/fneur.2015.00235
Hirbec H., Rassendren F., Audinat E. Microglia Reactivity: Heterogeneous Pathological Phenotypes // Methods. Mol. Biol. 2019. V. 2034. P. 41. https://doi.org/10.1007/978-1-4939-9658-2_4
Horenstein A.L., Chillemi A., Zaccarello G. et al. A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes // Oncoimmunology. 2013. V. 2. e26246. https://doi.org/10.4161/onci.26246
Horenstein A.L., Quarona V., Toscani D. et al. Adenosine Generated in the Bone Marrow Niche Through a CD38-Mediated Pathway Correlates with Progression of Human Myeloma // Mo.l Med. 2016. V. 22. P. 694. https://doi.org/10.2119/molmed.2016.00198
Huber B.R., Meabon J.S., Hoffer Z.S. et al. Blast exposure causes dynamic microglial/macrophage responses and microdomains of brain microvessel dysfunction // Neuroscience. 2016. P. V. 319. 206. https://doi.org/10.1016/j.neuroscience.2016.01.022
Illes P., Rubini P., Ulrich H., Zhao Y., Tang Y. Regulation of Microglial Functions by Purinergic Mechanisms in the Healthy and Diseased CNS // Cells. 2020. V. 9. P. 1108. https://doi.org/10.3390/cells9051108
Jacobson K.A., Müller C.E. Medicinal chemistry of adenosine, P2Y and P2X receptors // Neuropharmacology. 2016. V. 104. P. 31. https://doi.org/10.1016/j.neuropharm.2015.12.001
Jarrahi A., Braun M., Ahluwalia M. et al. Revisiting Traumatic Brain Injury: From Molecular Mechanisms to Therapeutic Interventions // Biomedicines. 2020. V. 8. P. 389. https://doi.org/10.3390/biomedicines8100389
Jassam Y.N., Izzy S., Whalen M., McGavern D.B., El Khoury J. Neuroimmunology of Traumatic Brain Injury: Time for a Paradigm Shift // Neuron. 2017. V. 95. P. 1246. https://doi.org/10.1016/j.neuron.2017.07.010
Jeong S.Y., Jeon R., Choi Y.K. et al. Activation of microglial Toll-like receptor 3 promotes neuronal survival against cerebral ischemia // J. Neurochem. 2016. V. 136. P. 851. https://doi.org/10.1111/jnc.13441
Jin W., Xu W., Chen J., Zhang X., Shi L., Ren C. Adenosine kinase facilitated astrogliosis-induced cortical neuronal death in traumatic brain injury // J. Mol. Histol. 2016. V. 47. P. 259. https://doi.org/10.1007/s10735-016-9670-7
Karasawa A., Kawate T. Structural basis for subtype-specific inhibition of the P2X7 receptor // Elife. 2016. V. 5. e22153. https://doi.org/10.7554/eLife.22153
Karmakar M., Katsnelson M.A., Dubyak G.R., Pearlman E. Neutrophil P2X7 receptors mediate NLRP3 inflammasome-dependent IL-1β secretion in response to ATP // Nat. Commun. 2016. V. 7. P. 10555. https://doi.org/10.1038/ncomms10555
Kelley N., Jeltema D., Duan Y., He Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation // Int. J. Mol. Sci. 2019. V. 20. P. 3328. https://doi.org/10.3390/ijms20133328
Kempuraj D., Ahmed M.E., Selvakumar G.P. et al. Mast cell activation, neuroinflammation, and tight junction protein derangement in acute traumatic brain injury // Mediat. Inflamm. 2020. 2020. P. 4243953. https://doi.org/10.1155/2020/4243953
Keystone E.C., Wang M.M., Layton M., Hollis S., McInnes I.B. D1520C00001 Study Team. Clinical evaluation of the efficacy of the P2X7 purinergic receptor antagonist AZD9056 on the signs and symptoms of rheumatoid arthritis in patients with active disease despite treatment with methotrexate or sulphasalazine // Ann. Rheum. Dis. 2012. V. 71. P. 1630. https://doi.org/10.1136/annrheumdis-2011-143578
Kigerl K.A., Gensel J.C., Ankeny D.P., Alexander J.K., Donnelly D.J., Popovich P.G. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord // J. Neurosci. 2009. V. 29. P. 13435. https://doi.org/10.1523/JNEUROSCI.3257-09.2009
Kim E., Lauterbach E.C., Reeve A. et al. Neuropsychiatric complications of traumatic brain injury: a critical review of the literature (a report by the ANPA Committee on Research) // J. Neuropsychiatr. Clin. Neurosci. 2007. V. 19. P. 106. https://doi.org/10.1176/jnp.2007.19.2.106
Kim S.W., Davaanyam D., Seol S.I., Lee H.K., Lee H., Lee J.K. Adenosine Triphosphate Accumulated Following Cerebral Ischemia Induces Neutrophil Extracellular Trap Formation // Int. J. Mol. Sci. 2020. V. 21. P. 7668. https://doi.org/10.3390/ijms21207668
Kimbler D.E., Shields J., Yanasak N., Vender J.R., Dhandapani K.M. Activation of P2X7 promotes cerebral edema and neurological injury after traumatic brain injury in mice // PLoS One. 2012. 7. e41229. https://doi.org/10.1371/journal.pone.0041229
Koizumi H., Arito M., Endo W. et al. Effects of tofacitinib on nucleic acid metabolism in human articular chondrocytes. Mod. Rheumatol. 2015. V. 25 P. 522. https://doi.org/10.3109/14397595.2014.995874
Koizumi S., Ohsawa K., Inoue K., Kohsaka S. Purinergic receptors in microglia: Functional modal shifts of microglia mediated by P2 and P1 receptors // Glia. 2013. V. 61. P. 47. https://doi.org/10.1002/glia.22358
Kumar V. Toll-like receptors in the pathogenesis of neuroinflammation // J. Neuroimmunol. 2019. V. 332. P. 16. https://doi.org/10.1016/j.jneuroim.2019.03.012
Latini S., Pedata F. Adenosine in the central nervous system: release mechanisms and extracellular concentrations // J. Neurochem. 2001. V. 79. P. 463. https://doi.org/10.1046/j.1471-4159.2001.00607.x
Lazarowski E.R. Vesicular and conductive mechanisms of nucleotide release. Purinergic Signal. 2012. V. 8. P. 359. https://doi.org/10.1007/s11302-012-9304-9
Li J., Ramenaden E.R., Peng J. et al. Tumor necrosis factor α mediates lipopolysaccharide-induced microglial toxicity to developing oligodendrocytes when astrocytes are present // J. Neurosci. 2008. V. 28. P. 5321. https://doi.org/10.1523/JNEUROSCI.3995-07.2008
Lipmann F. Adv. Enzymol. 1941. V. 1. P. 99. https://doi.org/10.1002/9780470122464.ch4
Liu X., Zhao Z., Ji R. et al. Inhibition of P2X7 receptors improves outcomes after traumatic brain injury in rats // Purinergic Signal. 2017. V. 13. P. 529. https://doi.org/10.1007/s11302-017-9579-y
Luongo L., Guida F., Imperatore R. et al. The A1 adenosine receptor as a new player in microglia physiology // Glia. 2014. V. 62. P. 122. https://doi.org/10.1002/glia.22592
Lusardi T.A. Adenosine neuromodulation and traumatic brain injury // Curr. Neuropharmacol. 2009. V. 7. P. 228. https://doi.org/10.2174/157015909789152137
Markowitz C.E., Spitsin S., Zimmerman V. et al. The treatment of multiple sclerosis with inosine // J. Altern. Complement Med. 2009. V. 15. P. 619. https://doi.org/10.1089/acm.2008.0513
Martinez F.O., Sica A., Mantovani A., Locati M. Macrophage activation and polarization // Front. Biosci. 2008. V. 13. P. 453. https://doi.org/10.2741/2692
Matute C. P2X7 receptors in oligodendrocytes: a novel target for neuroprotection // Mol Neurobiol. 2008. V. 38. P. 123. https://doi.org/10.1007/s12035-008-8028-x
McInnes K., Friesen C.L., MacKenzie D.E., Westwood D.A., Boe S.G. Mild Traumatic Brain Injury (mTBI) and chronic cognitive impairment: A scoping review // PLoS One. 2017. V. 12. e0174847. https://doi.org/10.1371/journal.pone.0174847
Meng F., Guo Z., Hu Y. et al. CD73-derived adenosine controls inflammation and neurodegeneration by modulating dopamine signaling // Brain. 2019. V. 142. P. 700. https://doi.org/10.1093/brain/awy351
Merighi S., Battistello E., Giacomelli L. et al. Targeting A3 and A2A adenosine receptors in the fight against cancer // Expert Opin. Ther. Targets. 2019. V. 23. P. 669–678. https://doi.org/10.1080/14728222.2019.1630380
Milior G., Morin-Brureau M., Chali F. et al. Distinct P2Y Receptors Mediate Extension and Retraction of Microglial Processes in Epileptic and Peritumoral Human Tissue // J. Neurosci. 2020. V. 40. P. 1373. https://doi.org/10.1523/JNEUROSCI.0218-19.2019
Morabito L., Montesinos M.C., Schreibman D.M. et al. Methotrexate and sulfasalazine promote adenosine release by a mechanism that requires ecto-5'-nucleotidase-mediated conversion of adenine nucleotides // J. Clin. Invest. 1998. V. 101. P. 295–300. https://doi.org/10.1172/JCI1554
Morote-Garcia J.C., Rosenberger P., Kuhlicke J., Eltzschig H.K. HIF-1-dependent repression of adenosine kinase attenuates hypoxia-induced vascular leak. Blood. 2008. V. 111. P. 5571. https://doi.org/10.1182/blood-2007-11-126763
Morra M., Zubiaur M., Terhorst C., Sancho J., Malavasi F. CD38 is functionally dependent on the TCR/CD3 complex in human T cells // FASEB J. 1998. V. 12. P. 581. https://doi.org/10.1096/fasebj.12.7.581
Murray P.J., Allen J.E., Biswas S.K. et al. Macrophage activation and polarization: Nomenclature and experimental guidelines // Immunity. 2014. V. 41. P. 14. https://doi.org/10.1016/j.immuni.2014.06.008
Mutch C.A., Talbott J.F., Gean A. Imaging Evaluation of Acute Traumatic Brain Injury // Neurosurg. Clin. N. Am. 2016. V. 27. P. 409. https://doi.org/10.1016/j.nec.2016.05.011
Nakajima K., Tohyama Y., Maeda S., Kohsaka S., Kurihara T. Neuronal regulation by which microglia enhance the production of neurotrophic factors for GABAergic, catecholaminergic, and cholinergic neurons // Neurochem Int. 2007. V. 50. P. 807–20. https://doi.org/10.1016/j.neuint.2007.02.006
Nedeljkovic N., Bjelobaba I., Lavrnja I. et al. Early temporal changes in ecto-nucleotidase activity after cortical stab injury in rat // Neurochem. Res. 2008. V. 33. P. 873. https://doi.org/10.1007/s11064-007-9529-0
Nguyen R., Fiest K.M., McChesney J. et al. The International Incidence of Traumatic Brain Injury: A Systematic Review and Meta-Analysis // Can. J. Neurol. Sci. 2016. V. 43. P.774. https://doi.org/10.1017/cjn.2016.290
Niemelä J., Ifergan I., Yegutkin G.G., Jalkanen S., Prat A., Airas L. IFN-beta regulates CD73 and adenosine expression at the blood-brain barrier // Eur. J. Immunol. 2008. V. 38. P. 2718. https://doi.org/10.1002/eji.200838437
Nimmerjahn A., Kirchhoff F., Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo // Science. 2005. V. 308. P. 1314. https://doi.org/10.1126/science.1110647
Ohsawa K., Irino Y., Nakamura Y., Akazawa C., Inoue K., Kohsaka S. Involvement of P2X4 and P2Y12 receptors in ATP-induced microglial chemotaxis // Glia. 2007. V. 55. P. 604. https://doi.org/10.1002/glia.20489
Olson J.K., Miller S.D. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs // J. Immunol. 2004. V. 173. P. 3916. https://doi.org/10.4049/jimmunol.173.6.3916
Palmer C., Roberts R.L., Young P.I. Timing of neutrophil depletion influences long-term neuroprotection in neonatal rat hypoxic-ischemic brain injury // Pediatr. Res. 2004. V. 55. P. 549. https://doi.org/10.1203/01
Pawson A.J., Sharman J.L., Benson H.E. et al. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucleic Acids Res. 2014. 42(Database issue):D1098-106. https://doi.org/10.1093/nar/gkt1143
Pedata F., Melani A., Pugliese A.M., Coppi E., Cipriani S., Traini C. The role of ATP and adenosine in the brain under normoxic and ischemic conditions // Purinergic Signal. 2007. V. 3. P. 299. https://doi.org/10.1007/s11302-007-9085-8
Pedata F., Dettori I., Coppi E. et al. Purinergic signalling in brain ischemia // Neuropharmacology. 2016. V. 104. P. 105. https://doi.org/10.1016/j.neuropharm.2015.11.007
Perry V.H., Nicoll J.A., Holmes C. Microglia in neurodegenerative disease // Nat. Rev. Neurol. 2010. V. 6. P. 193. https://doi.org/10.1038/nrneurol.2010.17
Quarona V., Zaccarello G., Chillemi A. et al. CD38 and CD157: a long journey from activation markers to multifunctional molecules // Cytometry B Clin. Cytom. 2013. V. 84. P. 207. https://doi.org/10.1002/cyto.b.21092
Relja B., Land W.G. Damage-associated molecular patterns in trauma // Eur. J. Trauma Emerg. Surg. 2020. V. 46. P. 751. https://doi.org/10.1007/s00068-019-01235-w
Robertson C.L., Bell M.J., Kochanek P.M. et al. Increased adenosine in cerebrospinal fluid after severe traumatic brain injury in infants and children: association with severity of injury and excitotoxicity // Crit. Care Med. 2001. V. 29. P. 2287. https://doi.org/10.1097/00003246-200112000-00009
Roszek K., Czarnecka J. Is Ecto-nucleoside Triphosphate Diphosphohydrolase (NTPDase)-based Therapy of Central Nervous System Disorders Possible? // Mini-Reviews Medicinal Chemistry. 2015. V. 15. P. 5. https://doi.org/10.2174/1389557515666150219114416
Roth T.L., Nayak D., Atanasijevic T., Koretsky A.P., Latour L.L., McGavern D.B. Transcranial amelioration of inflammation and cell death after brain injury // Nature. 2014. V. 505. P. 223. https://doi.org/10.1038/nature12808
Ruhal P., Dhingra D. Inosine improves cognitive function and decreases aging-induced oxidative stress and neuroinflammation in aged female rats. // Inflammopharmacology. 2018. V. 26. P. 1317. https://doi.org/10.1007/s10787-018-0476-y
Schilling M., Strecker J.K., Ringelstein E.B., Schäbitz W.R., Kiefer R. The role of CC chemokine receptor 2 on microglia activation and blood-borne cell recruitment after transient focal cerebral ischemia in mice // Brain Res. 2009. V. 1289. P. 79. https://doi.org/10.1016/j.brainres.2009.06.054
Schneider M., Prudic K., Pippel A. et al. Interaction of Purinergic P2X4 and P2X7 Receptor Subunits // Front. Pharmacol. 2017. V. 8. P. 860. https://doi.org/10.3389/fphar.2017.00860
Sciotti V.M., Van Wylen D.G. Increases in interstitial adenosine and cerebral blood flow with inhibition of adenosine kinase and adenosine deaminase // J. Cereb. Blood. Flow Metab. 1993. V. 13. P. 201. https://doi.org/10.1038/jcbfm.1993.24
Sheth S., Brito R., Mukherjea D., Rybak L.P., Ramkumar V. Adenosine receptors: expression, function and regulation // Int. J. Mol. Sci. 2014 V. 15. P. 2024. https://doi.org/10.3390/ijms15022024
Simon D.W., McGeachy M.J., Bayır H., Clark R.S., Loane D.J., Kochanek P.M. The far-reaching scope of neuroinflammation after traumatic brain injury // Nat. Rev. Neurol. 2017. V. 13. P. 171. https://doi.org/10.1038/nrneurol.2017.13
Sipe G., Lowery R., Tremblay M.È. et al. Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex // Nat. Commun. 2016. V. 7. P.10905. https://doi.org/10.1038/ncomms10905
Solini A., Chiozzi P., Morelli A., Fellin R., Di Virgilio F. Human primary fibroblasts in vitro express a purinergic P2X7 receptor coupled to ion fluxes, microvesicle formation and IL-6 release. // J. Cell Sci. 1999. V. 112. P. 297. PMID: .9885283
Sluyter R., Dalitz J.G., Wiley J.S. P2X7 receptor polymorphism impairs extracellular adenosine 5′-triphosphate-induced interleukin-18 release from human monocytes // Genes Immun. 2004. V. 5. P. 588. https://doi.org/10.1038/sj.gene.6364127
Sofoluwe A., Bacchetta M., Badaoui M., Kwak B.R., Chanson M. ATP amplifies NADPH-dependent and -independent neutrophil extracellular trap formation // Sci. Rep. 2019. V. 9. P.16556. https://doi.org/10.1038/s41598-019-53058-9
Sperlágh B., Vizi E.S., Wirkner K., Illes P. P2X7 receptors in the nervous system // Prog. Neurobiol. 2006. V. 78. P. 327. https://doi.org/10.1016/j.pneurobio.2006.03.007
Spitsin S., Hooper D.C., Leist T., Streletz L.J., Mikheeva T., Koprowskil H. Inactivation of peroxynitrite in multiple sclerosis patients after oral administration of inosine may suggest possible approaches to therapy of the disease // Mult. Scler. 2001. V. 7. P. 313. https://doi.org/10.1177/135245850100700507
Stock T.C., Bloom B.J., Wei N. et al. Efficacy and safety of CE-224,535, an antagonist of P2X7 receptor, in treatment of patients with rheumatoid arthritis inadequately controlled by methotrexate // J. Rheumatol. 2012. V. 39. P. 720. https://doi.org/10.3899/jrheum.110874
Talley Watts L., Sprague S., Zheng W. et al. Purinergic 2Y1 receptor stimulation decreases cerebral edema and reactive gliosis in a traumatic brain injury model // J. Neurotrauma. 2013. V. 30. P. 55. https://doi.org/10.1089/neu.2012.2488
Taruno A. ATP release channels // Int. J. Mol. Sci. 2018. V. 19. P. 808. https://doi.org/10.3390/ijms19030808
Teixeira F.C., Gutierres J.M., Soares M.S.P. et al. Inosine protects against impairment of memory induced by experimental model of Alzheimer disease: a nucleoside with multitarget brain actions // Psychopharmacology (Berl). 2020. V. 237. P. 811. https://doi.org/10.1007/s00213-019-05419-5
Trautmann A. Extracellular ATP in the immune system: more than just a “danger signal” // Sci. Signal. 2009. V. 2. pe6. https://doi.org/10.1126/scisignal.256pe6
Vazquez J.F., Clement H.W., Sommer O., Schulz E., van Calker D. Local stimulation of the adenosine A2B receptors induces an increased release of IL-6 in mouse striatum: an in vivo microdialysis study // J. Neurochem. 2008. V. 105. P. 904. https://doi.org/10.1111/j.1471-4159.2007.05191.x
Veres G., Radovits T., Seres L., Horkay F., Karck M., Szabó G. Effects of inosine on reperfusion injury after cardiopulmonary bypass // J. Cardiothorac. Surg. 2010. V. 5. P. 106. https://doi.org/10.1186/1749-8090-5-106
Vespa P., Bergsneider M., Hattori N. et al. Metabolic crisis without brain ischemia is common after traumatic brain injury: a combined microdialysis and positron emission tomography study // J. Cereb. Blood. Flow Metab. 2005. V. 25. P. 763. https://doi.org/10.1038/sj.jcbfm.9600073
Vincenzi F., Pasquini S., Borea P.A., Varani K. Targeting Adenosine Receptors: A Potential Pharmacological Avenue for Acute and Chronic Pain // Int. J. Mol. Sci. 2020. V. 21. P. 8710. https://doi.org/10.3390/ijms21228710
von Kügelgen I. Pharmacological profiles of cloned mammalian P2Y-receptor subtypes // Pharmacol. Ther. 2006 V. 110. P. 415. https://doi.org/10.1016/j.pharmthera.2005.08.014
Walker K.R., Tesco G. Molecular mechanisms of cognitive dysfunction following traumatic brain injury // Front Aging Neurosci. 2013. V. 5. P. 29. doi: Mutch .https://doi.org/10.3389/fnagi.2013.00029
Xiang Z., Chen M., Ping J. et al. Microglial morphology and its transformation after challenge by extracellular ATP in vitro // J. Neurosci. Res. 2006. V. 83. P. 91. https://doi.org/10.1002/jnr.20709
Zarrinmayeh H., Territo P. Purinergic receptors of the Central Nervous System: Biology, PET Ligands, and Their Applications. // Molecular Imaging. 2020. V. 19. P. 1. https://doi.org/10.1177/1536012120927609
Zhou A.M., Li W.B., Li Q.J., Liu H.Q., Feng R.F., Zhao H.G. A short cerebral ischemic preconditioning up-regulates adenosine receptors in the hippocampal CA1 region of rats // Neurosci. Res. 2004. V. 48. V. 397. https://doi.org/10.1016/j.neures.2003.12.010
Zimmermann H. Extracellular metabolism of ATP and other nucleotides // Naunyn Schmiedebergs Arch Pharmacol. 2000. V. 362. P. 299. https://doi.org/10.1007/s002100000309
Дополнительные материалы отсутствуют.
Инструменты
Успехи физиологических наук