

Успехи физиологических наук, 2023, T. 54, № 1, стр. 55-69
Патогенез посттравматического стрессового расстройства, терапевтические мишени
М. С. Лапшин a, *, М. В. Кондашевская b, **, В. В. Епишев a, ***, Н. А. Паточкина a, ****
a Южно-Уральский государственный университет
454080 Челябинск, Россия
b Научно-исследовательский институт морфологии человека им. академика А.П. Авцына”
ФГБНУ “Российский научный центр хирургии им. академика Б.В. Петровского”
117418 Москва, Россия
* E-mail: lapshin1982@yandex.ru
** E-mail: actual_probl@mail.ru
*** E-mail: epishevvv@susu.ru
**** E-mail: patochkinana@susu.ru
Поступила в редакцию 10.06.2022
После доработки 05.07.2022
Принята к публикации 20.08.2022
- EDN: GXGBAF
- DOI: 10.31857/S0301179823010058
Аннотация
В обзоре обобщены современные сведения литературы о механизмах патогенеза тяжелого, индуцируемого стрессом заболевания – посттравматического стрессового расстройства (ПТСР). Охарактеризованы происходящие при ПТСР гормональные, биохимические, генетические и морфофункциональные изменения в периферических органах и в ЦНС. Выяснилось, что у большинства исследователей сформировалось мнение о ведущей роли хронического воспаления при ПТСР. Приведены данные изучения действия противовоспалительных средств, имеющих узкую биохимическую направленность. Обзор завершается представлением гипотезы о том, что патогенез ПТСР следует рассматривать как интегративный воспалительный процесс периферических и центральных систем. Терапевтическим средством в таком случае, вероятнее всего, должно быть полифункциональное лекарственное средство. Судя по результатам экспериментов авторов, вероятнее всего это должны быть препараты фармакологической группы гепаринов.
ВВЕДЕНИЕ
Нервно-психические расстройства превратились в глобальную мировую проблему, затрагивающую каждого четвертого человека. Посттравматическое стрессовое расстройство (ПТСР) представляет собой тяжелый, инвалидизирующий синдром, который индуцируется экстраординарными стрессовыми событиями. Дальнейшее развитие болезни обусловлено активацией нейробиологических путей, связанных со стрессом. Существенной причиной сложности изучения механизмов патогенеза заболевания является достаточно длительный и неопределенный по времени период развития заболевания от момента стрессирования до проявления его симптомов. В 50% случаев симптомы ПТСР редуцируются в первые два года после травматических событий. Затем процесс выздоровления замедляется. Через шесть лет в трети случаев заболевание переходит в хроническую форму течения. У пострадавших от травматических событий впоследствии диагностируются тревожные, депрессивные и соматоформные расстройства, развивающиеся коморбидно с ПТСР или самостоятельно, вызывая профессиональную и социальную дезадаптацию с устойчивыми изменениями личностных черт [4, 58].
Наиболее часто ПТСР рассматривается как психотравмирующее последствие пребывания в боевой обстановке. Не менее актуальными пусковыми инцидентами являются стихийные, техногенные и социально-экономические катастрофы. К этому списку также следует добавить пандемию 2019–2022 гг., вызванную новым коронавирусом SARS-CoV-2, характеризующимся высоким уровнем контагиозности и смертности от заболевания, названного COVID-19 [24]. ПТСР представляет собой психическое состояние, отличающееся проявлением стойких тревожно-депрессивных, тревожно-фобических, обсессивных нарушений с формированием избегающего поведения, наличием многолетних переживаний психотравмирующей ситуации. Указанные симптомы всегда сопровождаются депрессией и/или тревогой [4, 58]. Важным шагом, обобщающим все многообразие клинических проявлений на угрожающие события, стало выделение особой диагностической группы – “посттравматические стрессовые расстройства” в Diagnostic and Statistical Manual of Mental Disorders-III (DSM-III, Диагностическое и Статистическое Руководство по Психическим Расстройствам). Основные характеристики признаков и симптомов ПТСР были разработаны M.J. Horowitz [42, 43]. В последующем эти характеристики ПТСР были внесены в МКБ-10 (Международная статистическая классификация болезней и проблем, связанных со здоровьем) в группу расстройств “Реакция на тяжелый стресс и нарушения адаптации”.
В мае 2019 г., в МКБ-11, была выделена особая категория расстройства – “комплексное посттравматическое расстройство” (КПТСР), отличающееся от (ПТСР) по ряду специфических характеристик [3, 30]. На современном этапе МКБ-11 включает два различных родственных состояния – посттравматическое стрессовое расстройство (ПТСР) (код 6B40) и комплексное ПТСР (КПТСР) (код 6B41), под общей категорией “Специфические расстройства, связанные со стрессом”. ПТСР характеризуется тремя группами симптомов, включая повторное переживание травмы, избегание травматических воспоминаний и постоянное чувство текущей угрозы, которое проявляется преувеличенным испугом, повышенной бдительностью. КПТСР – это более тяжелая форма ПТСР, отличающаяся от известного травматического синдрома пятью наиболее выраженными компонентами: флешбэками, токсичным чувством вины, пренебрежением своих интересов, внутренней критикой и социальной тревожностью [3, 30].
Для установления диагноза применяют международный опросник по травмам (ITQ), состоящий из 18 пунктов, содержащих 3 кластера симптомов, относящихся к ПТСР (повторное переживание, избегание и чувство угрозы) и 3 дополнительных кластера симптомов, относящихся к нарушению самоорганизации, типичных для КПТСР (аффективная дисрегуляция, нарушения в отношениях и негативная Я-концепция). Дополнительным инструментом для выявления симптомов КПТСР является “Структурированное интервью для расстройств в результате экстремального стресса” (SIDES), содержащее 40 вопросов, с помощью которых можно выявить критерии симптомов КПТСР. Используются и другие методы выявления КПТСР [3, 30]. Считается, что Большое депрессивное расстройство и Генерализированное тревожное расстройство в бóльшей степени связаны с КПТСР, чем с простой формой ПТСР. При установлении диагноза КПТСР, риск возникновения хронических заболеваний повышается примерно в два раза. В Великобритании выявлено, что КПТСР является более распространенным диагнозом, чем простая форма ПТСР. Тогда как исследования в Израиле установили, что простая форма ПТСР (9.0%) превышает КПТСР (2.6%) по распространенности. В то время, как исследования в США продемонстрировали другие показатели: ПТСР – 3.4% и КПТСР – 3.8% [4].
До настоящего времени не существует общепринятой теории механизмов патогенеза ПТСР и КПСР. Возможно, поэтому пока еще отсутствуют надежные методы лечения этих заболеваний [30]. Выделяют два основных метода лечения этих заболеваний: когнитивная терапия, эффективная в 40% случаев и фармакологическая терапия, которая еще менее действенна. До сих пор для коррекции ПТСР и КПТСР используются те же методы и препараты, которые применялись еще 30–40 лет назад. Сложность понимания патогенеза этих заболеваний заключается главным образом, в том, что в основе этих расстройств лежит широкий спектр механизмов, переплетение путей которых обычно приводит к декомпенсации различных систем и органов, порождая такие заболевания, как сердечно-сосудистые, желудочно-кишечные, аутоиммунные, эндокринные и др. Следовательно, ПТСР и КПТСР не ограничиваются психиатрической сферой и должны рассматриваться как системные заболевания [4, 58].
МЕХАНИЗМЫ ПАТОГЕНЕЗА ПОСТТРАВМАТИЧЕСКОГО СТРЕССОВОГО РАССТРОЙСТВА
Классическая концепция стресса связана с работами Ганса Селье, который выделял полезный, физиологический (эустресс), связанный со слабыми краткосрочными воздействиями и патологический (дистресс), вызванный сильными повреждающими воздействиями. Хотя патофизиологическая основа механизмов стресса до конца не изучена, данные постоянно показывают ключевую роль медиаторов активации гипоталамо-гипофизарно-адреналовой (ГГА) системы, индуцирующей выброс глюкокортикоидов и симпатоадреналовой (СА) системы, обусловливающей выброс адреналина и норадреналина при расстройствах, вызванных стрессом. При этом стрессоры дифференцированно влияют на ГГА и СА системы, а сила и результат реакции зависят от общего состояния гомеостаза организма – взаимодействия генов, состояния внутренней и окружающей среды, программирующих реакцию глюкокортикоидов, биогенных аминов и других биологически активных агентов [67, 75]. Реакция на стресс при ПТСР является хорошим примером такой дифференцированной реакции. Во-первых, общеизвестно, что симптомы ПТСР проявляются далеко не у всех субъектов (людей и животных), подвергавшихся одинаковому стрессу. То есть, выявляются устойчивые и неустойчивые к данному стрессу индивиды (особи) [59, 68]. Во-вторых, в то время, как большинство разновидностей стресса приводит к гиперактивации ГГА системы, развивающейся в результате десенситизации глюкокортикоидной отрицательной обратной связи, только при ПТСР формируется ее сенситизация. R. Yehuda и соавт. установили, что при ПТСР наблюдается инактивация ГГА системы, объясняющаяся развитием патологически быстро развивающейся отрицательной обратной связи в результате форсированного торможения выброса глюкокортикоидов – кортизола у людей и кортикостерона у лабораторных грызунов [75–77].
Глюкокортикоидные гормоны играют важную роль в развитии ПТСР. Неадекватное высвобождение глюкокортикоидов после стресса может привести к задержке восстановления и/или длительным нарушениям процессов интеграции памяти, нарушая обработку стрессовой информации. В работах, выполненных на людях, многие исследователи обнаружили, что более низкие уровни кортизола в остром периоде после травмы являются предиктором развития более тяжелых симптомов ПТСР [25, 71, 74, 78]. В большинстве экспериментальных работ популярным показателем развития ПТСР в настоящее время является снижение уровня глюкокортикоидных гормонов в крови [6, 62, 65, 80].
При моделировании ПТСР, в наших исследованиях было охарактеризовано морфофункциональное состояние надпочечников устойчивых и неустойчивых к стрессу крыс Вистар, обусловленному предъявлением запаха хищника (мочи кошки) [6]. У обоих фенотипов были выявлены признаки дисфункции адреналовых желез, выражающиеся в уменьшении толщины пучковой зоны коркового слоя. Морфологические изменения надпочечников сопровождались снижением концентрации кортикостерона и тестостерона в крови животных и в ткани коры надпочечников. Следовательно, наши данные позволяют конкретизировать причину снижения концентрации глюкокортикоидов при ПТСР, заключающуюся в редуцировании функциональной зоны коры надпочечников, клетки которой секретируют эти гормоны. Тем не менее, в отличие от неустойчивых животных, у устойчивого фенотипа крыс эти изменения были менее выражены. Необходимо подчеркнуть, что нами впервые было выявлено утолщение зоны, расположенной между клубочковой и пучковой зонами – интермедиальной зоны, а также высокие значения индекса функциональной активности стволовых клеток этой зоны у устойчивых к стрессу крыс. В совокупности полученные сведения позволяют сделать вывод, что у данного фенотипа крыс наблюдается восстановление морфофункционального состояния надпочечников, то есть продуктивная адаптация [6].
Эффект снижения уровня глюкокортикоидов при ПТСР во многом остается неразрешенной загадкой. Следуя классическим представлениям, можно было бы ожидать повышения уровня глюкокортикоидных гормонов, поскольку они являются необходимой частью механизма обратной связи и защиты организма, однако при ПТСР у людей и животных часто наблюдается низкий уровень глюкокортикоидных гормонов. Некоторые исследователи считают, что подавление секреции глюкокортикоидов при ПТСР происходит в результате нарушения провоспалительными цитокинами активности в системе ферментов 11β-гидроксистероиддегидрогеназ 1 и 2-го типа, участвующих в конвертации кортизона в кортизол и обратно [64, 66]. Эта гипотеза возникла на основании выявления тесной связи между признаками воспалительной реакции, такими как появление в кровотоке повышенных концентраций провоспалительных интерлейкинов (ИЛ-1, IL-6, TNF-a и др.) и снижением уровня глюкокортикоидов [64, 66]. Относительно недавно было доказано, что кроме хорошо известной реакции активации стрессором ГГА системы, запускающей быструю и интенсивную продукцию глюкокортикоидов, обнаружена стимуляция гипоталамуса провоспалительными интерлейкинами: IL-1, фактором некроза опухоли (TNF) и IL-6 [27]. Кроме того, установлено, что Toll-like рецепторы 2 и 4 (TLR2 и TLR4) в клетках коры надпочечников индуцируют продукцию глюкокортикоидов путем прямой активации [19]. В то же время известно, что глюкокортикоиды являются мощными регуляторами воспаления, способными подавлять афферентные сигнальные пути от многих сенсоров, тем самым угнетая продукцию медиаторов воспаления. Одновременно с этим, глюкокортикоиды воздействуют на гены, например, активируют гены, кодирующие ингибиторы TLR. Интересно отметить, что некоторые воспалительные сигналы могут отменять ряд ингибирующих эффектов глюкокортикоидов в отношении компонентов иммунной системы [37]. В связи с этим, появилась гипотеза, что глюкокортикоиды и компоненты воспалительных сигналов имеют систему механизмов взаиморегуляции, которая нарушается при ПТСР. В.П. Большаков и соавт. (2021) считают, что глюкокортикоиды в низких концентрациях способны сдвигать соотношение Т-хелперов/Т-супрессоров в сторону преимущественно Т-хелперной активности, индуцируя таким образом провоспалительный эффект [2].
Ранее воспалительной реакции придавали мало значения. Современные исследования позволили установить, что стресс-реакция, кроме традиционных систем (ГГА и симпатической), запускает реакцию воспаления, которая считается эволюционно закрепленной [37]. Резидентные клетки иммунной системы экспрессируют трансмембранные и цитоплазматические белки, которые функционируют как сенсоры “опасности”. Система сенсоров включает в себя рецепторы распознавания паттернов (PRR), рецепторы комплемента и Fc-рецепторы, расположенные на поверхности В-лимфоцитов, дендритных клеток, макрофагов, служащие для специфичного распознавания и связывания с фрагментом молекулы антитела, называемым Fc. PRR связывают патоген-ассоциированные молекулярные структуры (PAMP), которые являются эволюционно закрепленными молекулярными сигналами от микробов, сопряженными с повреждением молекулярными структурами (DAMP), являющимися эндогенными клеточными продуктами, которые высвобождаются при повреждении тканей. После активации PRR, тканевые макрофаги, тучные клетки и стромальные клетки начинают секретировать провоспалительные медиаторы, включая липидные медиаторы, вазоактивные амины, а также интерлейкины. Интерлейкины связываются со своими рецепторами на поверхности соседних клеток, что часто включает экспрессию еще большего количества цитокинов [37].
Способность глюкокортикоидов подавлять воспалительную реакцию широко известна и используется в клинической практике. В нормальной физиологической ситуации глюкокортикоиды тормозят все компоненты воспалительной реакции на всех стадиях ее развития, независимо от вызвавших ее причин. Избыточное, либо недостаточное содержание в крови глюкокортикоидов (независимо от причин сдвига), а также степень активации глюкокортикоидных и минералокортикоидных рецепторов вызывают ряд серьезных осложнений, в том числе воспаление [2]. Провоспалительные эффекты глюкокортикоидов в более высоких, чем в норме концентрациях были охарактеризованы в некоторых экспериментальных моделях при индуцировании стрессором повышения уровня эндогенного кортикостерона, или с применением экзогенного гормона до введения ЛПС. В обоих случаях провоспалительный ответ в виде повышения экспрессии мРНК, TNF-α, IL-1β и IL-6 наблюдался как в макрофагах печени, так и в клетках микроглии гиппокампа [31].
Рассмотренные сведения о возможном провоспалительном действии глюкокортикоидов как в низких, так и высоких концентрациях у хронически стрессированных животных, наводят на мысль о существовании противовоспалительного диапазона концентраций кортикостерона. Следует отметить, что глюкокортикоидные рецепторы (ГР) занимают центральное место в регуляции уровня экспрессии и механизмов действия глюкокортикоидов. В отсутствие гормона его рецептор удерживается в цитоплазме клетки в комплексе с несколькими молекулярными шаперонами (Hsp70 и Hsp90), а после связывания с гормоном, переходит в клеточное ядро, где взаимодействует со специфическими участками ДНК (GREs – glucocorticoid responsive elements) генов-мишеней глюкокортикоидов, что приводит к активации, либо репрессии этих генов [56]. На людях с ПТСР и на животных моделях показано, что одним из важных компонентов мультибелкового комплекса, необходимого для придания ГР гормон-связывающей конформации, удержания его в цитоплазме клетки и обеспечения защиты от протеолитической деградации, является белок FKBP5 (FKBP51, иммунофилин), связанный с шаперонами Hsp90 и Hsp70. В некоторых работах показано, что шапероны Hsp70 и Hsp90 способны активировать, либо инактивировать ГР [15]. При сцеплении гормона с рецептором, FKBP5 замещается другим иммунофилином – FKBP4 (FKBP52), после чего в комплексе с ГР перемещается в ядро клетки [51]. L. Herrmann и соавт. в работах на животных и на человеке обнаружили доказательства способности FKBP51 модулировать экспрессию своего антагониста FKBP52 [39]. Установлено, что высокие уровни FKBP51 связаны с резистентностью к ГР и снижением способности индивидуума (особи) справляться со стрессом. Кроме того, распространенные аллельные варианты гена FKBP5 обусловливают повышение риска развития таких аффективных расстройств, как ПТСР [23]. Индуцированная глюкокортикоидами активация ГР уже через 1.5 ч приводит к повышению экспрессии FKBP51 и отмене опосредованного ГР-подавления провоспалительного ответа. Более того, FKBP51 потенцирует сигнальный каскад NF-κB, контролирующий очень большую группу генов, отвечающих за процесс воспаления, пролиферацию клеток, апоптоз и являющийся компонентом других провоспалительных путей, например сигнального пути TNF-α и Toll-подобных рецепторов [46]. Все это означает, что различные факторы, запускающие воспаление, вызовут более сильный провоспалительный ответ в условиях повышенного уровня FKBP51. В целом ряде работ показано, что FKBP51 может выступать в качестве регулятора функции ГР, а комплекс ГР-FKBP51 может быть диагностическим биомаркером и потенциальной терапевтической мишенью для предотвращения или лечения ПТСР [45, 51, 63].
Хорошо известно, что ГР обнаруживаются практически во всех ядерных клетках, во всех органах человека и животных. Особенно высока плотность рецепторов в мозге, в частности в гиппокампе. Белок FKBP51 экспрессируется также во всех органах и клетках, следовательно, провоспалительный ответ может сформироваться как на периферии, так и в ЦНС. Термин “нейровоспаление”, вошедший в употребление в 1990-х г., можно определить как стереотипную неспецифическую реакцию нервной ткани, для которой характерна активация тканевых макрофагов (микроглии) и астроцитов, сопровождающаяся увеличением ими экспрессии провоспалительных цитокинов и активацией системы комплемента. Локальное контролируемое нейровоспаление может возникать в норме, оно важно для оптимизации нейропластичности при адаптации [16]. Однако, чаще нейровоспаление привлекает к себе внимание исследователей, занимающихся изучением патогенеза ПТСР, как патологический неконтролируемый хронический процесс. Нейровоспаление считается причиной депрессивно-тревожных расстройств и нарушений памяти, в первую очередь памяти о страхе. К проблеме поиска лекарственных средств подходят с разных сторон, в частности, в настоящее время ведутся работы по изучению влияния ряда веществ на комплекс ГР-FKBP51. H. Li и соавт. создали пептид, который разрушал связывание ГР-FKBP51, что обусловливало нормализацию поведенческих и молекулярных механизмов, измененных у стрессированных мышей [51]. J.J. Sabbagh и соавт., проведя скрининг 1280 фармакологически активных соединений, идентифицировали три соединения, которые восстанавливали опосредованное FKBP51 подавление активности ГР без непосредственной их активации. Выбранные соединения относились к антипаркинсоническим препаратам бензтропинового ряда, которые обладали способностью разрушать комплекс FKBP51/GR/Hsp90 [63].
Кроме указанных выше механизмов, провоспалительный ответ при ПТСР может быть индуцирован и другими эффектами глюкокортикоидов. Результаты экспериментов Q. Gong и соавт. показали, что при ПТСР кортикостерон способен негативно влиять на функции нейронов, нарушая энергетический обмен и физиологическую деятельность митохондрий. А именно: кортикостерон подавляет экспрессию белков NADH-дегидрогеназы (убихинон) (Nduf), включающего подмножество, комплекса убихинол–цитохром с-редуктазы (Uqcc) и митохондриального рибосомного белка (Mrp), тем самым значительно дезорганизуя процесс окислительного фосфорилирования, что, в конечном счете, обусловливает нарушение газообмена и энергообеспечение нейронов [35]. В работах других авторов обнаружился еще один митохондриальный эффект кортикостерона – способность снижать экспрессию гена Mrp. Предполагается, что этот эффект может быть вовлечен в механизмы тревожного поведения, депрессии и нарушение когнитивных функций [55]. Хорошо известно, что любой дефект процессов нормального функционирования митохондрий может привести к аномальной продукции активных форм кислорода, что индуцирует окислительный стресс. Стресс обусловливает высвобождение промежуточных продуктов метаболизма: АТФ и АДФ, продуктов цикла Кребса, кислорода, АФК, при этом состояние стресса поддерживается пуринергической передачей сигналов. В совокупности указанные нарушения вызывают апоптоз, некроз, усиление экспрессии провоспалительных интерлейкинов и, как результат, развитие нейровоспаления.
Как было указано выше, одновременно с ГГА системой, на стресс реагирует симпатоадреналовая (СА) система, индуцирующая выброс адреналина и норадреналина из клеток мозгового вещества надпочечников, а также из периферических нервных окончаний. Адренергические рецепторы катехоламинов присутствуют на иммунных клетках, и их активность зависит от сопутствующих изменений уровней этих нейротрансмиттеров. Катехоламины передают сигналы в первую очередь через β2-адренорецепторы. Установлено, что несколько генов, связанных с иммунным воспалительным ответом, сцеплены с реакцией на стресс, приводящей к развитию ПТСР [67]. Все новые и новые экспериментальные данные указывают на одновременную активацию провоспалительных сигнальных путей не только по оси ГГА, но также периферической и центральной ренин-ангиотензиновой системы (РАС). Еще одной причиной развития нейровоспаления при ПТСР является эксайтотоксичность – патологический процесс, возникающий при перепроизводстве основного возбуждающего нейромедиатора – глутамата. Установлено, что при ПТСР наблюдается, снижение уровня важнейшего тормозного нейромедиатора ЦНС – гамма-аминомасляной кислоты (ГАМК) и резкое возрастание уровня глутамата, обусловленное влиянием глюкокортикоидных гормонов [36]. Избыточный выброс в постсинаптическую щель глутамата индуцирует гиперактивацию постсинаптических ионотрофных рецепторов глутамата (NMDA и AMPA). Основной функцией этих рецепторов является регуляция содержания ионов кальция (Са2+), калия, натрия, хлора во внеклеточном и внутриклеточном пространстве. Результатом избыточной активации данных рецепторов является форсированный вход Са2+ в клетки и высвобождение этого медиатора из внутриклеточных депо с последующей стимуляцией активации ряда протеолитических ферментов (фосфолипаз, эндонуклеаз, протеаз), разрушающих цитозольные структуры. Указанные процессы обусловливают повышение синтеза оксида азота и возрастание процессов перекисного окисления липидов (ПОЛ) с последующим развитием окислительного стресса, нарушением синтеза нейротрофических факторов, что индуцирует запуск апоптоза и гибели клеток. Согласно классификации “Комитета по клеточной гибели” (Nomenclature Committee on Cell Death), следует различать типичные и нетипичные формы гибели клеток [48]. Для мозга типичными формами гибели клеток являются апоптоз, некроз и аутофагия. Эксайтотоксическую гибель нейронов относят к нетипичной форме гибели клеток, при которой наблюдаются как апоптоз, так и некроз, а в некоторых условиях аутофагия [48]. У гибнущих клеток происходит нарушение целостности клеточных мембран и неконтролируемое высвобождение продуктов клеточной гибели в межклеточное пространство. Это индуцирует воспалительный ответ окружающих тканей [1].
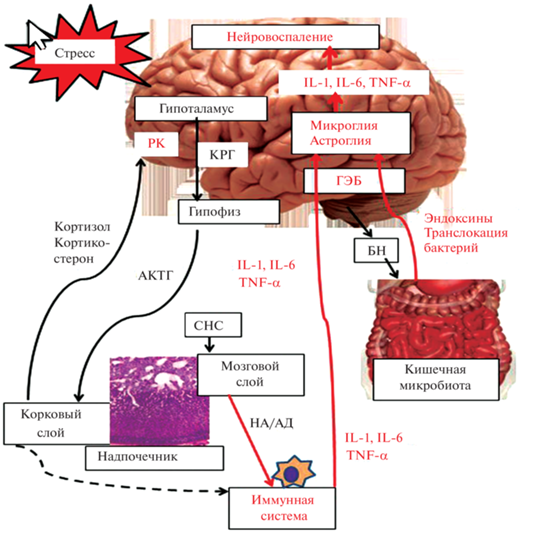
Понимание патогенеза ПТСР не может быть полным без изучения функциональной связи между кишечной микробиотой и функциями мозга. Еще со времен Г. Селье известно, что при стрессе изменяется морфофункциональное состояние ЖКТ. Ученые признали связь между мозгом и кишечником уже больше ста лет назад, но эту связь рассматривали в зависимости “сверху вниз” (мозг → ЖКТ → микробиота), тогда как в настоящее время исследователи рассматривают связь “снизу вверх” (микробиота → ЖКТ → мозг), то есть изучают влияние состояния ЖКТ с его микрофлорой на функции мозга [7, 32, 33]. Исследование оси микробиота → ЖКТ → мозг внесло свои коррективы в переосмысление патогенеза ПТСР. Предполагается, что состояние ЖКТ прямо, или косвенно влияет на настроение, когнитивные процессы и поведение, являющиеся ключевыми компонентами психического здоровья. В настоящее время установлено, что сообщения между мозгом и микробиотой ЖКТ двунаправлены через нервный, эндокринный и иммунный пути. Микробиота кишечника участвует в широком спектре физиологических процессов, включая прямое воздействие на мозг, например, путем производства нейроактивных метаболитов [21]. Дисбиоз микробиоты и связанное с этим повышение проницаемости стенки кишечника, отягчаемое иммунными проблемами, являются источником хронического вялотекущего воспаления, сопряженного с нервно-психическими расстройствами и нейровоспалением [26, 32]. В работах J.S. Bajaj и соавт. (2019) при изучении микробиоты ЖКТ ветеранов боевых действий с диагнозом ПТСР выявлено значительно большее количество патобионтов (Enterococcus и Escherichia/Shigella) и меньшее число аутохтонных родов (Lachnospiraceaeae и Ruminococcaceae). Измененное состояние микробиоты коррелировало с расстройствами когнитивных функций [14]. Несмотря на то, что механизмы, которые связывают дисбиоз кишечника с восприимчивостью к стрессу еще малоизученны, некоторые исследователи уже получили сведения о том, что можно передать поведенческий “тревожный” фенотип между двумя линиями мышей (BALB/c против NIH Swiss) посредством переноса фекальной микробиоты [17]. Концепция обеспечения устойчивости к стрессу посредством специфической модуляции кишечной микробиоты может стать многообещающим направлением для новых исследований лечения ПТСР. Предположительно, терапия будет направлена на увеличение разнообразия микробиоты (рис. 1).
Рис. 1.
Стресс-индуцированные механизмы развития нейровоспаления и воспаления при посттравматическом стрессовом расстройстве. КРГ – кортикотропин-рилизинг гормон, АКТГ – адренокортикотропный гормон, РК – рецепторы кортизола (кортикостерона), ГЭБ – гематоэнцефалический барьер, БН – блуждающий нерв, СНС – симпатическая нервная система, НА – норадреналин, АД – адреналин, IL-1, IL-6, TNF-α – провоспалительные интерлейкины. Сплошные стрелки – активирующие процессы, пунктирные стрелки – тормозные процессы, красные стрелки – процессы активации воспаления.
Следует отметить, что ЖКТ оснащен самой большой популяцией тучных клеток в организме млекопитающих. Тучные клетки (мастоциты) являются фундаментальными элементами кишечного барьера, так как они регулируют функции эпителия, его целостность и обновляемость, модулируют врожденную и адаптивную реакцию иммунного ответа слизистой оболочки, поддерживают нервно-иммунные взаимодействия, являющиеся ключевыми для функционирования ЖКТ [7, 13]. Поддержание гомеостаза слизистой оболочки мастоцитами происходит посредством регуляции обновляемости эпителия и продукции этими клетками IgA. Тучные клетки способствуют дифференцировке В-клеток в IgA-продуцирующие плазматические клетки в кишечном компартменте через продукцию IL-6, а также клеточно-клеточные взаимодействия – через путь CD40L–CD40. На базальном уровне проницаемость и миграция клеток парацеллюлярного и трансклеточного эпителия жестко регулируется протеазами, высвобождаемыми мастоцитами [7]. В экспериментальных работах показано, что снижение клеточной миграции и проницаемости эпителия вызывает изменение морфологии и функции ворсин. Жесткая регуляция кишечной проницаемости представляет собой центральный механизм в поддержании здоровья, профилактике и лечении многих заболеваний человека [13].
Тучные клетки вносят основной вклад в выработку иммунной толерантности слизистой оболочки, секретируя IL-9, который индуцирует выброс фермента – индоламиновой 2,3-диоксигеназы дендритными клетками. Кроме того, известно, что иммунная толерантность слизистой оболочки кишечника поддерживается влиянием IgG и его рецептора FcyRIIb, расположенного на мембране тучных клеток, на приобретение невосприимчивости к пищевым аллергенам, путем ингибирования продукции антител, опосредованной IgE, связывающимся с рецепторами FcεRI, также расположенными на тучных клетках [18]. Вместе с тем, стимулируя продукцию IgA В-клетками/плазматическими клетками через экспрессию IL-6 и CD40–CD40L, тучные клетки могут способствовать бактериальному гомеостазу. Нарушение функции мастоцитов в значительной степени вовлечено в возникновение и развитие многих воспалительных заболеваний ЖКТ. Имеются сведения, что модуляция микробиоты мастоцитами способствует нейропротекции ЦНС, сказывается на поведении и положительно влияет при нейродегенеративных процессах [7, 34, 61]. В работах P.F. Kuan и соавт. (2019) при изучении экспрессии генов, участвующих в активации и координации тучных клеток при ПТСР, обнаружено нарушение регуляции этих процессов [49]. Таким образом, управление микрофлорой кишечника и тучными клетками может стать новым методом лечения ПТСР.
МЕХАНИЗМЫ ИЗМЕНЕНИЯ ПОВЕДЕНИЯ И КОГНИТИВНЫХ ПРОЦЕССОВ ПРИ ПТСР
Ученые отмечают, что в основе формирования патофизиологических структурных преобразований в мозге и, соответственно, поведения, лежит гибель нейронов и нарушение нейрогенеза при ПТСР [29]. Исследования этих процессов послужили основой создания нейротрофической гипотезы нервно-психических расстройств, согласно которой изменение уровня мозгового нейротрофического фактора (BDNF) является ключевым механизмом формирования депрессивных расстройств и ПТСР. BDNF – это белок, принадлежащий к классу цитокинов, семейству факторов роста и подсемейству нейротрофинов, он выявляется в глиальных и нейрональных клетках. Повышение его активности, наблюдаемое при психотерапии или фармакотерапии у лиц с депрессией и ПТСР, коррелирует с улучшением памяти и обучения. BDNF играет до сих пор недооцененную, многофакторную роль, как регулятора, так и мишени передачи сигналов гормонов стресса в головной мозг. Современные исследования выявили новые данные о роли BDNF в поведенческой нейробиологии и нейропсихиатрии о консолидации и угасании памяти о страхе при ПТСР. Показано, что гормоны стресса взаимодействуют с эндогенной передачей сигналов на ген BDNF и его высокоаффинную рецепторную тропомиозин-родственную киназу B (TrkB), изменяя гомеостаз мозга. BDNF и TrkB в высокой степени экспрессируются в областях мозга, связанных с функциями памяти, включая гиппокамп, миндалевидное тело и префронтальную кору. Установлено, что глюкокортикоиды, воздействуя на BDNF, обусловливают усиление памяти о страхе. В исследованиях на людях и лабораторных грызунах показано, что при замене в BDNF одной единицы функционального полиморфизма Val66Met (rs6265) аминокислоты валина (Val) на метионин (Met) в кодоне 66, происходит нарушение внутриклеточного транспорта, негативное изменение памяти, появляется тревожность, нарушаются функции гиппокампа [28]. В результате была предложена гипотеза стресс-чувствительности BDNF, которая утверждает, что влияние на эндогенную активность таких факторов, как полиморфизм Val66Met BDNF, может способствовать управлению чувствительностью индивида (особи) к стрессу, травме и риску развития стресс-индуцированных расстройств [41, 57].
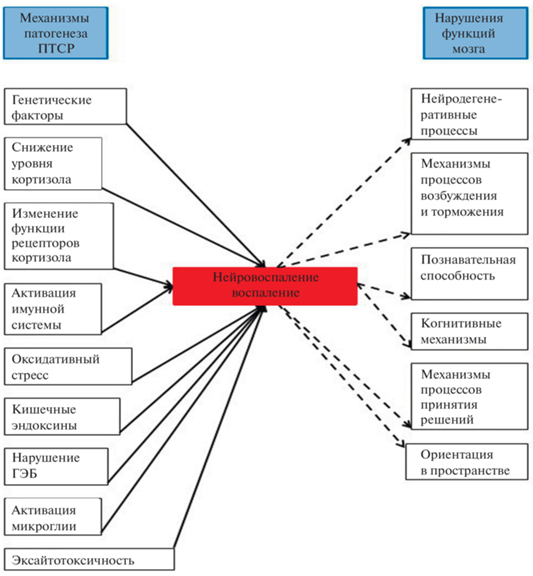
В предыдущей главе были охарактеризованы биохимические маркеры воспаления, развивающегося как на периферии, так и в ЦНС, играющего основополагающую роль в патогенезе ПТСР. Одним из важнейших эндогенных факторов, играющих центральную роль в запуске множества механизмов повреждения ЦНС, является наступающее на той или иной стадии патологического процесса повышение проницаемости гематоэнцефалического барьера (ГЭБ). В работах, проводимых на человеке и животных, C. Menard и соавт. показали, что при стрессе происходит снижение уровня эндотелиального белка плотных контактов Claudin-5, что приводит к интенсификации проницаемости периферического провоспалительного IL-6. В связи с этим, возникло предположение, что нарушение ГЭБ происходит в строго фиксированных областях, в которые клетки ЦНС привлекают IL-6, необходимый для синаптической пластичности зон, включившихся в патофизиологический процесс [54]. Это предположение подтвердилось в работах D.B. McKim и соавт., установивших, что тревожное поведение мышей, подвергавшихся стрессу, обусловлено активацией локальных зон микроглии, стимулирующих рекрутирование периферических моноцитов, экспрессирующих провоспалительный IL-1β [53]. При этом было определено, что активированная область микроглии избирательно усиливала экспрессию хемокина CCL2, привлекающего моноциты, которые прикреплялись к рецепторам IL-1R1 нейроваскулярных эндотелиальных клеток. Показано, что IL-1β, производимый рекрутированными моноцитами, усиливал тревожное поведение мышей, поскольку повторяющийся стресс активировал центры оценки угрозы в ЦНС мышей, что пространственно совпадало с зонами активации микроглии и эндотелиальной стимуляции рекрутирования моноцитов [53]. Накопление продуктов воспаления и токсинов в таких зонах, приводит к нейротоксичности, окислительному стрессу, нарушению выработки нейромедиаторов, расстройству межнейрональных и системных отношений. Эти процессы являются причиной нарушений выработки нейромедиаторов, уменьшения в объеме некоторых структур мозга, вызывая изменение поведения на тревожно/депрессивное и ухудшение когнитивных функций и других симптомов ПТСР (рис. 2) [29].
Рис. 2.
Связь воспаления с поведением и нарушениями функции ЦНС при посттравматическом стрессовом расстройстве. ГЭБ – гематоэнцефалический барьер. Сплошные стрелки – механизмы, приводящие к развитию воспаления, пунктирные стрелки – нарушения ЦНС, индуцируемые воспалением.
Изучая механизмы развития патогенеза ПТСР, исследователи наиболее часто отмечают нарушение в механизмах угашения памяти о стрессорных событиях [44, 72]. Прогресс в изучении патогенеза, профилактики и лечения ПТСР достигнут во многом благодаря моделированию этого расстройства на животных. В настоящее время стресс, обусловленный предъявлением запаха хищника (мочи кошки) лабораторным грызунам, считается наиболее адекватной моделью для изучения ПТСР-подобного состояния. В наших экспериментах по изучению поведенческих изменений при моделировании ПТСР и острого стресса (ОС), было установлено, что при ПТСР-подобном состоянии у крыс Вистар наблюдается снижение уровня кортикостерона в сыворотке крови, тогда как при ОС – повышение, что характерно для этих моделей [5, 70, 76]. Тестирование крыс в приподнятом крестообразном лабиринте (ПКЛ), считающемся одной из наиболее этологически “богатых” и чувствительных моделей, позволило установить, что по сравнению с контролем число заходов в открытые рукава ПКЛ и время нахождения в них было снижено как при ПТСР-подобном состоянии, так и при ОС, что расценивается как выраженный признак тревожно/депрессивного состояния [12]. Нюансы этого состояния нам удалось установить, применяя метод регистрации 23 разновидностей психоэмоциональных проявлений, демонстрируемых лабораторными грызунами во многих экспериментальных средах [9, 10, 47]. При моделировании ПТСР наблюдается увеличение числа контекстных реакций, свидетельствующее о затруднении при ориентации в пространстве. Поведенческое зацикливание, наблюдающееся при этом, вероятнее всего объясняется нарушениями функции механизмов ЦНС, участвующих в процессах концентрации внимания, а также принятия решения при выборе направления движения. Следует подчеркнуть, что аналогичными изменениями характеризовалось психоэмоциональное состояние людей с диагнозом ПТСР [11]. В отличие от крыс группы ПТСР, при моделировании ОС, обнаруживалось облегчение процесса ориентации в пространстве, что свидетельствует о сохранности и, даже повышенной активности отмеченных выше механизмов ЦНС. Имеются сведения, что в ряде случаев острый стресс способствует оптимизации механизмов памяти и ускорению процессов обучения, что свидетельствует о повышении концентрации внимания и оптимальной ориентации в пространстве. S. Уе и соавт. объясняют это влиянием кортикостерона на функции кислоточувствительных ионных каналов через посредство активации сигнального пути протеинкиназы С, которая выявляется в различных тканях млекопитающих, однако в мозге ее концентрация является наибольшей [73]. Обращал на себя внимание тот факт, что число активно оборонительных проявлений у группы животных ОС было не столь высоким, как при ПТСР-подобном состоянии.
Кроме изучения поведения крыс Вистар при моделировании ПТСР и ОС, нами были количественно охарактеризованы возбудительно-тормозные процессы ЦНС. В нашей работе установлено, что для ПТСР-подобного состояния было характерно резкое снижение всех показателей возбуждения (длительности, силы и интенсивности), а также интенсивности торможения, тогда как остальные показатели торможения (длительность и сила) значительно увеличивались. При остром стрессе остались незатронутыми показатели поведения, отражающие длительность возбуждения и мало измененными – силы возбуждения. Однако в целом, в нашем эксперименте при моделировании ПТСР и ОС, была выявлена значительная недостаточность в системе возбуждения и выраженная избыточность в системе торможения, интенсивность функционирования механизмов которых в значительной степени определяется соотношением уровней активности глутаматергических и ГАМКергических нейронов ЦНС, а также рецепторов соответствующих медиаторов [5]. Интерес к роли воспаления при нервно-психических расстройствах обусловил исследования, связанные с блокадой воспаления, как потенциальной стратегией лечения [69]. Наши эксперименты по использованию курсового введения нефракционированного гепарина (НФГ) при моделировании ПТСР продемонстрировали перспективность применения малых доз НФГ как полифункционального средства, нормализующего состояние возбудительно-тормозных механизмов ЦНС [8]. Поскольку хорошо известно, что гепарин обладает не только антикоагулянтным действием, но и хорошо выраженным противовоспалительным и многими другими эффектами [40]. Необходимо подчеркнуть, что введение НФГ при моделировании ПТСР-подобного состояния привело к нормализации сопряженной совокупности механизмов возбуждения и торможения, а введение гепарина контрольным крысам не отразилось на этих процессах. Среди возможных механизмов антистрессорного действия препаратов семейства гепаринов следует отметить результаты, полученные Renqi Li и соавт., которые установили, что низкомолекулярный гепарин (НМГ) при ЛПС-индуцированной активации глии в гиппокампе способствовал снижению уровня провоспалительного цитокина IL-1b, МДА (маркер ПОЛ и оксидативного стресса), Toll-подобного рецептора 4, индуцибельной циклооксигеназы-2, а также повышению уровня нейротрофического фактора мозга. Этими авторами установлено, что введение НМГ оказывало защитное действие от ЛПС-индуцированных гиппокамп-зависимых когнитивных нарушений [52]. Ряд исследователей считают, что механизмы, лежащие в основе изменений в ЦНС при ПТСР, во многом аналогичны ЛПС-индуцированным и связаны с уязвимостью к стрессу гиппокампа [22]. В нашей работе продемонстрирована перспективность использования малых доз гепарина в качестве эффективного средства при воздействии экстремальных факторов (рис. 3).
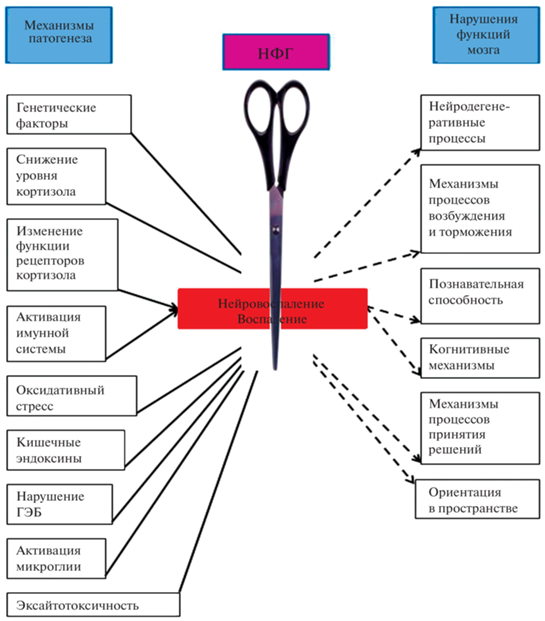
ЗАКЛЮЧЕНИЕ
Посттравматическое стрессовое расстройство (ПТСР) обычно развивается после перенесенной экстраординарной психологической, и/или тяжелой физической травмы, обусловливая у пациентов формирование инвалидизирующей тревоги и кошмарных воспоминаний. Хотя диагноз основывается на психопатологических критериях, ПТСР часто сопровождается сопутствующими соматическими заболеваниями и “ускоренным биологическим старением”. В данное время общепризнанно, что ПТСР представляет собой коморбидное заболевание со значительной степенью резистентности к лечению. Разработка новых, более эффективных терапевтических методов затруднена из-за значительного разнообразия подходов к изучению молекулярных механизмов, лежащих в основе ПТСР.
В настоящем обзоре обобщены современные концепции о молекулярных механизмах и предлагаемых терапевтических средствах борьбы с последствиями стрессового расстройства. Если ранее уделялось мало внимания воспалению, то в современных работах детально охарактеризованы биологически активные агенты, участвующие в развитии воспаления на периферии и в ЦНС. Поскольку ПТСР характеризуется избыточным или недостаточным уровнем глюкокортикоидных гормонов, которые считаются основными гормонами стресса, ингибирующими развитие воспаления, бóльшая часть исследователей занимается изучением биохимических компонентов, влияющих на экспрессию этих гормонов и активацию их рецепторов (ГР). Установлено, что про- или противовоспалительное действие глюкокортикоидов в значительной степени зависит от состояния и концентрации компонентов мультибелкового комплекса, необходимого для придания ГР гормон-связывающей конформации – иммунофилина FKBP51 и шаперонов Hsp90 и Hsp70. Показано, что комплекс ГР-FKBP51 может быть диагностическим биомаркером и потенциальной терапевтической мишенью для предотвращения или лечения ПТСР.
Исследователи, имеющие разную специализацию, выделяют различные терапевтические мишени, сходясь во мнении лишь в том, что при воспалении, как на периферии, так и в ЦНС (нейровоспаление) появляется избыточное количество провоспалительных цитокинов, сопровождающееся снижением уровня противовоспалительных цитокинов. Для нормализации содержания цитокинов, в качестве лекарственных препаратов предлагается множество монотерапевтических средств, имеющих сугубо узкую направленность действия.
В настоящем обзоре впервые представлен материал исследований, указывающих на специфическую локальную взаимосвязь структурных компонентов ЦНС и периферии, осуществляемую через региональные нарушения ГЭБ. В связи с этим, авторы обзора предлагают рассматривать патологический процесс при ПТСР преимущественно как воспалительный интегративный процесс периферических и центральных систем. Исходя из выдвинутой гипотезы, можно предположить, что терапевтическим средством лечения вероятнее всего, будет полифункциональный лекарственный препарат. Например, препараты фармакологической группы гепаринов, которые способны проходить через ГЭБ и обладают свойствами адаптогенов. Многочисленные свойства гепаринов были хорошо изучены при лечении коронавирусной инфекции COVID-19. В случае применения гепаринов в профилактических и, особенно, малых дозах, побочные негативные эффекты проявляются редко, либо вообще не проявляются.
Список литературы
Архипов В.И., Капралова М.В., Першина Е.В. Эксайтотоксичность и экспериментальные подходы к нейропротекции // Современные проблемы науки и образования. 2013. № 5. С. 486.
Большаков А.П., Третьякова Л.В., Квичанский А.А., Гуляева Н.В. Глюкокортикоиды в нейровоспалении гиппокампа: доктор Джекилл и мистер Хайд // Биохимия. 2021. Т. 86. Вып. 2. С. 186–199.
Кадыров Р.В., Венгер В.В. Комплексное посттравматическое стрессовое расстройство: современные подходы к определению понятия, этиологии, диагностика и психотерапия // Психолог. 2021. № 4. С. 45–60. https://doi.org/10.25136/2409-8701.2021.4.35811
Казенная Е.В. Современные зарубежные исследования посттравматического стрессового расстройства и его лечения эффективными психотерапевтическими методами у взрослых // Современная зарубежная психология. 2020. Т. 9. № 4. С. 110–119. https://doi.org/10.17759/jmfp.2020090410
Кондашевская М.В. Сравнительный анализ гормональных и поведенческих изменений в моделях посттравматического стрессового расстройства и остром стрессе // Российский физиологический журн. им. И.М. Сеченова. 2019. Т. 105. № 7. С. 879–887. https://doi.org/10.1134/S0869813919070045
Кондашевская М.В., Комелькова М.В., Цейликман В.Э. и др. Новые морфофункциональные критерии профиля устойчивости при моделировании посттравматического стрессового расстройства – триггера дисфункции надпочечников // Доклады РАН. Науки о Жизни. 2021. Т. 501. С. 28–33. https://doi.org/10.31857/S2686738921060056
Кондашевская М.В. Экосистема тучных клеток – ключевой полифункциональный компонент организма животных и человека. М.: Группа МДВ. 2019. 99 с. ISBN 978-5-906748-08-9.
Кондашевская М.В. Гепарин в модуляции основных свойств центральной нервной системы при экспериментальном посттравматическом стрессовом расстройстве. Новый взгляд на механизмы патогенеза и лечения // Бюллетень экспериментальной биологии и медицины. 2019. Т. 168. № 7. С. 12–16.
Кондашевская М.В., Цейликман В.Э., Манухина Е.Б. и др. Нарушение морфофункционального состояния надпочечников при экспериментальном посттравматическом стрессовом расстройстве у крыс: корреляция с поведенческими маркерами // Росс. физиол. журн. им. И.М. Сеченова. 2017. Т. 103. № 7. С. 808–818.
Никольская К.А., Шпинькова В.Н., Доведова Е.Л., Сергутина А.В., Герштейн Л.М. Типология познавательной деятельности в нейрохимических показателях мозга животных // Электрон. науч. журн. “Исследовано в России”. 2007. Т. 16. № 060207. С. 150–179.
Тушкова К.В., Бундало Н.Л. Реактивная и личностная тревожность у мужчин и женщин при посттравматическом стрессовом расстройстве различной степени тяжести. // Сибирское медицинское обозрение. 2013. Т. 3. № 81. С. 89–93.
Цейликман В.Э., Лапшин М.С., Комелькова М.В. и др. Динамика изменения содержания ГАМК, катехоламинов и активности МАО-А при экспериментальном посттравматическом стрессовом расстройстве у крыс // Росс. физиол. журн. им. И.М. Сеченова. 2018. Т. 104. № 2. С. 156–163.
Albert-Bayo M., Paracuellos I., Gonzlez-Cfstro A.M. et al. Intestinal mucosal mast cells: key modulators of barrier function and homeostasis // Cells. 2019. V. 8. № 2. P. E135. https://doi.org/10.3390/cells8020135
Bajaj J.S., Sikaroodi M., Fagan A., Heuman D., Gilles H., Gavis E.A., Fuchs M., Gonzalez-Maeso J., Nizam S., Gillevet P.M., Wade J.B. Posttraumatic stress disorder is associated with altered gut microbiota that modulates cognitive performance in veterans with cirrhosis // Am. J. Physiol. Gastrointest. Liver Physiol. 2019. V. 317(5). P. G661-G669. https://doi.org/10.1152/ajpgi.00194.2019
Baker J.D., Ozsan I., Ospina S.R., Gulick D., Blair L.J. Hsp90 heterocomplexes regulate steroid hormone receptors: from stress response to psychiatric disease // Int. J. Mol. Sci. 2019. V. 20. P. 79. https://doi.org/10.3390/ijms20010079
Bartsch T., Wulff P. The hippocampus in aging and disease: From plasticity to vulnerability // Neuroscience. 2015. V. 309. P. 1–16. https://doi.org/10.1016/j.neuroscience.2015.07.084
Bercik P., Denou E., Collins J., Jackson W., Lu J., Jury J. et al. The intestinal microbiota affect central levels of brain-derived neurotropic factor and behavior in mice // Gastroenterology. 2019. V. 141. P. 599–609, 609.e591–593.
Burton O.T., Tamayo J.M., Stranks A.J. et al. Allergen-specific IgG antibody signaling through FcγRIIb promotes food tolerance // J. Allergy Clin. Immunol. 2018. V. 141. № 1. P. 189–201.e3. https://doi.org/10.1016/j.jaci.2017.03.045
Cain D.W., Cidlowski J.A. Immune regulation by glucocorticoids // Nature Reviews Immunology. 2017. V. 17. P. 233–247. https://doi.org/10.1038/nri.2017.1
Carobrez A.P., Bertoglio L.J. Ethological and temporal analyses of anxiety-like behavior: the elevated plus-maze model 20 years on // Neurosci. Biobehav. Rev. 2005. V. 8. № 29. P. 1193–1205. https://doi.org/10.1016/j.neubiorev.2005.04.017
Cathomas F., Murrough J.W., Nestler E.J., Han M.H., Russo S.J. Neurobiology of Resilience: Interface Between Mind and Body Biol Psychiatry. 2019. V. 86(6). P. 410–420. https://doi.org/10.1016/j.biopsych.2019.04.011
Chao L.L., Tosun D., Woodward S.H., Kaufer D., Neylan T.C. Preliminary Evidence of Increased Hippocampal Myelin Content in Veterans with Posttraumatic Stress Disorder // Front. Behav. Neurosci. 2015. V. 9. P. 333. https://doi.org/10.3389/fnbeh.2015.00333
Criado-Marrero M., Rein T., Binder E.B., Porter J.T., Koren J. 3rd, Blair L.J. Hsp90 and FKBP51: complex regulators of psychiatric diseases // Philos. Trans. R Soc. Lond. B Biol. Sci. 2018. V. 373(1738). P. 20160532. https://doi.org/10.1098/rstb.2016.0532
d'Ettorre G., Ceccarelli G., Santinelli L., Vassalini P. et al. Post-Traumatic Stress Symptoms in Healthcare Workers Dealing with the COVID-19 Pandemic: A Systematic Review // Int. J. Environ. Res. Public. Health. 2021. V. 18. № 2. P. 601. https://doi.org/10.3390/ijerph18020601
Delahanty D., Raimonde A., Spoonster E. Initial posttraumatic urinary cortisol levels predict subsequent PTSD symptoms in motor vehicle accident victims // Biol. Psychiatry. 2000. V. 48. P. 940–947. https://doi.org/10.1016/S0006-3223(00)00896-9
Dodiya H.B., Forsyth C.B., Voigt R.M. et al. Chronic stress-induced gut dysfunction exacerbates Parkinson’s disease phenotype and pathology in a rotenone-induced mouse model of Parkinson’s disease // Neurobiol Dis. 2020. V. 135. P. 104352. https://doi.org/10.1016/j.nbd.2018.12.012
Dunn A.J. Cytokine activation of the HPA axis // Ann. N.Y. Acad. Sci. 2000. V. 917. P. 608–617. https://doi.org/10.1111/j.1749-6632.2000.tb05426.x
Egan M.F., Kojima M., Callicott J.H. et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function // Cell. 2003. V. 112. P. 257–269. https://doi.org/10.1016/s0092-8674(03)00035-7
Felger J.C. Imaging the Role of Inflammation in Mood and Anxiety-related Disorders // Curr Neuropharmacol. 2018. V. 16. № 5. P. 33–558. https://doi.org/10.2174/1570159X15666171123201142
Ford J.D, Courtois C.A. Complex PTSD and borderline personality disorder // Borderline Personal Disord Emot Dysregul. 2021. V. 8. № 1. P. 16. https://doi.org/10.1186/s40479-021-00155-9
Frank M.G., Miguel Z.D., Watkins L.R., Maier S.F. Prior exposure to glucocorticoids sensitizes the neuroinflammatory and peripheral inflammatory responses to E. coli lipopolysaccharide // Brain Behav. Immun. 2010. V. 24. P. 19–30. https://doi.org/10.1016/j.bbi.2009.07.008
Ganci M., Suleyman E., Butt H. et al. The role of the brain-gut-microbiota axis in psychology: The importance of considering gut microbiota in the development, perpetuation, and treatment of psychological disorders // Brain Behav. 2019. V. 9. № 11. P. e01408. https://doi.org/10.1002/brb3.1408
Gellhorn E. Hypothalamus, sino-aortic reflexes and activity of the gut // Acta. Neuroveg. (Wien). 1959. V. 19. № (3–4). P. 221–234. https://doi.org/10.1007/BF01227097
Girolamo F., Coppola C., Ribatti D. Immunoregulatory effect of mast cells influenced by microbes in neurodegenerative diseases // Brain Behav. Immun. 2017. № 65. P. 68–89. https://doi.org/10.1016/j.bbi.2017.06.017
Gong Q., Yan X.J., Lei F. et al. Proteomic profiling of the neurons in mice with depressive-like behavior induced by corticosterone and the regulation of berberine: pivotal sites of oxidative phosphorylation // Mol. Brain. 2019. V. 12. № 1. P. 118. https://doi.org/10.1186/s13041-019-0518-4
Groc L., Choquet D., Stephenson F. et al. NMDA receptor surface trafficking and synaptic subunit composition are developmentally regulated by the extracellular matrix protein Reelin // J. Neurosci. 2007. V. 27. № 38. P. 10165–75. https://doi.org/10.1523/JNEUROSCI.1772-07.2007
Guiducci C., Gong M., Xu Z. et al. TLR recognition of self-nucleic acids hampers glucocorticoid activity in lupus // Nature. 2010. V. 465. P. 937–941. https://doi.org/10.1038/nature09102
Hartmann J., Dedic N., Pöhlmann M.L. et al. Forebrain glutamatergic, but not GABAergic, neurons mediate anxiogenic effects of the glucocorticoid receptor // Mol. Psychiatry. 2017. V. 22. № 3. P. 466–475. https://doi.org/10.1038/mp.2016.87
Herrmann L., Ebert T., Rosen H., Novak B., Philipsen A., Touma C., Schreckenbach M., Gassen N.C., Rein T., Schmidt U. Analysis of the cerebellar molecular stress response led to first evidence of a role for FKBP51 in brain FKBP52 expression in mice and humans // Neurobiol. Stress. 2021. V. 15. P. 100401. https://doi.org/10.1016/j.ynstr.2021.100401
Hogwood J., Pitchford S., Mulloy B., Page C., Gray E. Heparin and non-anticoagulant heparin attenuate histone-induced inflammatory responses in whole blood // PLoS One. 2020. V. 15. № 5. P. e0233644. https://doi.org/10.1371/journal.pone.0233644
Hori H., Itoh M., Yoshida F. et al. The BDNF Val66Met polymorphism affects negative memory bias in civilian women with PTSD // Sci. Rep. 2020. V. 10. № 1. P. 3151. https://doi.org/10.1038/s41598-020-60096-1
Horowitz M., Becker S. Cognitive Response to Stressful Stimuli // Arch. Gen. Psychiatry. 1971. V. 25. № 5. P. 419-28. https://doi.org/10.1001/archpsyc.1971.01750170035007
Horowitz M., Wilner N., Kaltreider N., Alvarez W. Signs and Symptoms of Posttraumatic Stress Disorder // Archives of General Psychiatry. 1980. V. 37. № 1. P. 85–92. https://doi.org/10.1001/archpsyc.1980.01780140087010
Huang F.L., Li F., Zhang W.J. et al. Brd4 participates in epigenetic regulation of the extinction of remote auditory fear memory // Neurobiol. Learn Mem. 2021. V. 179. P. 107383. https://doi.org/10.1016/j.nlm.2021.107383
Jiang A., Zhou C., Samsom J., Yan S., Yu D.Z., Jia Z.P., Wong A.H.C., Liu F. The GR-FKBP51 interaction modulates fear memory but not spatial or recognition memory // Prog Neuropsychopharmacol Biol. Psychiatry. 2022. V. 119. P. 110604. https://doi.org/10.1016/j.pnpbp.2022.110604
Kästle M., Kistler B., Lamla T., Bretschneider T., Lamb D. et al. FKBP51 modulates steroid sensitivity and NFκB signalling: A novel anti"inflammatory drug target // Eur. J. Immunol. 2018. V. 48. 1904–1914. https://doi.org/10.1002/eji.201847699
Kondashevskaya M.V., Ponomarenko E.A. Features of behavioral changes accompanied by decreases in corticosterone levels in post-traumatic stress disorder. Experimental application of novel models and test methods // Neurosci. And Behav. Physiol. 2018. V. 48. № 5. P. 521–527.
Kroemer G., Galluzzi L., Vandenabeele P. et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009 // Cell Death Differ. 2009. V. 16. № 1. P. 3–11. https://doi.org/10.1038/cdd.2008.150
Kuan P.F., Yang X., Clouston S., Ren X., Kotov R., Waszczuk M., Singh P.K., Glenn S.T., Gomez E.C., Wang J., Bromet E., Luft B.J. Cell type-specific gene expression patterns associated with posttraumatic stress disorder in World Trade Center responders // Transl. Psychiatry. 2019. V. 9(1). P. 1. https://doi.org/10.1038/s41398-018-0355-8
Lee B., Shim I., Lee H., Hahm D.H. Effect of oleuropein on cognitive deficits and changes in hippocampal brain-derived neurotrophic factor and cytokine expression in a rat model of post-traumatic stress disorder // J. Nat. Med. 2018. V. 72. № 1. P. 44–56. https://doi.org/10.1007/s11418-017-1103-8
Li H., Su P., Lai T.K., Jiang A., Liu J., Zhai D., Campbell C.T., Lee F.H., Yong W., Pasricha S., Li S., Wong A.H., Ressler K.J., Liu F. The glucocorticoid receptor-FKBP51 complex contributes to fear conditioning and posttraumatic stress disorder // J. Clin. Invest. 2020. V. 130(2). P. 877–889. https://doi.org/10.1172/JCI130363
Li R., Tong J., Tan Y. et al. Low molecular weight heparin prevents lipopolysaccharide induced-hippocampus-dependent cognitive impairments in mice // Int. J. Clin. Exp. Pathol. 2015. V. 8. № 8. P. 8881–8891.
McKim D.B., Weber M.D., Niraula A., Sawicki C.M., Liu X., Jarrett B.L. et al. Microglial recruitment of IL-1beta-producing monocytes to brain endothelium causes stress-induced anxiety // Mol. Psychiatry. 2018. V. 23. P. 1421–1431. https://doi.org/10.1038/mp.2017.64
Menard C., Pfau M.L., Hodes G.E., Kana V., Wang V.X., Bouchard S. et al. Social stress induces neurovascular pathology promoting depression // Nature neuroscience. 2017. V. 20. P. 1752–1760. https://doi.org/10.1038/s41593-017-0010-3
Mouchiroud L., Houtkooper R.H., Moullan N., Katsyuba E., Ryu D., Cantó C. et al. The NAD+/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling // Cell. 2013. V. 154(2). P. 430. https://doi.org/10.1016/j.cell.2013.06.016
Meijsing S.H. Mechanisms of glucocorticoidregulated gene transcription // Adv. Exp. Med. Biol. 2015. V. 872. P. 59–81. https://doi.org/10.1007/978-1-4939-2895-8_3
Notaras M., van den Buuse M. Neurobiology of BDNF in fear memory, sensitivity to stress, and stress-related disorders // Mol. Psychiatry. 2020. V. 25. № 10. P. 2251–2274. https://doi.org/10.1038/s41380-019-0639-2
Oroian B.A., Ciobica A., Timofte D., Stefanescu C., Serban I.L. New Metabolic, Digestive, and Oxidative Stress-Related Manifestations Associated with Posttraumatic Stress Disorder // Oxid Med. Cell Longev. 2021. V. 2021. P. 5599265. https://doi.org/10.1155/2021/5599265
Osorio C., Probert T., Jones E. et al. Adapting to Stress: Understanding the Neurobiology of Resilience // Behav. Med. 2017. V. 43. № 4. P. 307–322. https://doi.org/10.1080/08964289.2016.1170661
Poterucha T.J., Libby P., Goldhaber S.Z. More than an anticoagulant: Do heparins have direct anti-inflammatory effects? // Thromb Haemost. 2017. V. 117. № 3. P. 437–444. https://doi.org/10.1160/TH16-08-0620
Renga G., Moretti S., Oikonomou V. et al. IL-9 and Mast Cells Are Key Players of Candida albicans Commensalism and Pathogenesis in the Gut // Cell Rep. 2018. V. 23. № 6. P. 1767–1778. https://doi.org/10.1016/j.celrep.2018.04.034
Ridker P.M. Inflammatory biomarkers and risks of myocardial infarction, stroke, diabetes, and total mortality: implications for longevity // Nutr. Rev. 2007. V. 65. № 12. Pt 2. S253–259. https://doi.org/10.1111/j.1753-4887.2007.tb00372.x
Sabbagh J.J., Cordova R.A., Zheng D., Criado–Marrero M., Lemus A., Li P., Baker J.D., Nordhues B.A., Darling A.L., Martinez-Licha C., Rutz D.A., Patel S., Buchner J., Leahy J.W., Koren J. 3rd, Dickey C.A., Blair L.J. Targeting the FKBP51/GR/Hsp90 Complex to Identify Functionally Relevant Treatments for Depression and PTSD // ACS Chem Biol. 2018. V. 13(8). P. 2288–2299. https://doi.org/10.1021/acschembio.8b00454
Sarapultsev A., Sarapultsev P., Dremencov E. et al. Low glucocorticoids in stress-related disorders: the role of inflammation // Stress. 2020. V. 23. № 6. P. 651–661. https://doi.org/10.1080/10253890.2020.1766020
Seetharaman S., Fleshner M., Park C.R., Diamond D.M. Influence of daily social stimulation on behavioral and physiological outcomes in an animal model of PTSD // Brain Behav. 2016. V. 6(5). P. e00458. https://doi.org/10.1002/brb3.458
Somvanshi P.R., Mellon S.H., Yehuda R. et al. Role of enhanced glucocorticoid receptor sensitivity in inflammation in PTSD: insights from computational model for circadian-neuroendocrine-immune interactions // Am. J. Physiol. Endocrinol. Metab. 2020. V. 319. № 1. E48–66. https://doi.org/10.1152/ajpendo.00398.2019
Sugama S., Kakinuma Y. Stress and brain immunity: Microglial homeostasis through hypothalamus-pituitary-adrenal gland axis and sympathetic nervous system // Brain Behav. Immun. Health. 2020. V. 7. P. 100 111. https://doi.org/10.1016/j.bbih.2020.100111
Tang W., Hu T., Hu B. et al. Prevalence and correlates of PTSD and depressive symptoms one month after the outbreak of the COVID-19 epidemic in a sample of home-quarantined Chinese university students // J. Affect Disord. 2020. V. 274. P. 1–7. https://doi.org/10.1016/j.jad.2020.05.009
Toft H., Lien L., Neupane S.P., Abebe D.S., Tilden T., Wampold B.E., Bramness J.G. Cytokine concentrations are related to level of mental distress in inpatients not using anti-inflammatory drugs // Acta Neuropsychiatr. 2020. V. 32(1). P. 23–31. https://doi.org/10.1017/neu.2019.36
Wang Q., Yu K., Wang J. et al. Predator stress-induced persistent emotional arousal is associated with alterations of plasma corticosterone and hippocampal steroid receptors in rat // Behav. Brain Res. 2012. V. 230. № 1. P. 167-74. https://doi.org/10.1016/j.bbr.2012.01.051
Witteveen A.B., Huizink A.C., Slottje P., Bramsen I., Smid T., Van Der Ploeg H.M. Associations of cortisol with posttraumatic stress symptoms and negative life events: A study of police officers and firefighters // Psychoneuroendocrinology. 2010. V. 35. P. 1113–1118. https://doi.org/10.1016/j.psyneuen.2009.12.013
Yabuki Y., Fukunaga K. Clinical Therapeutic Strategy and Neuronal Mechanism Underlying Post-Traumatic Stress Disorder (PTSD) // Int. J. Mol. Sci. 2019. V. 20. № 15. P. 3614. https://doi.org/10.3390/ijms20153614
Ye S., Yang R., Xiong Q. et al. Acute stress enhances learning and memory by activating acid-sensing ion channels in rats // Biochem. Biophys. Res. Commun. 2018. V. 498. № 4. P. 1078–1084. https://doi.org/10.1016/j.bbrc.2018.03.122
Yehuda R., Bierer L.M. Transgenerational transmission of cortisol and PTSD risk // Prog. Brain Res. 2008. V. 167. P. 121-35. https://doi.org/10.1016/S0079-6123(07)67009-5
Yehuda R., Flory J., Pratchett L. et al. Putative biological mechanisms for the association between early life adversity and the subsequent development of PTSD // Psychopharmacology. 2010. V. 212. № 3. P. 405-17. https://doi.org/10.1007/s00213-010-1969-6
Yehuda R., Koenen K., Galea S., Flory J. The role of genes in defining a molecular biology of PTSD // Disease Markers. 2011. V. 30. № 2–3. P. 67–76. https://doi.org/10.3233/DMA-2011-0794
Yehuda R., Neylan T., Flory J., McFarlane A. The use of biomarkers in the military: From theory to practice // Psychoneuroendocrinology. 2013. V. 389. P. 1912–1922. https://doi.org/10.1016/j.psyneuen.2013.06.009
Yehuda R., Seckl J. Minireview: Stress-related psychiatric disorders with low cortisol levels: a metabolic hypothesis // Endocrinology. 2011. V. 15212. P. 4496-503. https://doi.org/10.1210/en.2011-1218
Zass L.J., Hart S.A., Seedat S., Hemmings S.M., Malan–Müller S. Neuroinflammatory genes associated with post-traumatic stress disorder: implications for comorbidity // Psychiatr Genet. 2017. V. 27. № 1. P. 1–16. https://doi.org/10.1097/YPG.0000000000000143
Zoladz P.R., Del Valle C.R., Smith I.F., Goodman C.S., Dodson J.L., Elmouhawesse K.M., Kasler C.D., Rorabaugh B.R. Glucocorticoid Abnormalities in Female Rats Exposed to a Predator-Based Psychosocial Stress Model of PTSD // Front Behav. Neurosci. 2021. V. 15. P. 675 206. https://doi.org/10.3389/fnbeh.2021.675206
Дополнительные материалы отсутствуют.
Инструменты
Успехи физиологических наук