

Высокомолекулярные соединения (серия А), 2019, T. 61, № 1, стр. 67-81
ВЛИЯНИЕ КОМПЛЕКСА ЦИНК–ПОРФИРИН НА СТРУКТУРУ И СВОЙСТВА УЛЬТРАТОНКИХ ВОЛОКОН ПОЛИ(3-ГИДРОКСИБУТИРАТА)
С. Г. Карпова a, А. А. Ольхов b, c, А. В. Кривандин a, О. В. Шаталова a, А. В. Лобанов c, А. А. Попов b, А. Л. Иорданский c
a Институт биохимической физики им. Н.М. Эмануэля Российской академии наук
119991 Москва, ул. Косыгина, 4, Россия
b Российский экономический университет им. Г. В. Плеханова
117997 Москва, Стремянный пер., 36, Россия
c Институт химической физики им. Н.Н. Семенова Российской академии наук
119991 Москва, ул. Косыгина, 4, Россия
Поступила в редакцию 21.05.2018
После доработки 15.10.2018
Принята к публикации 29.10.2018
Аннотация
Изучено влияние малых добавок комплекса цинк–тетрафенилпорфирин (1–5%) на структуру и свойства ультратонких волокон на основе поли(3-гидроксибутирата). Использованы методы дифференциально-сканирующей калориметрии, рентгенодифракционного анализа, зондовый метод ЭПР, сканирующая электронная микроскопия, ИК-спектроскопия. Показано, что при добавлении в волокна порфиринового комплекса увеличивается доля паракристаллических структур и замедляется молекулярная подвижность в аморфных областях полимера, а степень кристалличности не меняется. Отжиг волокон при 140°C приводит к росту содержания паракристаллических структур и степени кристалличности волокон.
Фибриллярные полимерные носители биологически активных веществ в настоящее время находят широкое применение в биологии и медицине в качестве матриц пролонгированного действия, матриксов для клеточной инженерии, трансдермальных терапевтических систем, микрокапсулированных лекарственных форм и т.д. [1–3].
Один из эффективных экономически и наиболее технологичных способов создания фибриллярных матриц на основе ультратонких волокон – метод электростатического формования полимерных растворов и расплавов [2, 3]. Созданию биодеградируемых и биорезорбируемых нетканых волокнистых материлов посвящено много современных исследований [4]. Волокнистые нетканые материалы на основе биополимеров создают условия для свободной миграции и пролиферации клеток в трехмерном пространстве матрикса, что обеспечивает высокий уровень интегрирования материала с живыми тканями организма [5]. Среди биополимеров, применяющихся в стоматологии, травматологии, ортопедии, клеточной инженерии и хирургии, одним из самых распространенных является поли(3-гидроксибутират) (ПБГ) [6]. Этот биополимер, синтезируемый микроорганизмами, обладает высокой биосовместимостью, способностью к быстрой биорезорбции без образования токсичных продуктов и повышенной стойкостью к окислительной деструкции [7–9].
Как показали многочисленные исследования, морфология полимерного волокна существенно влияет на комплекс физико-механических и диффузионных свойств, кинетику биодеградации [10]. На надмолекулярную структуру волокна влияют не только молекулярные характеристики полимера, технологические параметры электроформования (концентрация полимера, температура и т.д.), но и введение в формовочный раствор веществ различной химической природы [11, 12]. Эти вещества благодаря наличию в них химически активных полярных функциональных групп вступают в межмолекулярное взаимодействие с биополимером. В результате такого взаимодействия может происходить как торможение, так и ускорение процессов кристаллизации, ориентации или релаксации полимерных макромолекул [13]. Вследствие изменения скорости данных процессов может изменяться геометрия волокон. Проблема формирования волокон определенной морфологии является одной из актуальных и практически значимых на сегодняшний день.
В наших работах мы рассматривали структурообразование волокнистых материалов на основе ПГБ, содержащих дипиридамол [14, 15], хитозан [16], наночастицы диоксида титана и кремния [17], комплексы железо (III)–хлорпорфирин [18]. Было показано влияние низкомоле-кулярных веществ на структуру кристаллической и аморфной фаз волокон ПГБ.
Комплексы металлов с тетрафенилпорфиринами обладают уникальными фотокаталитическими и антимикробными свойствами [19, 20]. В связи с этим создание нетканых высокопористых волокнистых биополимерных матриц для гигиены, терапии бактериальных инфекций и т.п. является практически важным направлением. Для формирования матриц с заданными свойствами необходимо установить взаимосвязь структуры низкомолекулярных комплексов, их концентрации в полимерном растворе со структурно-динамическими параметрами волокнистого материала.
Цель настоящей работы – изучение влияния комплекса цинк–тетрафенилпорфирин (ZnТФП) на надмолекулярную структуру ультратонких волокон ПГБ, получаемых методом электроформования, а также исследование действия на эти волокна тепла и озона как возможных факторов модификации их структуры и свойств.
ОБЪЕКТЫ И МЕТОДЫ ИССЛЕДОВАНИЯ
Для получения волокон ПГБ использовали природный биоразлагаемый полимер полигидроксибутират серии 16F, синтезированный микробиологическим методом компанией “Biomer ®” (Германия). Средневязкостная молекулярная масса ПГБ составляла 2.06 × 105, плотность ρ = = 1.248 г/см3, Тпл = 176°С, степень кристалличности ~63%.
В качестве модифицирующего вещества для создания волокнистых матриксов с антисептическими свойствами применяли комплекс цинка с тетрафенилпорфирином.
Готовили формовочные растворы ПГБ и ПГБ–ZnТФП в хлороформе при температуре 60°C с помощью автоматической магнитной мешалки. Концентрация ПГБ в растворе 7 мас. %, содержание ZnТФП 1, 3 и 5% от массы ПГБ. Волокна получали методом электроформования с помощью однокапиллярной лабораторной установки с диаметром капилляра 0.1 мм при напряжении электрического тока 12 кВ, расстоянии между электродами 18 см, электропроводности раствора 10 мкСм/см [21]. Исследованные в работе образцы представляли собой нетканый материал из этих волокон толщиной ~0.1–0.2 мм.
Рентгенодифракционное исследование структуры волокон ПГБ выполняли в Центре рентгенодифракционного анализа ИБХФ РАН. Интенсивность рентгеновского рассеяния измеряли “на просвет” в области малых и больших углов рассеяния на дифрактометре с линейным координатным (позиционно-чувствительным) детектором [22, 23] (излучение CuKα, расстояние образец–детектор 110 мм) и “на отражение“ по рентгенооптической схеме Брэгга–Брентано на дифрактометре HZG4 (“Freiberger Prisionsmechanik”, Германия) с графитовым монохроматором дифрагировавшего пучка (излучение CuKα).
Из интенсивности рентгеновского рассеяния, измеренной на дифрактометре с координатным детектором, вычитали интенсивность фонового рассеяния и получали “чистую” интенсивность рентгеновского рассеяния, которую использовали для расчета степени кристалличности образцов как в работе [24], проводя численное интегрирование интенсивностей в интервале S = 0.8–4.3 нм–1, где S = 2sin(θ)/λ, θ – половина угла рассеяния, λ – длина волны рентгеновского излучения (0.1542 нм для CuKα).
Средние размеры кристаллитов Lhkl рассчитывали из дифрактограмм, полученных методом Брэгга–Брентано (в этом случае инструментальное уширение дифракционных линий было значительно меньше, чем при измерении на дифрактометре с координатным детектором), по формуле Селякова–Шеррера
(1)
${{L}_{{hkl}}} = {\text{ }}\lambda {\text{/}}({{\beta }_{{hkl}}} \cdot {\text{cos}}{{\theta }_{{hkl}}})$Здесь Lhkl – средний размер кристаллитов в направлении, перпендикулярном кристаллографическим плоскостям (hkl); λ – длина волны рентгеновского излучения, βhkl и θhkl – интегральная ширина (в радианах в шкале 2θ), исправленная на инструментальное уширение, и половина угла рассеяния для дифракционной линии (hkl) соответственно. Интегральную ширину перекрывающихся дифракционных линий получали при помощи аналитического разложения экспериментальной интенсивности на сумму этих линий, используя программу EasyPlot for Windows ver. 2.22-5 (Spiral Software & MIT) и аппроксимируя профиль линий функциями псевдо-Войта (линейная комбинация функций Гаусса и Лоренца). Инструментальное уширение дифракционных линий учитывали с помощью дифракционной линии поликристаллического корунда (012).
При обработке малоугловых дифрактограмм после вычитания интенсивности фонового рассеяния вводили коллимационную поправку, как в работах [22, 23], и интенсивность умножали на S2 (поправка на разориентацию кристаллитов в образцах). Дифрактограммы анализировали модифицированным методом Цванкина как в работе [25], в результате чего получали среднюю толщину кристаллитов в направлении полимерных цепей и степень кристалличности в стопке кристаллитов (в фибрилле).
ЭПР-спектры (Х-диапазона) регистрировали на автоматизированном спектрометре ЭПР-В (Институт химической физики РАН, Москва). Микроволновая мощность в резонаторе не превышала 7 мВт, что позволяло избежать эффектов насыщения. При записи спектров амплитуда модуляции всегда была существенно меньше ширины резонансной линии и не больше 0.5 Гс. В качестве зонда использовали стабильный нитроксильный радикал ТЕМПО. Радикал вводили в волокна из газовой фазы при температуре 50°C. Концентрация радикала в полимере не превышала 10–3 моль/л. Экспериментальные спектры спинового зонда в области медленных движений (τ > > 10–10 с) анализировали в рамках модели изотропного броуновского вращения с помощью программы, описанной в работе [26]. При моделировании спектров применяли следующие главные значения g-тензора и тензора сверхтонкого взаимодействия радикала: gxx = 2.0096, gyy = 2.0066, gzz = 2.0025, Ахх = 7.0 Гc, Ayy = 5.0 Гc, Azz = 35.0 Гc. Величину Azz определяли экспериментально из ЭПР-спектров нитроксильного радикала в полимере при 77 К, она не сильно отличалась от значения, приведенного в работе [27].
Время корреляции вращения зонда τ в области быстрых вращений (5 × 10–11 < τ < 10–9 с) находили из спектров ЭПР по формуле [28]
(2)
$\quad\tau = {\text{ }}\Delta {{Н }_{ + }} \times [{{({{I}_{ + }}{\text{/}}{{I}_{--}})}^{{0.5}}}--1] \times 6.65 \times {{10}^{{--10}}},$Исследование образцов методом ДСК осуществляли на приборе DSC 204 F1 фирмы “Netzsch” в среде аргона при скорости нагревания 10 град/мин. Среднестатистическая ошибка измерения тепловых эффектов ±3%. Энтальпию плавления рассчитывали по программе “NETZSCH Proteus Thermal Analisis 4.8.4” по стандартной методике [29]. Пики разделяли с помощью программного обеспечения “NETZSCH Peak Sepa-ration 2006.01”. Вычисления проводили комбинированным методом Гаусса–Ньютона, в котором метод Marquardt соединен с оптимизацией длины шага итерации [30]. Ошибка составляет 2%.
Геометрию волокнистых материалов исследовали с помощью сканирующего электронного микроскопа “Hitachi TM-3000” (Япония) при ускоряющем напряжении 20 кВ. На поверхность образца нетканого волокнистого материала напыляли слой золота толщиной 100–200 Å.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Влияние концентрации ZnТФП на геометрические параметры волокна
В предыдущих работах [14–18] показано, что малые концентрации веществ различной химической природы влияют на геометрию полимерного волокна (диаметр, наличие утолщений по длине волокна). Форма волокна в первую очередь зависит от вязкости и электропроводности формовочного полимерного раствора. При добавлении в раствор сложного полиэфира (ПГБ) полярных веществ в малых концентрациях между полимером и данными веществами образуются водородные связи [16]. При этом меняются скорость кристаллизации, ориентации и релаксации полимерных макроцепей в процессе формирования твердого волокна из струи формовочного раствора [6]. На рис. 1 показано, что добавление в раствор ПГБ комплекса ZnТФП приводит к существенному изменению морфологии волокна. У волокна из ПГБ (рис. 1а) наблюдается чередование цилиндрических и веретенообразных участков. Наличие утолщений в структуре волокна может объясняться низкими значениями электропроводности и поверхностного натяжения полимерного формовочного раствора [17]. Средний диаметр цилиндрических участков волокна составляет 1–3 мкм, а веретеноподобные элементы имеют максмальный диаметр ~10 мкм и протяженность 20–30 мкм.
Рис. 1.
Микрофотографии волокнистых материалов ПГБ (а), ПГБ–1% ZnТФП (б), ПГБ–3% ZnТФП (в) и ПГБ–5% ZnТФП (г).

При добавлении в раствор 1–5% ZnТФП веретеноподобные элементы в структуре волокна пропадают полностью (рис. 1б, 1в). При введении 1% порфиринового комплекса появляются волокна со средним диаметром 1.5, 3.0 и 5.0 мкм. При повышении концентрации ZnТФП формируются волокна со средним диаметром 3 мкм, а волокна с диаметром 1.5 и 6.0 мкм практически полностью исчезают. Исчезновение веретеноподобных структур можно объяснить увеличением электропроводности формовочных растворов при введении полярных комплексов ZnТФП, проявляющих также парамагнитные свойства. С ростом проводимости раствора вытягивание капли раствора под действием электростатической силы происходит более интенсивно, что влечет за собой выравнивание волокон по толщине. Причиной возникновения волокон разного диаметра при электроформовании является эффект расщепления первичной струи формовочного раствора на несколько более тонких струй. В процессе электростатического формирования волокна из полимерного раствора с низкой электропроводностью в межэлектродном пространстве часто возникает коронный разряд. Коронный разряд, появляющийся на поверхности формируемой первичной струи прядильного раствора, уменьшает в ней концентрацию электрических зарядов. При добавлении в формовочный раствор электропроводящих частиц может возникнуть ток утечки, ослабляющий внешнее электрическое поле и приводящий к развороту первичной струи поперек силовых линий поля, что создает условия для расщепления первичной струи на множество дочерних [18]. На этой стадии процесса возможны еще и последовательные расщепления дочерних струй. Разделение струи на продольные элементы зависит от баланса вязкости, поверхностного натяжения и плотности электрического заряда в объеме струи [9]. При повышении концентрации комплекса возрастает вязкость формовочного раствора за счет взаимодействия полярных молекул комплекса с ПГБ. Поверхностное натяжение также увеличивается, и первичная струя практически не расщепляется. Диаметр волокон ПГБ при концентрации комплекса ZnТФП, равной 3–5%, составляет 3–4 мкм (рис. 1в).
Таким образом, можно полагать, что расщепление первичной струи, приводящее к образованию геометрических неоднородностей волокна, обусловлено неустойчивостью системы, когда концентрация полярных частиц в формовочном растворе достаточно мала (до 1%), и стабилизацией формовочной струи при росте концентрации ZnTФП.
Рентгенодифракционное исследование структуры волокон ПГБ–ZnТФП (большие углы рассеяния)
Дифрактограммы волокон ПГБ без ZnТФП и волокон, содержащих 1, 3 и 5% ZnТФП, полученные в области больших углов рассеяния “на просвет“, были идентичны. На рис. 2 приведены две такие дифрактограммы – для волокон ПГБ без ZnТФП (рис. 2а, кривая 1) и волокон, содержащих 5% ZnТФП (рис. 2б, кривая 1).
Рис. 2.
Дифрактограммы материала из волокон ПГБ без ZnТФП (а) и с 5% ZnТФП (б), полученные “на просвет” до отжига (1) и после отжига при 140°С в течение 3 ч (2). Интенсивности нормированы так, чтобы иметь одинаковые значения в минимумах между дифракционными максимумами.
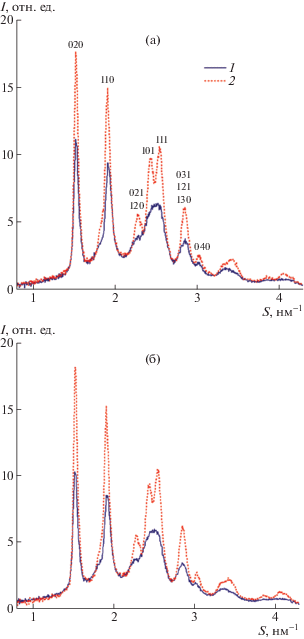
В результате отжига дифрактограммы образцов без ZnТФП и с ZnТФП изменялись одинаково (дифракционные линии становились более узкими и интенсивными) и были после отжига идентичными (рис. 2, кривые 2).
При исследовании материала из волокон ПГБ “на отражение” методом Брэгга–Брентано были получены дифрактограммы, похожие на дифрактограммы, снятые “на просвет”. На рис. 3 показаны такие дифрактограммы для волокон ПГБ без ZnТФП до и после отжига (кривые 1 и 2 соответственно). Дифрактограммы образцов, содержащих ZnТФП до и после отжига аналогичны дифрактограммам, приведенным на рис. 3.
Рис. 3.
Дифрактограммы материала из волокон ПГБ, полученные методом Брэгга– Брентано до отжига (1) и после отжига при 140°С в течение 3 ч (2).
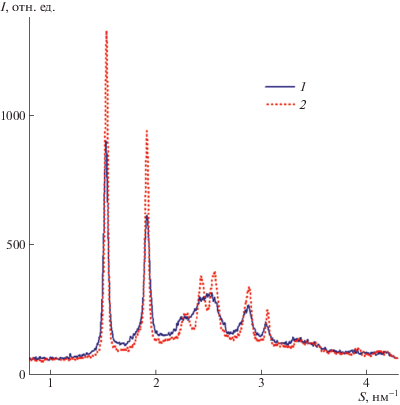
Сравнение дифрактограмм одних и тех же образцов, полученных “на просвет” (рис. 2) и “на отражение” (рис. 3), а также их сопоставление с дифрактограммой порошкового образца ПГБ [24], показывает, что исследованные образцы материала из волокон ПГБ не имеют выраженной ориентации (текстуры) кристаллитов. Некоторые различия в соотношении интенсивностей дифракционных максимумов на дифрактограммах, снятых “на просвет” и “на отражение”, скорее всего, объясняются разными угловыми факторами при этих методах съемки. Учитывая это обстоятельство и то, что волокна ПГБ, по данным сканирующей электронной микроскопии, лежат преимущественно в плоскости материала, можно сделать следующий вывод: кристаллиты в самих волокнах не имеют заметной преимущественной ориентации. Выраженную ориентацию кристаллитов ПГБ мы наблюдали раньше при исследовании пленок ПГБ, полученных из раствора ПГБ в хлороформе [31].
Положение дифракционных максимумов (линий) на дифрактограммах образцов материала из волокон ПГБ, как и в исследованных нами раньше порошковых образцах ПГБ фирмы “Biomer” [24] и пленках, полученных из раствора ПГБ в хлороформе [31], соответствует кристалличес-кой решетке этого полимера с орторомбической элементарной ячейкой (a = 0.576 нм, b = 1.320 нм, c = 0.596 нм, пространственная группа Р212121) [32].
В табл. 1 приведены параметры элементарной ячейки кристаллической решетки ПГБ, рассчитанные для исследованных образцов по угловому положению дифракционных линий (020), (110), (101) и (111) на дифрактограммах, полученных “на просвет” (рис. 2а). Для образцов, исследованных до отжига, параметр c не был определен по причине большой ширины и перекрытия дифракционных линий (101) и (111). Как следует из табл. 1, ни ZnТФП, ни отжиг существенно не влияют на параметры элементарной ячейки кристаллической решетки ПГБ.
Таблица 1.
Параметры элементарной ячейки (a, b, c) кристаллической решетки волокон ПГБ при разном содержании ZnТФП (точность ±0.002 нм)
| Содержание ZnТФП , % | a, нм | b, нм | c, нм | a, нм | b, нм | c, нм |
|---|---|---|---|---|---|---|
| до отжига | после отжига | |||||
| 0 | 0.571 | 1.315 | – | 0.573 | 1.316 | 0.590 |
| 1 | 0.571 | 1.314 | – | 0.572 | 1.313 | 0.593 |
| 3 | 0.571 | 1.315 | – | 0.572 | 1.314 | 0.592 |
| 5 | 0.571 | 1.313 | – | 0.572 | 1.313 | 0.594 |
Степень кристалличности, найденная из дифрактограмм, полученных “на просвет”, для всех образцов с точностью до 1% равна 45% до отжига и 58% после отжига (табл. 2). Отметим, что рассчитанная по дифрактограммам степень кристалличности может заметно отличаться от ее истинного значения [33]. Для нас было важно не столько определить истинные значения степени кристалличности исследуемых образцов, сколько проанализировать влияние на этот параметр добавок ZnТФП. Полученные результаты (табл. 2) однозначно свидетельствуют о том, что степень кристалличности волокон ПГБ не меняется при добавках 1, 3 и 5% ZnТФП и одинаково возрастает для всех образцов при отжиге.
Таблица 2.
Степень кристалличности волокон ПГБ при разном содержании ZnТФП по данным рентгеновской дифракции в больших углах (точность ±1%)
| Содержание ZnТФП, % | Степень кристалличности ξ, % | |
|---|---|---|
| до отжига | после отжига | |
| 0 | 45 | 58 |
| 1 | 45 | 59 |
| 3 | 45 | 58 |
| 5 | 44 | 58 |
Для анализа влияния добавки ZnТФП и отжига на размер кристаллитов в материале из волокон ПГБ были использованы дифрактограммы, полученные методом Брэгга–Брентано, поскольку инструментальное уширение дифракционных линий было значительно меньше, чем при измерениях “на просвет” на дифрактометре с координатным детектором. Результаты расчета средних размеров кристаллитов по дифракционным линиям (020), (101) и (111) показывают (табл. 3), что кристаллиты ПГБ в волокнах без ZnТФП и в волокнах, содержащих 5% ZnТФП, имеют до отжига одинаковые размеры: L020 = 26–27 нм, L101 и L111 примерно равны 5–6 нм (определить точнее L101 и L111 для этих образцов не представляется возможным в силу большой ширины и перекрытия дифракционных линий (101) и (111)). Средние размеры (толщину) кристаллитов δ в направлении оси с можно получить из величин Lhkl как δ = = Lhklcos(Ωhkl), где Ωhkl – угол между осью с и направлением, перпендикулярным плоскостям (hkl), который можно рассчитать, зная параметры кристаллической решетки ПГБ. Используя найденные значения L101, L111 (табл. 3) и Ω101 = 46°, Ω101 = 48.5° [31], приходим к среднему значению δ ≈ 4 нм.
Таблица 3.
Средние размеры кристаллитов Lhkl и средняя толщина кристаллитов в направлении полимерных цепей δ для волокон ПГБ по данным рентгеновской дифракции в больших углах
| Полимер | L020, нм (±2 нм) | L101, нм (±0.5 нм) | L111, нм (±0.5 нм) | δ, нм (±0.5 нм) |
|---|---|---|---|---|
| ПГБ | 26.1/39.3 | 5.6/9.4 | 5.6/10.3 | 3.8/6.7 |
| ПГБ–5%ZnТФП | 26.5/35.6 | 5.4/9.0 | 5.4/9.8 | 3.7/6.4 |
Примечание: Здесь и в табл. 4 в числителе – до отжига, в знаменателе – после отжига.
В результате отжига волокон ПГБ без ZnТФП и волокон, содержащих 5% ZnТФП, размеры кристаллитов увеличиваются примерно на одну и ту же величину: L020 до 36–39 нм, L101 и L111 до 9–10 нм. При этом толщина кристаллитов δ возраcтает примерно до 6–7 нм. Значения L020, L101, L111 и δ для отожженных образцов (табл. 3) близки к значениям, полученным в работе [24] для порошковых образцов ПГБ фирмы “Biomer”.
Малоугловое рентгенодифракционное исследование волокон ПГБ–ZnТФП
На малоугловых дифрактограмах волокон ПГБ без ZnТФП и волокон ПГБ, содержащих 1, 3 и 5% ZnТФП, наблюдается дифракционный максимум, которому соответствует большой период D = (Smax)–1, равный 5.4–5.7 нм (табл. 4). Для примера на рис. 4 приведены малоугловые дифрактограммы волокон ПГБ без ZnТФП (рис. 4а, кривая 1) и волокон ПГБ, содержащих 3% ZnТФП (рис. 4б, кривая 1). Близкое значение большого периода (6.1 нм) мы наблюдали раньше в ориентированных пленках ПГБ, полученных из раствора ПГБ в хлороформе [25].
Таблица 4.
Большой период D, среднее расстояние между центрами кристаллитов С, средняя толщина кристаллитов δ и степень кристалличности k для волокон ПГБ по данным малоугловой рентгеновской дифракции (точность ±5–7%)
| Содержание ZnТФП, % | D, нм | δ, нм | С, нм | k, % |
|---|---|---|---|---|
| 0 | 5.6/10.9 | 4.0/6.9 | 6.0/11.5 | 67/60 |
| 1 | 5.4/9.5 | 3.8/6.5 | 5.7/10.5 | 67/62 |
| 3 | 5.7/10.7 | 4.0/6.7 | 6.1/11.0 | 65/61 |
| 5 | 5.4/9.6 | 3.8/6.5 | 5.7/10.5 | 67/62 |
Рис. 4.
Малоугловые дифрактограммы материала из волокон ПГБ без ZnТФП (а) и содержащих 3% ZnТФП (б) до отжига (1) и после отжига при 140°С в течение 3 ч (2).
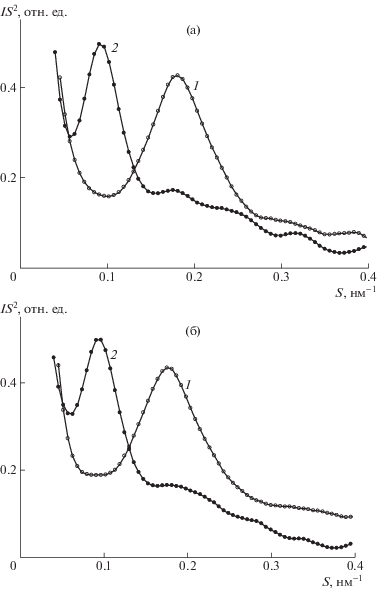
В результате отжига волокон ПГБ малоугловой дифракционный максимум сдвигается в сторону меньших углов рассеяния (рис. 4) и большой период D становится равным ~10 нм (табл. 4). Разброс полученных значений D для отожженных образцов (~15%) находится в пределах точности метода. На рис. 4 приведены малоугловые дифрактограммы отожженных волокон ПГБ без ZnТФП (рис. 4а, кривая 2) и волокон ПГБ с добавкой 3% ZnТФП (рис. 4б, кривая 2).
Результаты анализа малоугловых дифрактограмм методом Цванкина показывают (табл. 4), что для всех образцов толщина кристаллитов ПГБ до отжига составляет ~4 нм и возрастает при отжиге до ~7 нм. Эти значения находятся в согласии с результатами анализа ширины дифракционных линий в области больших углов рентгеновского рассеяния (табл. 3). Степень кристалличности образцов до и после отжига по данным малоугловой дифракции составляет 65–67% и 60–62% соответственно (табл. 4). Различия в этих значениях находятся в пределах точности метода. Отметим, что по данным рентгеновской дифракции в области больших углов степень кристалличности волокон ПГБ при отжиге изменяется от ~45 до ~58% (табл. 2). Различия в степени кристалличности, полученной в результате анализа рентгеновской дифракции в области больших и малых углов рассеяния, можно объяснить тем, что в первом случае степень кристалличности относится ко всему образцу, а во втором – только к тем областям, которые состоят из упорядоченных кристаллитов.
Термофизические характеристики кристаллической фазы композиций ПГБ–ZnТФП
Рассмотрим теперь влияние ZnТФП на кристаллическую фазу волокон ПГБ, используя метод ДСК (рис. 5). Следует отметить, что концентрация порфирина в аморфных областях полимера значительно выше, чем исходно заданные значения 1, 3 и 5%, поскольку эти значения рассчитаны на всю массу образца, а частицы ZnТФП находятся только в аморфной прослойке, доля которой уменьшается (как будет показано ниже) с ростом концентрации комплекса в полимере.
Рис. 5.
Термограммы плавления исходного ПГБ с 3% порфирина. Сплошная кривая – первое нагревание, штриховая – второе.
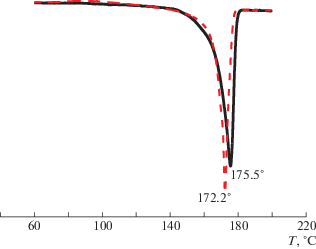
На рис. 6 представлены степени кристалличности образцов χ в зависимости от концентрации порфирина. Видно, что χ увеличивается при добавлении ZnТФП, причем более заметно при введении 1% порфирина. Термограммы как первого, так и второго сканирования имеют правильную симметричную форму, одну температуру плавления, что свидетельствует о достаточно однородной кристаллической фазе. Степени кристалличности приведены в табл. 5. Важно отметить, что величины χ, найденные методом ДСК, дают информацию как о доле кристаллической фазы, так и о паракристаллических структурах (структурах из выпрямленных цепей с двумерным порядком) в волокне. Полученные результаты свидетельствуют о том, что порфирин оказывает пластифицирующий эффект на кристаллизацию ПГБ в процессе электроформования волокна. При введении порфирина растет доля паракристаллических структур (содержание кристаллитов при этом, по данным рентгенодифракционного исследования, не меняется). Межмолекулярное расстояние и подвижность цепей при добавлении порфирина, увеличиваются, и процесс ориентации протекает все более эффективно. Важно отметить, что диапазон плавления кристаллитов лежит в области от 140–184°C.
Рис. 6.
Зависимость степени кристалличности χ от содержания порфирина в волокне: 1 – исходный образец, 2, 3 – образец после отжига при 140°C в течение 0.5 (2) и 3 ч (3).
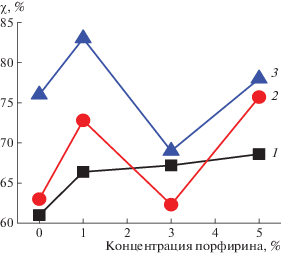
Таблица 5.
Степень кристалличности χ,% и температура плавления Тпл ультратонких волокон смесей ПГБ – ZnТФП по данным ДСК
| Смесь | χ, % | Тпл, °С |
|---|---|---|
| ПГБ | 61.0/60.3 | 175.5/172.3 |
| (63)/(55) | (176)/(172) | |
| (76.0)*/(61.4)* | (176)*/(172)* | |
| ПГБ–1% ZnТФП | 66.4/64.3 | 175.4/171.8 |
| (73)/ (65.7) | (174.2)/(171) | |
| (83)*/(66.4)* | (176)*/(171)* | |
| ПГБ–3% ZnТФП | 67.2/61.0 | 175.3/173.0 |
| (62.3)/(54.3) | (175)/(171) | |
| (68.6)*/(57.0)* | (176)*/(170)* | |
| ПГБ–5% ZnТФП | 68.6/67.2 | 177.0/172.6 |
| (75.7)/(64.3) | (176)/(172) | |
| (78.0)*/(65.7)* | (176)*/(170)* |
Волокна ПГБ имеют неравновесную структуру, и процесс кристаллизации в них незавершен. Для приведения системы в термодинамическое равновесие используют метод термического отжига при температурах ниже температуры плавления. Мы подвергали образцы отжигу при 140°C в течение 0.5 и 3 ч и затем резко охлаждали. В табл. 5 и на рис. 6 представлены полученные данные. В волокнах ПГБ и в волокнах ПГБ с 1% порфирина в ходе отжига степень кристалличности возрастает, причем тем сильнее, чем больше время отжига. Это соответствует данным рентгенодифракционного анализа (табл. 2), согласно которым степень кристалличности после отжига в течение 3 ч увеличивается от 45 до 58%. Для волокон ПГБ с 3% порфирина степень кристалличности снижается после отжига в течение 0.5 ч и повышается после 3 ч отжига. Термограмма для всех волокон имеет один симметричный пик плавления. Исключение составляет образец ПГБ–3% ZnТФП, термограмма которого содержит два пика плавления, что обусловлено, на наш взгляд, образованием паракристаллических структур с дефектами. Наличие двух пиков можно объяснить внедрением частиц порфирина в паракристаллические структуры, плавление которых начинается при 140°С. Порфирин проникает в паракристаллические структуры и фиксируется в них вследствие образования водородных связей между полярными молекулами ZnТФП и концевыми гидроксильными и карбонильными группами основной цепи ПГБ, а также вследствие резкого охлаждения волокна после отжига. Молекулы порфирина остаются в паракристаллических областях, что препятствует процессу уплотнения полимера, в результате степень кристалличности снижается. Наличие водородных связей между компонентами было нами ранее обнаружено в пленках на основе систем ПГБ–полиамид [34] и ПГБ–полиуретан [35]. Чем выше концентрация порфирина, тем в большей степени должны протекать процессы аморфизации паракристаллических областей. При введении 1% ZnТФП доля паракристаллических структур после отжига наиболее высокая, поскольку концентрация порфирина небольшая и, следовательно, доля деформированных порфирином паракристаллических областей низкая. Добавление 3% порфирина вызывает резкое снижение доли паракристаллических структур, после отжига в течение 0.5 и 3 ч, что обусловлено высокой долей деформированных порфирином паракристаллических областей. Однако при добавлении 5% ZnTФП наблюдается повышение χ (относительно образца ПГБ с 3% порфирина). По нашему мнению, такие изменения объясняются тем, что при больших концентрациях порфирин коалесцирует и образует кластеры большего размера. Вследствие этого проникновение порфирина в расплавленные паракристаллические области затруднено, и деформации порфирином подвергается более низкая доля паракристаллических структур. В результате отжиг волокон приводит к росту степени кристалличности и доли паракристаллических структур (рис. 6).
Совокупность результатов исследований особенностей геометрической структуры волокон и их термофизического поведения позволяет полагать, что при отсутствии порфирина в ПГБ образуются веретеноподобные неравновесные структуры. По мере повышения концентрации порфирина, эти структуры исчезают; цилиндрические тонкие волокна теперь характеризуются более совершенной и равновесной структурой с более высокой долей паракристаллических областей. Наиболее сильные изменения имеют место при добавлении 1% ZnTФП. Второе сканирование исследуемых волокон указывает на достаточно высокую скорость релаксации системы, о чем свидетельствуют узкие, симметричные пики плавления (рис. 5). Отжиг при 140°С обусловливает резкий рост доли кристаллитов и паракристаллических структур в волокне. Добавление в волокно 3% ZnTФП вызывает резкое снижение доли паракристаллических структур.
Динамические характеристики аморфной фазы ультратонких волокон ПГБ–ZnТФП
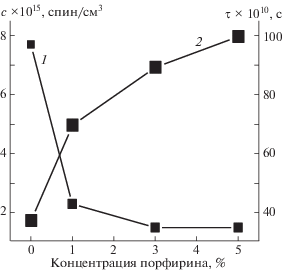
Структура аморфных областей в значительной степени определяется долей кристаллических и паракристаллических образований. При добавлении к ПГБ малого количества ZnТФП растет содержание паракристаллических структур, что приводит к изменению структурного и динамического состояния аморфных областей. Молекулярную динамику таких областей удобно исследовать методом ЭПР с помощью стабильных радикалов. ЭПР-спектры радикала ТЕМПО в матрице ПГБ имеют сложный вид и представляют собой суперпозицию двух спектров, соответствующих двум популяциям радикалов с различающимися временами корреляции τ1 и τ2, где τ1 характеризует молекулярную подвижность в более плотных, а τ2 в менее плотных аморфных областях (рис. 7). С использованием специальной программы NLSL, аналогичной процедуре, описанной ранее [26], произведена оценка соотношения концентраций различных по плотности областей и рассчитаны времена корреляции в плотных и рыхлых областях из экспериментальных спектров. Результаты таких расчетов представлены в табл. 6. На рис. 8 приведены спектры быстрой и медленной составляющих волокна ПГБ. Из данных табл. 6 видно, что с повышением концентрации порфирина в волокне доля плотных областей возрастает в ∼ 6 раз. Время корреляции в плотных областях также увеличивается. Причем наиболее резкое изменение τ с ростом концентрации порфирина происходит при добавлении 1% ZnТФП, при дальнейшем повышении концентрации этого препарата наблюдается более плавное увеличение времени корреляции (табл. 6). Важно отметить, что аналогичным образом изменяется χ с ростом концентрации добавки (рис. 6). В работе рассчитано время корреляции τ2 по спектрам ЭПР (5 × 10–11 < τ2 < 10–9 с). Зависимость τ2 от концентрации порфирина также носит нелинейный характер (рис. 9). По-видимому, увеличение времени корреляции в смесевых композициях обусловлено замедлением молекулярной подвижности вследствие уплотнения аморфной фазы. Рост доли паракристаллических структур всегда сопровождается увеличением доли выпрямленных цепей в аморфной прослойке и, как следствие, замедлением молекулярной подвижности. Такие изменения приводят к уменьшению концентрации радикала. Равновесную концентрацию радикала, адсорбированного в одинаковых по массе образцах волокон ПГБ–ZnТФП, рассчитывали с использованием программного обеспечения фирмы “Bruker–winer and simfonia”. Результаты показывают, что замедление молекулярной подвижности происходит симбатно с изменением концентрации радикала в волокне (рис. 9).
Рис. 7.
Спектры ЭПР нитроксильного радикала ТЕМПО образцов ПГБ–ZnТФП. Содержание ZnТФП 0 (1), 1 (2), 3 (3) и 5% (4).
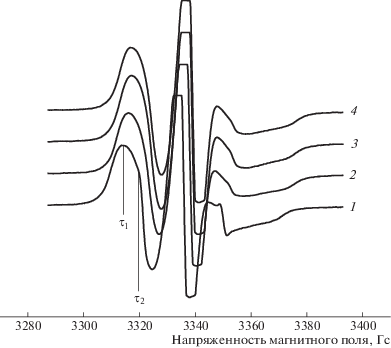
Таблица 6.
Время корреляции τ и доля аморфных областей α в волокнах ПГБ–порфирин
| Содержание ZnТФП,% | τ × 1010, с | τ × 1010, с (после отжига 140°С) | α1/α2, % | |||
|---|---|---|---|---|---|---|
| плотная область | рыхлая область | плотная область | рыхлая область | до отжига | после отжига | |
| 0 | 80.7 | 5.7 | 73.0 | 6.0 | 3.1 | 2.6 |
| 1 | 107.0 | 6.0 | 56.6 | 7.5 | 6.2 | 1.6 |
| 3 | 117.0 | 6.1 | 59.3 | 7.9 | 11.0 | 1.5 |
| 5 | 125.0 | 6.9 | 58.0 | 8.1 | 19.0 | 1.5 |
Рис. 8.
Экспериментальные (1) и теоретические (2) спектры ЭПР нитроксильного радикала ТЕМПО в образце ПГБ, а также медленная (3) и быстрая (4) составляющие спектра.
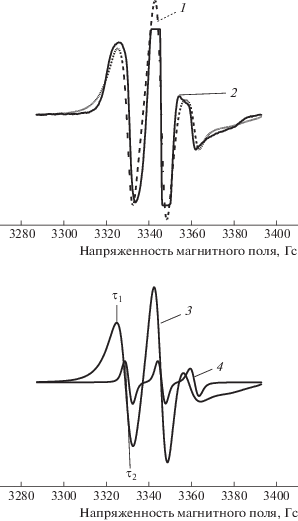
Рис. 9.
Зависимости концентрации радикала с (1) и времени корреляции τ (2) в образцах ПГБ–ZnТФП от концентрации ZnТФП.
Представляет интерес характеристика молекулярной подвижности после отжига при 140°С, когда система находится в достаточно равновесном состоянии. Радикал вводили в волокна после отжига. На рис. 10а приведено отношение τ/τ0 (τ0 – исходное, τ – текущее значение) в зависимости от времени отжига при 140°С для изучаемых композиций. Уже на первых минутах отжига происходит резкое снижение времени корреляции и только после отжига в течение 30 мин этот процесс резко замедляется. Рост степени кристалличности и содержания паракристаллических структур при отжиге, как отмечалось выше, обусловливает сильное увеличение плотности цепей в аморфной фазе, и радикал теряет способность проникать в наиболее плотные аморфные структуры. Концентрация радикала в волокнах также резко уменьшается после отжига в течение 2 ч. Так, например, в волокнах ПГБ концентрация радикала составляла 8 × 1015, а после отжига в течение 2 ч – 1.4 × 1015 спин/г. Аналогичная картина наблюдается во всех изучаемых волокнах. Были рассчитаны также времена корреляции после отжига в течение 2 ч (табл. 6). Видно, что подвижность радикала в плотных областях и доля плотных аморфных областей резко уменьшаются при введении порфирина в волокна ПГБ. Однако с ростом концентрации порфирина эти величины меняются чрезвычайно слабо.
Рис. 10.
Зависимости параметра τ/τ0 (а) и параметра $I_{ + }^{1}{\text{/}}I_{ + }^{2}$ (б) от времени отжига волокна при 140°С. Содержание ZnТФП 0 (1), 1 (2), 3 (3) и 5% (4).
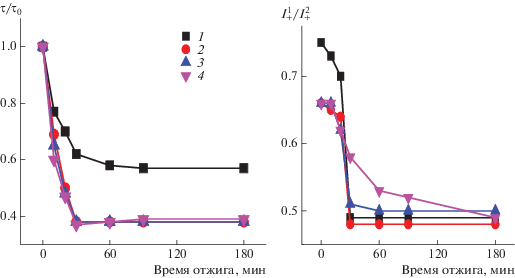
Таким образом, можно сделать вывод, что введение ZnТФП в ПГБ приводит к уплотнению аморфной фазы полимера в процессе формирования волокна и соответственно к замедлению молекулярной подвижности. Доля плотных областей возрастает и, как следствие, уменьшается концентрация радикала в волокне. Отжиг при 140°С вызывает повышение степени кристалличности и содержания паракристаллических структур, а также резкое уплотнение аморфных областей. Зонд теряет способность сорбироваться в достаточно плотных прослойках и в результате подвижность зонда, а, следовательно, и молекулярная подвижность цепей резко возрастает.
Наблюдаемые изменения сегментальной подвижности при отжиге образцов мы объясняем двумя параллельными процессами. Первый процесс связан с образованием межмолекулярных водородных связей между ПГБ и комплексами ZnТФП. С повышением температуры вероятность образования таких связей увеличивается. Соответственно происходит уплотнение аморфных областей и замедление молекулярной подвижности в волокнах. Второй процесс состоит в росте доли предельно выпрямленных цепей. Природа докристаллизации заключается в том, что сегменты со степенью выпрямленности β больше критического значения $h{\text{/}}L = \sqrt 2 {\text{/}}k$ (h – расстояние между концами цепей, L – длина цепи, k – длина сегмента) [36] при отжиге стремятся принять предельно выпрямленную конформацию. В связи с этим при повышенных температурах мы наблюдаем увеличение степени кристалличности и содержания паракристаллических структур в волокнах и рост доли аморфной фазы с высокой степенью выпрямленности цепей. Как результат, концентрация радикала уменьшается и повышается его подвижность, а значит, и молекулярная подвижность, так как проникновение радикала в плотные аморфные прослойки затруднено. В волокнах ПГБ эффект снижения времени корреляции не столь значителен, поскольку в них имеет место только процесс распрямления и уплотнения макромолекул.
В работе также получены данные об изменении отношения амплитуд медленной и быстрой составляющей молекулярной динамики в спектре ЭПР $I_{ + }^{1}{\text{/}}I_{ + }^{2}$ с увеличением времени отжига (рис. 10б). Как видно, доля медленной составляющей уменьшается, что подтверждает вывод о сорбции радикала в более рыхлых областях волокна.
Воздействие озонолиза на динамику вращения радикала в волокнах композиции ПГБ–ZnТФП
При использовании биомедицинских материалов одновременно с механическим и температурным воздействиями на их структуру и сегментальную подвижность влияет озон. Существует два источника его появления в атмосфере. Во-первых, озон образуется при работе мощных электрических приборов, обеспечивающих жизнедеятельность пациентов как в процессе хирургических операций, так и при терапии или мониторинге в стационаре. Во-вторых, озон в некоторых случаях продолжает использоваться при стерилизации медицинских изделий. Несмотря на повседневный контакт данного агрессивного соединения с полимерами, его влияние на их морфологические и динамические характеристики остается малоисследованной областью полимерного материаловедения.
В работе изучали влияние озонного воздействия на смесевые композиции при температуре 25°С (рис. 11). Озонолиз сложным образом влияет на структуру полимера. Происходят два параллельных процесса: сшивка макромолекул и их распрямление, что обусловливает рост жесткости и, как следствие, замедление молекулярной подвижности. Кроме того, увеличивается подвижность цепей из-за их деструкции и перехода в более свернутую конформацию. Природа роста жесткости цепей заключается в том, что сегменты с высокой степенью выпрямленности β после окисления стремятся принять предельно выпрямленную конформацию. Данные ранних работ [37, 38] показали, что самопроизвольная доориентация имеет место в трех случаях: 1) петли макромолекул на границе кристаллит–аморфная фаза окисляются с наиболее высокой скоростью из-за высоких напряжений и, имея большие значения β, распрямляются при озонном воздействии; 2) макромолекулы, жесткость которых возрастает вследствие образования полярных кислородсодержащих групп в боковых цепях и физической межмолекулярной сшивки при озонолизе; число сегментов в молекуле при этом уменьшается (как результат, растет величина β) и появляется тенденция к распрямлению; 3) цепи после озонной деструкции с достаточно высокой степенью выпрямленности. Указанные три составляющие обусловливают рост τ. Параллельно происходят деструкция макромолекул и их переход в свернутую конформацию, в результате увеличивается молекулярная подвижность. На первых стадиях окисления преобладают процессы, обусловливающие замедление подвижности цепей, при более длительном озонолизе (более 50 мин) – процессы разрушения структуры волокна.
Рис. 11.
Зависимость времени корреляции τ от длительности озонного воздействия. Содержание ZnТФП 0 (1), 1 (2), 3 (3) и 5% (4).
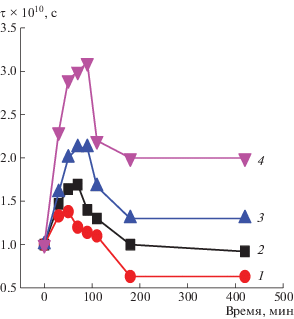
На рис. 11 видно, что чем выше концентрация порфирина, тем больше эффект замедления подвижности радикала, а следовательно, молекулярной подвижности в волокне. Наблюдаемые изменения мы объясняем ростом степени кристалличности и доли паракристаллических структур по мере повышения концентрации ZnТФП, когда увеличивается доля трехмерных кристаллитов (доля складок на границе кристаллит–аморфная фаза), а также доля паракристаллических структур, что приводит к более эффективной физической сшивке цепей при озонолизе. Аналогичные результаты были получены при озонолизе полиэтилена, полипропилена и смесей ПГБ с полиуретаном [37, 38].
Для подтверждения изложенных выше механизмов, приводящих к замедлению подвижности радикала, а, значит, и молекулярной подвижности в аморфной фазе, проводили исследования методом ИК-спектроскопии образцов. В табл. 7 представлены данные по изменению площади пиков при 1179 (аморфная составляющая) и 1276 см–1 (кристаллическая составляющая), нормированных на площадь полосы при 1720 см–1 для исходных волокон ПГБ с разным содержанием порфирина и образцов после озонолиза в течение 30 мин. Представленные данные свидетельствуют о повышении содержания паракристаллических структур, во-первых, с ростом концентрации порфирина в волокне, и, во-вторых, на начальной стадии озонного окисления.
Озоно-кислородное воздействие вызывает увеличение времени корреляции зонда в волокнах, а, следовательно, замедление молекулярной подвижности на ранних стадиях окисления (до 50 мин), что связано с физической сшивкой макромолекул и с процессами доориентации цепей. При более глубоких степенях окисления преобладают процессы деструкции цепей, что обеспечивает рост подвижности зонда и молекулярной подвижности в аморфной фазе волокон.
В настоящей работе на основе структурно-динамических исследований методами ЭПР, ДСК, рентгенодифракционного анализа и ИК-спектроскопии охарактеризовано влияние лекарственного препарата ZnТФП на кристаллическую и паракристаллическую фазу, а также на сегментальную подвижность макромолекул в аморфных областях ультратонких волокон ПГБ. Введение малых концентраций порфирина (1, 3, 5%), обусловливает рост доли паракристаллических структур и связанное с ним изменение динамики вращения зонда в аморфных областях.
Добавка в материал ПГБ 1, 3 и 5% ZnТФП не меняет кристаллическую структуру ПГБ (параметры элементарной ячейки кристаллической структуры, степень кристалличности, размеры кристаллитов, большой период и степень кристалличности в фибрилле). При отжиге образцов при 140°С наблюдаются значительные изменения структуры: растет степень кристалличности, размеры кристаллитов и большой период. При содержании порфирина 5% степень кристалличности выше, чем в ПГБ–3% ZnТФП, что объясняется, на наш взгляд, образованием агрегатов ZnТФП большего размера, которые плохо проникают в расплавленные кристаллические области.
При озонном воздействии (до 50 мин) на волокна изучаемой смеси резко возрастает время корреляции, что обусловлено увеличением доли выпрямленных цепей и усилением межмолекулярного взаимодействия вследствие накопления кислородсодержащих групп. При более глубоком окислении подвижность радикала, а, значит, и молекулярная подвижность, повышается, что связано с интенсивной деструкцией макромолекул.
Авторы выражают глубокую благодарность Н.Г. Шилкиной за предоставленные данные ДСК.
Измерения кристалличности проводили методом ДСК на приборе DSC204 F1 фирмы “Netzsch”, Германия (Центр коллективного пользования Института биохимической физики РАН “Новые материалы и технологии”).
Список литературы
Xie G., Wang Y., Han X., Gong Y., Wang J. et al. // Ind. Eng. Chem. Res. 2016. V. 55. № 26. P. 7116.
Schiffman J.D., Schauer C.A. // Polym. Revs. 2008. V. 48. № 2. P. 317.
Kong L., Ziegler G.R. // Biomacromolecules. 2012. V. 13. P. 2247.
Perale G., Hilborn J. Bioresorbable Polymers for Biomedical Applications. Amsterdam; Boston; London: Elsevier, 2017.
Kulkarni A., Bambole V.A., Mahanwar P.A. // Polymer-Plastics Technology and Engineering. 2010. V. 49. № 5. P. 427.
Трегер Ю.А., Морозов К.А., Дасаева Г.С., Фролкова А.К. // Тонкие химические технологии. 2018. Т. 13. № 4. С. 26.
Wang Y., Pan J., Han X., Sinka C., Ding L. // Biomaterials. 2008. V. 29. № 3. P. 393.
Mergaert J., Webb A., Anderson C., Wouters A., Swings J. // Appl. Environ. Microbiol. 1993. V. 59. № 10. C. 3233.
Michalak M., Kwiecień M., Kawalec M., Kurcok P. // RSC Adv. 2016. V. 6. P. 12809.
Жаркова И.И., Староверова О.В., Воинова В.В., Андреева Н.В., Шушкевич А.М., Склянчук Е.Д., Кузьмичева Г.М., Беспалова А.Е., Акулина Е.А., Шайтан К.В., Ольхов А.А. // Биомед. химия. 2014. Т. 60. № 5. С. 553.
Karpova S.G., Ol’khov A.A., Iordanskii A.L., Shilkina N.S., Popov A.A. // Russ. J. Phys. Chem. B. 2016. V. 10. № 4. P. 687.
Hu Y., Sato H., Zhang J., Noda I., Ozaki Y. // Polymer. 2008. V. 49. P. 19.
Ольхов А.А., Иорданский А.Л., Староверова О.В., Гумаргалиева К.З., Склянчук Е.Д., Аббасов Т.А., Ищенко А.А., Роговина С.З., Берлин А.А. // Хим. волокна. 2015. № 5. С. 8.
Karpova S.G., Ol’khov A.A., Shilkina N.G., Popov A.A., Filatova A.G., Kucherenko E.L., Iordanskii A.L. // Polymer Science A. 2017. V. 59. № 1. P. 58.
Iordanskii A.L., Ol’khov A.A., Karpova S.G., Kucherenko E.L., Kosenko R.Yu., Rogovina S.Z., Chalykh A.E., Berlin A.A. // Polymer Science A. 2017. V. 59. № 3. P. 343.
Karpova S.G., Ol’khov A.A., Iordanskii A.L., Lomakin S.M., Shilkina N.G., Popov A.A., Gumargalieva K.Z., Berlin A.A. // Polymer Science A. 2016. V. 58. № 1. P. 76.
Olkhov A.A., Staroverova O.V., Bonartsev A.P., Zharkova I.I., Sklyanchuk E.D., Iordanskii A.L., Rogovina S.Z., Berlin A.A., Ishchenko A.A. // Polymer Science D. 2015. V. 8. № 2. P. 100.
Karpova S.G., Olkhov A.A., Bakirov A.V., Shilkina N.G., Popov A.A. // Russ. J. Phys. Chem. B. 2018. V. 12. № 1. P. 142.
Глазкова М.Е., Агеева Т.А., Николаева О.И., Румянцева Ю.В., Койфман О.И. // Изв. вузов. Химия и хим. технология. 2011. № 3. С. 104.
Филимонов Д.А., Самолетов О.В., Базанов М.И., Семейкин А.С., Петров А.В. // Электрохим. энергетика. 2009. Т. 9. № 2. С. 91.
Филатов Ю.Н. Электроформование волокнистых материалов (ЭФВ-процесс). М.: Нефть и газ, 1997.
Krivandin A.V., Solov’eva A.B., Glagolev N.N., Shatalova O.V, Kotova S.L. // Polymer. 2003. V. 44/19. P. 5789.
Krivandin A.V., Fatkullina L.D., Shatalova O.V., Goloshchapov A.N., Burlakova E.B. // Russ. J. Phys. Chem. B. 2013. V. 7. № 3. P. 338.
Shibryaeva L.S., Shatalova O.V., Krivandin A.V., Tertyshnaya Yu.V., Solovova Yu.V. // Russ. J. Appl. Chem. 2017. V. 90. № 9. P. 1443.
Krivandin A.V., Shatalova O.V., Iordanskii A.L. // Polymer Science B. 1997. V. 39. № 11. P. 1865.
Budil D.E., Lee S., Saxena S., Freed J.H. // J. Magn. Res. A. 1996. V. 120. P. 155.
Тимофеев В.П., Мишарин А.Ю., Ткачев Я.В. // Биофизика. 2011. Т. 56. № 3. С. 420.
Бучаченко А.Л., Вассерман А.М. Стабильные радикалы. М.: Химия, 1973.
Stephen Z.D. Handbook of Thermal Analysis and Calorimetry, Applications to Polymers and Plastics. Amsterdam: Elsevier, 2002. V. 3.
Opfermann J. // Rechentechnik – Datenverarbeitung. 1985. V. 23. № 3. P. 26.
Krivandin A.V., Shatalova O.V., Iordanskii A.L. // Polymer Science B. 1997. V. 39. № 3. P. 543.
Yokouchi M., Chatani Y., Tadokoro H., Teranishi K., Tani H. // Polymer. 1973. V. 14. № 6. P. 267.
Vainshtein B.K. Diffraction of X-rays by chain molecules. Amsterdam: Elsevier, 1966.
Ольхов А.А., Панкова Ю.Н., Гольдштрах М.А., Косенко Р.Ю., Маркин В.С., Ищенко А.А., Иорданский А.Л. // Материаловедение. 2015. № 10. С. 20.
Olkhov A.A., Markin V.S., Kosenko R.Yu., Gol’dshtrakh M.A., Iordanskii A.L. // Russ. J. Appl. Chem. 2015. V. 88. № 2. P. 308.
Озерин А.Н. Дисс. … канд. хим. наук. М.: НИФХИ им. Карпова, 1977.
Карпова С.Г., Попов А.А., Заиков Г.Е. // Высокомолек. соед. А. 1991. Т. 33. № 5. С. 931.
Karpova S.G., Iordanskii A.L., Popov A.A., Chvalun S.N., Berlin A.A. // Russ. J. Phys. Chem. B. 2012. V. 6. № 1. P. 72.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия А)