

Высокомолекулярные соединения (серия А), 2019, T. 61, № 2, стр. 139-151
ТЕРМИЧЕСКИЕ СВОЙСТВА ПОРИСТОГО ПОЛИЛАКТИДА
Л. С. Шибряева a, b, *, В. Г. Крашенинников c, В. Н. Горшенев a
a Институт биохимической физики им. Н.М. Эмануэля Российской академии наук
119334 Москва, ул. Косыгина, 4, Россия
b Федеральный научный агроинженерный центр ВИМ
109428 Москва, 1-й Институтский пр-д, 5, Россия
c Институт химической физики им. Н.Н. Семенова Российской академии наук
119991 Москва, ул. Косыгина, 4, Россия
* E-mail: lyudmila.shibryaeva@yandex.ru
Поступила в редакцию 23.04.2018
После доработки 26.09.2018
Принята к публикации 10.10.2018
Аннотация
Методами дифференциальной сканирующей калориметрии и термогравиметрии изучены процессы плавления и термодеструкции пористого полилактида. Поры сформированы из раствора азо-бис-изобутиронитрила в ацетоне и углекислого аммония в водно-ацетоновой смеси в условиях термостимулированного нагрева наполненного полимера, а также в среде сверхкритического диоксида углерода. Показано, что порообразователи разрушают кристаллические области полимера, а также понижают температуру и теплоту плавления кристаллитов в зависимости от пластифицирующего действия на полимерную матрицу, метода порообразования и природы порофора. Порообразование полилактида приводит к уменьшению начальной температуры термоокислительной деструкции полимера и изменению механизма процесса.
ВВЕДЕНИЕ
В настоящее время для производства имплантатов с целью замены поврежденного участка костной ткани широко используют полимерные биоматериалы [1–5]. Особый интерес к таким материалам отмечается в технологии, основанной на помещении клеток, полученных из тканей пациента, на трехмерный носитель (скаффолд), где происходит образование ткани за счет роста и дифференциации клеток [6–12]. Как показали результаты ряда исследований [13–15], в случае применения данной технологии для достижения наилучших результатов необходимо наличие пористости в материале. Изучение физических и биологических свойств образцов пористого материала на основе сверхвысокомолекулярного полиэтилена, с целью их использования при замещении дефектов костной ткани, показало, что остеокондуктивные свойства исследуемого материала достигаются в случае наличия открытых проходных пор размером более 100 мкм [15]. Все материалы создают оптимальные условия для беспрепятственного врастания соединительной и костной ткани в имплантат в случае, когда средний размер диаметра микропор в толще полимера составляет 100–250 мкм, при этом пористость материала равна в среднем 45–50% [16]. Условием применения пористых материалов в хирургии наряду с макропористостью (поры 30–700 мкм) является наличие микропористой структуры с порами близкими к 0.01–0.1 мкм [17].
Функциональная нагрузка пористости состоит в том, что поры, способствуя прорастанию ткани и циркуляции биологических жидкостей в организме, обеспечивают питание, клеточное распространение и формирование капилляров [18, 19].
Одной из задач значится сохранение материалом имплантата при наличии пористости высоких физико-механических (прежде всего прочностных) параметров, требуемых для его эксплуатации. Известно, что процесс порообразования, оказывая влияние на форму и размеры пор, вид распределения пор в полимерной матрице, плотность полимера и т.д. трансформирует надмолекулярную структуру полимера. Поэтому при изготовлении имплантатов необходимо учитывать закономерности изменения структуры полимера под влиянием формирующихся пор.
Другой важной задачей является проблема термо- и термоокислительной стойкости пористого материала. После получения изделий требуемой формы пористый имплантат подвергают термической обработке. При этом его выдерживают при температуре 50–75°С в течение 3–28 ч, или при 80–130°С в течение 0.5–1.0 ч [17]. Кроме того, для получения изделий из биодеградируемых имплантатов, например для остеосинтеза в челюстно-лицевой хирургии с целью регенерации костной ткани, используют методы переработки полимеров в расплаве, такие как литьевое прессование, литье под давлением и другие. Таким образом, в ходе технологии переработки, предусматривающей высокотемпературное воздействие или длительное термостатирование на пористый полимер для имплантата, он может подвергаться термодеструкции или термоокислительному процессу по радикально-цепному механизму.
Цель настоящей работы состояла в изучении зависимости теплофизических параметров, термо- и термоокислительной стойкости от структуры пористого материала на примере биодеградируемого полилактида, широко используемого для производства имплантатов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе применяли гранулы полилактида (ПЛА) марки 4032 D компании “Nature Works” (США) с Mw = 1.7 × 105, d = 1.27 г/см3. Исследовали образцы в виде пленок и пластин. Пленки толщиной 1.2–1.4 мм получали прессованием гранул полилактида при 200°С, после чего выдерживали материал в форме в течение 5 мин. На выходе имели оптически прозрачные пленки с температурой плавления 173°С. Для создания пористой структуры применяли две технологии порообразования: во-первых, обработку в условиях термостимулированного нагрева (термообработку) пленок полилактида, предварительно насыщенных порофорами из растворов в органических растворителях, во-вторых, обработку полимера в среде сверхкритического диоксида углерода. В первом методе в качестве порофоров использовали азо-бис-изобутиронитрил (АИБН) и углекислый аммоний (NH4)2CO3. Порофоры в пленки ПЛА вводили из 10%-ного раствора АИБН в ацетоне и 1 моль/л раствора (NH4)2CO3 в водно-ацетоновой смеси (массовое соотношение 3 : 10). Для насыщения порофорами образцы полимера выдерживали в растворах при комнатной температуре в течение суток. После этого на образец воздействовали СВЧ-излучением 1 минуту для удаления органического растворителя. Затем ПЛА подвергали термообработке: образец, насыщенный АИБН, прогревали при 150°С в течение 10 мин, образец, наполненный (NH4)2CO3 – при 200°С в течение 5 мин. В результате термообработки образцов происходило разложение АИБН или углекислого аммония с газовыделением, приводя тем самым к образованию открытых пор в массе полимера. Для порообразования с помощью сверхкритического диоксида углерода (сверхкритический CO2) использовали метод, ранее разработанный и примененный в работе [20] для стирола и хитозана. С целью повышения растворимости СО2 полимер обрабатывали сверхкритическим диоксидом углерода совместно с газообразующими порофорами АИБН и (NH4)2CO3. Для АИБН осуществляли обработку при температуре его разложения (90°С), для (NH4)2CO3 – при температуре перехода полилактида в вязкотекучее состояние (162°С). Таким образом, сформировалась разветвленная протяженная структуры из нано- и микропор. Плотность полученных пористых изделий составляла 0.4–0.8 г/см3.
Для контроля за наполнением пленок ПЛА порофорами азо-бис-изобутиронитрилом и углекислым аммонием, а также их удалением из пористого образца, использовали ИК-спектроскопию и дифференциально сканирующую калориметрию. Спектры снимали на фурье-спектрометре фирмы “Perkin-Elmer”. Контролировали наличие характеристических полос поглощения связей –N=N– в АИБН и связей NH в (NH4)2CO3 при 1530 и 3300–3500 см–1 соответственно. Для порофоров определяли тепловые эффекты с помощью ДСК (модель DSC-204 F1 фирмы “Netzsch”, Германия) при скорости нагревания 10 град/мин в диапазоне температуры 30–190°С в токе аргона. Методом ДСК также изучали тепловые эффекты в полимере, определяли теплоту и температуру плавления исходных и пористых образцов ПЛА. Для точного нахождения параметров в регистрируемые величины вводили поправки по параметрам пика плавления эталона. В качестве эталона использовали образец индия (температура и теплота плавления составляют Тm = 156.7°С и ΔHm = = 28.58 Дж/г). Скорость сканирования изменяли в интервале 4–32 град/мин. Навеску образца варьировали в пределах 2–8 мг. По теплоте плавления определяли степень кристалличности ПЛА из соотношения $\chi = (\Delta {{H}_{m}}{\text{/}}\Delta H_{m}^{*})$ × 100%, где ΔHm – теплота, поглощенная при плавлении образца в расчете на единицу массы ПЛА, $\Delta H_{m}^{*}$ = 106 Дж/г – теплота плавления единичной массы кристаллов ПЛА [21].
Процесс термо- и термоокислительной деструкции изучали с помощью термогравиметрического анализа на термомикровесах TG 209 F1 Iris (“Netzsch”) в динамических условиях нагревания на воздухе и в токе аргона. Навески образцов составляли 5–8 мг, анализ проводили при скорости нагревания 20 град/мин.
Точность определения температуры плавления ПЛА в случае ДСК и ТГА составляла ±1 и ±2 oC соответственно. Среднеквадратичные отклонения экспериментальных значений площади пиков плавления и термодеструкции разных образцов (не менее 10) лежали в пределах 10%.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Исследование поверхности пористых образцов, проведенное с помощью оптической микроскопии, показало четко выраженные различия в пористой структуре полимерной матрицы, полученной в результате термостатирования образцов, наполненных порофорами из растворов углекислого аммония и АИБН, а также в среде сверхкритического углекислого газа (рис. 1).
Рис. 1.
Оптические микрофотографии поверхности пористых образцов ПЛА, полученных для порофора (NH4)2CO3 методом термообработки (а) и в среде сверхкритического СО2 (б).

Структура пористого материала, сформированного порообразователями с термообработкой (рис. 1а), характеризуется крупными порами шарообразной формы, достигающими в диаметре 300 мкм. Обработка ПЛА сверхкритического СО2 с порофорами демонстрирует тонкую пористую структуру, на фоне которой наблюдается появление некоторых углублений в виде тонких волокон (рис. 1б). Размеры пор достигают порядка ≤30 мкм. Следует отметить, что в обеих технологиях порообразования происходит формирование пор в объеме полимера, что соответствует оптимальным условиям развития клеток, а значит, удовлетворяет требованиям приготовления материалов для имплантатов. Здесь важно установить влияние строения пор на структуру и свойства полимерной матрицы.
Теплофизические параметры пористого полилактида
Закономерности влияния порообразования на теплофизические параметры ПЛА были изучены с помощью ДСК. Термо- и теплофизические параметры ПЛА являются высокочувствительными показателями структуры кристаллических областей [22]. На рис. 2 представлены термограммы ДСК углекислого аммония (кривая 1) и АИБН (кривая 2) в твердом состоянии, а также ПЛА, наполненного порофорами. Последние термограммы получены при первичном нагревании ПЛА с введенными в полимерную матрицу (NH4)2CO3 из водно-ацетоновой смеси (кривая 1') и АИБН из ацетонового раствора (кривая 2') после удаления из полимера растворителей. На этом же рисунке приведены термограммы пористых образцов ПЛА, полученные термостатированием после разложения и удаления из ПЛА порофоров (NH4)2CO3 (кривая 1'') и АИБН (кривая 2''), а также исходного ПЛА (кривая 3). Анализ приведенных термограмм показывает, во-первых, различный характер тепловых эффектов для твердых углекислого аммония и АИБН; во-вторых, форма и температурные области пиков тепловых эффектов этих веществ изменяются при их введении в полимерную матрицу. На термограмме углекислого аммония наблюдается широкий эндотермический пик в интервале температуры 100–165°С с максимумом при ~158°С, относящийся к его разложению (рис. 2, кривая 1). У АИБН присутствуют два пика – малоинтенсивный эндотермический пик его плавления при ~108°С и интенсивный экзотермический пик с максимумом Tm при 120°С, характерный для его разложения (рис. 2, кривая 2). Вместе с тем, в полимерной матрице у (NH4)2CO3 пик разложения смещается в низкотемпературную область (в интервал 40–135°С), при этом Tm составляет ~80°С. Пик плавления АИБН также сдвигается в сторону низких значений температуры (до Тm = 50°С). Узкий пик разложения расширяется в интервале 100–140°С, Тm ~ 123°С (кривые 1', 2'). Очевидно такие изменения в форме пиков тепловых эффектов и, следовательно, теплофизических параметров порофоров, вызваны взаимодействием между порофорами и полимерной матрицей на молекулярном уровне.
Рис. 2.
а – Термограммы углекислого аммония (1) и АИБН (2) в твердом состоянии; б – термограммы первичного плавления полимерной матрицы с введенными в нее порофорами (NH4)2CO3 (1') из водно-ацетонового и АИБН (2') из ацетонового растворов; в – эндотермы плавления пористых образцов ПЛА, приготовленных термообработкой порофоров, полученных после удаления из них (NH4)2CO3 (1'') и АИБН (2''); 3 – эндотерма плавления чистого ПЛА.
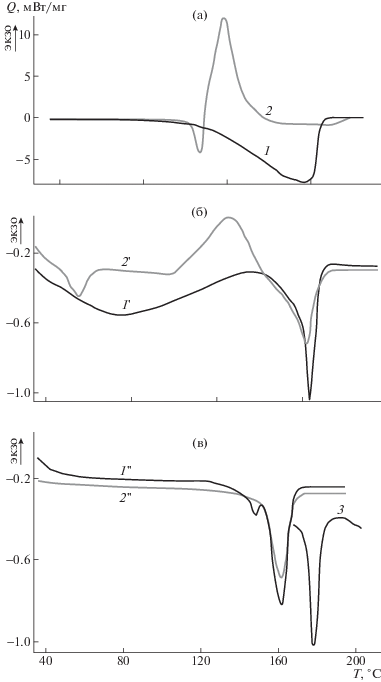
Важно отметить отсутствие пиков тепловых эффектов, относящихся к порофорам, на термограммах пористых образцов. Удаление порофоров из массы пористого ПЛА подтверждают данные ИК-спектроскопии. Участки термограмм (рис. 2), демонстрирующие эндотермы плавления полимера, для образцов, наполненных порофорами (кривые 1', 2'), а также пористых (кривые 1'', 2'') после термостатирования и удаления порофоров, показывают явно выраженное отличие от исходного ПЛА (кривая 3). Эндотермический пик исходного ПЛА с температурой в максимуме Tm = = 173°С, характерный для плавления его кристаллитов, на термограммах наполненного и пористого ПЛА значительно смещается в низкотемпературную область. Причем смещение наблюдается после наполнения образцов порофорами и достигает ~10–12°С (табл. 1). Порообразование вызывает незначительные изменения температурной области плавления полимера по сравнению с наполненными образцами. Помимо уменьшения температуры плавления у наполненного порофорами и пористого ПЛА уменьшается теплота плавления, а значит, и степень его кристалличности, т.е. происходит аморфизация полимера. Причем наполнение образцов порофорами приводит к большей аморфизации ПЛА, чем порообразование наполненного полимера (табл. 1).
Таблица 1.
Теплофизические параметры образцов пористого полилактида
| Образец | Температура начала плавления Т1, °С | Температура максимума пика плавления Tm, °С | Теплота плавления, Дж/г | Степень кристал-личности, % | Энергия активации плавления ПЛА, кДж/моль |
|---|---|---|---|---|---|
| ПЛА | 145 | 173 | 62 | 58 | 430 ± 10 |
| ПЛА, насыщенный АИБН | 141 | 162 | 51 | 48 | 290 ± 20 |
| Пористый ПЛА, термообработка с АИБН | 145 | 161 | 45 | 42 | 370 ± 15 |
| ПЛА, насыщенный (NH4)2CO3 | 138 | 163 | 54 | 51 | 260 ± 20 |
| Пористый ПЛА, термообработка с (NH4)2CO3 | 134 | 161+плечо 148 | 49 | 46 | 240 ± 15 |
| Пористый ПЛА, сверхкритический СО2 с АИБН | 140 | 165 | 51 | 48 | 380 ± 15 |
| Пористый ПЛА сверхкритический СО2 с (NH4)2CO3 | 132 | 147 | 37 | 35 | 330 ± 15 |
Можно полагать, что введение порофоров в большей степени влечет изменение структуры полимера вследствие пластифицирующего действия порофора, либо растворителя, из которого вводили порофор. Сравнение термограмм плавления, полученных для образцов с разной концентрацией порофора, после удаления из них растворителя позволяет предположить, что определяющая роль в изменении температуры и теплоты плавления ПЛА принадлежит действию порофора. На рис. 3 для образцов ПЛА, наполненных (NH4)2CO3 до 3 и 10% (кривые 1 и 2 соответственно), четко видно, что увеличение концентрации порофора (о чем свидетельствует увеличение интенсивности пика его разложения) понижает температуру, а также теплоту плавления кристаллических областей ПЛА (кривые 1', 2').
Рис. 3.
Термограммы распада углекислого аммония в образцах ПЛА, наполненных до концентрации 3 (1) и 10% (2), а также соответствующие им эндотермы первичного плавления кристаллитов полимера (1', 2') и кривые термогравиметрии (3, 4).
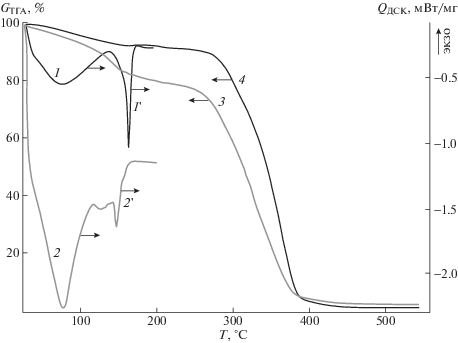
Кривые термогравиметрии на рис. 3 демонстрируют, что рост концентрации углекислого аммония в ПЛА приводит к понижению температуры начала и росту скорости разрушения полимерной матрицы. К моменту начала плавления кристаллитов увеличивается доля полимера, подвергнутого термоокислительной деструкции (рис. 3, кривые 3 и 4). По-видимому, в ходе получения термогравиметрической кривой может происходить разложение порофора с порообразованием, что может инициировать процесс деструкции ПЛА, и тем с бóльшей скоростью, чем выше концентрация порофора.
Термограммы плавления образцов ПЛА, в которых порообразование осуществлено воздействием сверхкритическим углекислым газом, представлены на рис. 4. Сравнение этих эндотерм с эндотермами плавления пористых образцов, приготовленных по методу термостатирования наполненного полимера, демонстрирует одинаковый характер изменения теплофизических параметров ПЛА. Наблюдается падение температуры и теплоты плавления кристаллических структур (табл. 1). При этом различаются количественные показатели. В случае порообразования методом сверхкритического CO2, смещение пика плавления ПЛА в сторону низких значений температуры для образца с (NH4)2CO3 больше, чем для образца с АИБН и больше, чем в случае порообразования термообработкой. У пористого образца, полученного с помощью сверхкритического СО2 с АИБН, сдвиг пика плавления в низкотемпературную область несколько меньше, чем у полученного термообработкой наполненного порофором (табл. 1). Аналогичным образом изменяется степень кристалличности полимера. По-видимому, это обусловлено условиями порообразования. В случае применения (NH4)2CO3, температура воздействия на этот порофор в обоих методах порообразования была значительно выше, чем на АИБН. Аморфизация полимера зависит от скорости охлаждения расплава пористой матрицы, в случае сверхкритического СО2 с (NH4)2CO3 охлаждение полимера происходит на стадии декомпрессии при быстром удаление молекул газа. Возможно, полимер не успевает кристаллизоваться, что способствует его аморфизации.
Рис. 4.
Термограммы плавления пористых образцов ПЛА, полученных методом сверхкритического CO2 с (NH4)2CO3 (1) и АИБН (2).
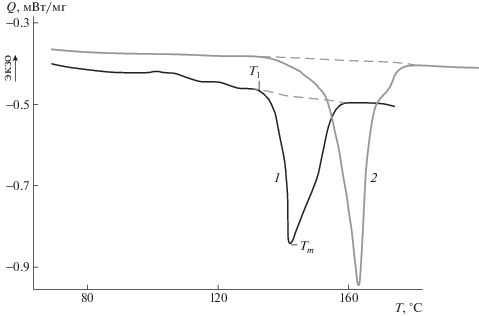
Для изучения особенности структуры кристаллитов, формирующихся в пористых образцах ПЛА, применена термокинетическая модель, в основе которой лежит анализ кривых, характеризующих распределение кристаллитов по температуре плавления [24]. Такие кривые были получены из температурных зависимостей для степени превращения кристаллитов ПЛА в расплав (dXm(T)/dT) в ходе их плавления, в соответствии с методом анализа теплофизических данных ДСК, полученных в неизотермическом режиме (режим сканирования по температуре с постоянной скоростью), предложенным в работе [24]. При этом учтены представления об особенностях фазового перехода – плавления, включающего плавление ламеллярных структур и переход макроцепей в расплав [25, 26]. С целью исключения влияния стадии перехода макромолекул в расплав после разрушения ламелей, в работе рассмотрен температурный интервал, ограничивающий плавление ламеллярных структур. Полному разрушению ламелей соответствует температурный интервал от начальной температуры плавления T1 до максимума пика плавления Tm (см. рис. 4).
Степень превращения кристаллитов в расплав для момента времени ti была определена с помощью соотношения
(1)
${{X}_{m}} = \frac{{\int\limits_{{{t}_{0}}}^{{{t}_{i}}} {\left( {\frac{{d\Delta {{H}_{m}}(t)}}{{dt}}} \right)dt} }}{{\int\limits_{{{t}_{0}}}^{{{t}_{\infty }}} {\left( {\frac{{d\Delta {{H}_{m}}(t)}}{{dt}}} \right)dt} }},$Кривые, отражающие характер распределения кристаллитов по температуре плавления, получены из выражения
Здесь dXm(T)/dT – изменение степени превращения кристаллитов при плавлении в выбранном интервале температуры; Hm – теплота, поглощенная при плавлении; b = dT/dt – скорость нагревания.
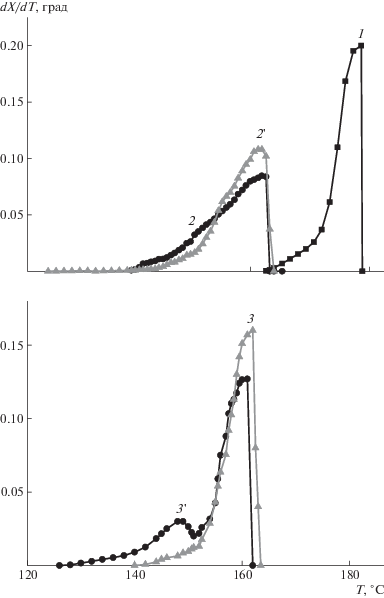
На рис. 5 представлены кривые, полученные с помощью уравнения (1), характеризующие распределение кристаллических структур ПЛА по температуре плавления в исходном образце, наполненных порофорами образцах и после порообразования в них. Видно, что порообразование как методом термообработки образцов ПЛА, наполненных порофорами, так и методом сверхкритического CO2 в среде порофоров приводит к обогащению кристаллических областей полимера низкоплавкими фракциями кристаллитов. Причем закономерности формирования кристаллитов зависят и от природы порофора, и от условий порообразования. Порообразование термообработкой образца ПЛА, наполненного порофором АИБН, влечет уменьшение содержания фракции кристаллитов с более низкой температурой Тm = 145°C, но при этом несколько увеличивается фракция кристаллитов с Тm ~ 160°С (кривые 2, 2'). У образца ПЛА с (NH4)2CO3 содержание низкоплавкой фракции увеличивается, а высокоплавкой – понижается (кривые 3, 3'). В кристаллических областях пористого образца, полученного из сверхкритического CO2 с АИБН, преобладают кристаллиты с Tm = 163°C, у образца с (NH4)2CO3 – c Tm = 145°С.
Рис. 5.
Кривые распределения кристаллических структур ПЛА по температуре плавления в образцах чистого полимера (1), наполненных порофорами АИБН (2), (NH4)2CO3 (3) и пористых образцов, полученных термообработкой c АИБН (2') и (NH4)2CO3 (3').
В работе была проведена оценка энергии активации плавления кристаллитов Ea, в связи с чем было использовано уравнение Киссенджера [27] для неизотермического процесса плавления кристаллитов ПЛА. Уравнение основано на зависимости величины температуры плавления от скорости нагревания полимера:
(2)
${{Е }_{a}} = {\text{ }}\left[ {R{{Т }_{1}}{{Т }_{2}}{\text{/}}\left( {{{Т }_{2}}--{{Т }_{1}}} \right)} \right]{\text{ln}}\left( {{{V}_{2}}{\text{/}}{{V}_{1}}} \right),$С помощью уравнения (2) были определены Еа по величине сдвига максимальной температуры плавления Tm, с изменением скорости нагревания в интервале 4–32 град/мин. Для контроля за изменением температуры использовали эталон In.
Полученные величины Еа представлены в табл. 1. Как видно, насыщение полимера порофором снижает энергию активации плавления кристаллитов, что указывает на формирование низкоплавких кристаллитов с мелкой структурой или высокой дефектностью [23]. Порообразование в случае термостатирования изменяет величину Еа в зависимости от природы порофора. У АИБН после термообработки в пористом образце данная величина растет, что соответствует характеру изменения кривой распределения по температуре плавления и указывает на совершенствование структуры кристаллитов. Следует также отметить более высокое значение Еа у образцов пористого ПЛА, полученных в среде сверхкритического CO2. Причем в последней технологии порообразования проявляется природа наполнителя, значение Еа в случае АИБН несколько выше, чем у углекислого аммония.
Термоокисление пористых образцов полилактида
Известно, что надмолекулярная структура полимера определяет его реакционную способность по отношению к высокотемпературному воздействию и кислороду. В связи с медицинской направленностью применения изучаемого полимера, установление влияния пористой структуры на термоокислительную стойкость ПЛА является важным. Для решения этой проблемы в работе был использован метод термогравиметрии. Получены ТГ-кривые потери массы и их первые производные по температуре (кривые ДТГ) для исходного, модифицированных порофорами и пористых образцов ПЛА в воздушной среде (рис. 6).
Рис. 6.
Кривые потери массы (1–7) и их первые производные (1 '–7 ') в ходе термодеструкции в аргоне (1, 1 '), термоокислительной деструкции в воздушной среде (2, 2')–(7, 7') для исходного ПЛА (1, 1', 2, 2 '), порофора (NH4)2CO3 (3, 3 '), а также образцов ПЛА, наполненного этим порофором до 3 (4, 4 ') и 10 мас. % (5, 5 '), и пористого ПЛА, полученного термообработкой образцов, наполненных (NH4)2CO3 (6, 6 ') и АИБН (7, 7 ').
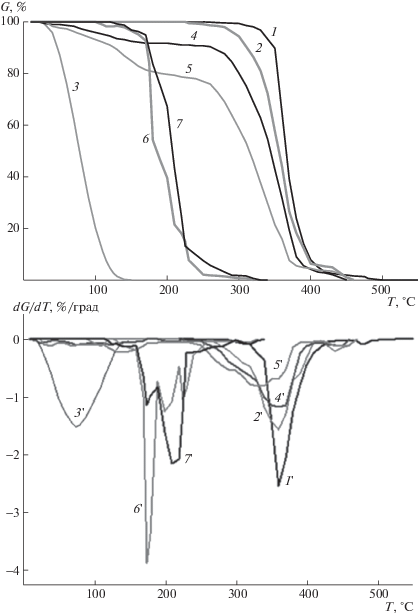
Сравнительная оценка их термоокислительной стабильности проведена по величинам начальных значений температуры деструкции Tн, определяемых по ДТГ-кривой, в качестве температуры, при которой скорость процесса достигает 1% потери массы образца в минуту; по температурным интервалам (Tн–Tк) потери массы, по максимальным величинам скорости и температуры полураспада образцов.
Для более детального исследования влияния пористой структуры на термоокислительную стойкость ПЛА в работе изучали особенности разрушения ПЛА в наполненных и пористых образцах.
Показателями механизма процесса в полимерных материалах являются эффективная энергия активации ET, предэкспоненциальный фактор A и порядок реакции n. В работе эти кинетические параметры были получены термокинетическим методом анализа ТГ-кривых.
При обработке результатов термогравиметрии сделаны следующие допущения. Химическая реакция разложения схематически изображается как
Расчеты кинетических параметров по ТГ-кривой основаны на формальном кинетическом уравнении, хорошо описывающим кинетику разложения твердых веществ:
где W – масса образца, вступившая в реакцию; k – константа скорости, n – порядок реакции; t – время.Зависимость константы скорости реакции от температуры описывается уравнением Аррениуса:
Здесь A – предэкспоненциальный множитель, T – абсолютная температура (K).
Исходя из этих допущений, формально кинетическое уравнение принимает вид:
Для его решения применен дифференциальный метод установления кинетических параметров (Freeman и Carroll) в виде уравнения:
Порядок реакции для каждого из исследованных образцов был определен из зависимости [27]:
(3)
$\frac{{\Delta \ln \frac{{dW}}{{dt}}}}{{\Delta \ln W}} = f\left[ { - \frac{{\Delta \left( {\frac{1}{T}} \right)}}{{R\Delta \ln W}}} \right]$Величины n, полученные из зависимости (3), даны в табл. 2. Они составляют величину близкую к единице.
Таблица 2.
Кинетические параметры деструкции образцов пористого полилактида
| Образец | Температурные интервалы пика потери массы ПЛА (Tк–Tн), °C | Температура потери 50% массы полимера T1/2, °C | Потеря массы ПЛА*, % | Кинетические параметры деструкции, определенные с помощью модели [27] | ||
|---|---|---|---|---|---|---|
| ET, кДж/моль | A, л/ моль с | n | ||||
| ПЛА в аргоне | 400–300 | 360 | 51 | 260.0 ± 20.0 | 3.6 × 1019 | 1.0 |
| ПЛА на воздухе | 400–250 | 360 | 31 | 206.4 ± 40.0 | 9.0 × 1016 | 1.1 |
| ПЛА, наполненный (NH4)2CO3 до 10% на воздухе** | 380–210 | 330 | 16 | 120.9 ± 15.0 | 1.4 × 109 | 1.1 |
| 180–110 | 140 | 6 | 55.8 ± 15.0 | 2.2 ×104 | 1.0 | |
| 70–40 | – | 4 | 10.0 ± 2.5 | 3.9 × 10–2 | 1.0 | |
| (NH4)2CO3 на воздухе*** | 140–30 | 72 | – | 35.0 ± 2.5 | 4.9 × 102 | 0.9 |
| Пористый ПЛА, термообработка (NH4)2CO3 на воздухе | 250–135 | 225 | 19 | 192.0 ± 15.0 | 5.0 × 1016 | 1.0 |
| 205 | 25 | 157.0 ± 15.0 | – | 1.0 | ||
| 175 | 77 | 97.6 ± 10.0 | 8.2 × 1013 | 0.9 | ||
| Пористый ПЛА, термообработка АИБН на воздухе | 300–150 | 215 | 43 | 180.5 ± 20.0 | 1.0 × 1013 | 0.9 |
| 170 | 14 | 120.9 ± 20.0 | 1.4 × 109 | 1.0 | ||
Для описания процесса деструкции, протекающего при одной постоянной скорости нагревания образца и n = 1, уравнение (3) может быть представлено линейной зависимостью в полулогарифмических координатах:
где наклон линии равен ЕT; параметр A – определен экстраполяцией кривой на логарифмическую ось при нулевом значении 104/RT.С помощью линейной зависимости (4) представлены кинетические кривые, характеризующие процессы деструкции исследуемых образцов, а также рассчитаны кинетические параметры процессов для температурных интервалов, соответствующих потере массы образцов. Данные расчетов представлены в табл. 2.
Прежде всего сопоставление кривых ТГ и ДТГ, полученных в инертной атмосфере (рис. 6, кривые 1, 1') и в воздушной среде (рис. 6, кривые 2, 2') для исходного ПЛА показало, что воздушная атмосфера оказывает влияние на процесс разрушения полимера, ускоряет распад его полимерных цепей, о чем свидетельствует понижение температуры начала деструкции на ~50°С, уширение пика ДTГ-кривой в низкотемпературную область при том, что максимальная скорость полураспада полимера уменьшается в ~1.6 раза.
Таким образом, очевидно изменение характера деструкции. Для исходного ПЛА замена инертной среды на воздушную вызывает уменьшение величин эффективной энергии активации и предэкспоненты (табл. 2). Обнаруженные изменения формы кривых TГ и ДTГ, кинетических параметров распада ПЛА и их величины при смене инертной на воздушную среду, скорее всего, указывают на кислородную активизацию разрушения связей С–С в основных цепях полимера по радикально-цепному механизму.
Анализ кривых TГ и ДTГ для образцов модифицированного ПЛА (рис. 6, кривые 4 и 5), полученных в воздушной атмосфере, показал, что введение порофоров значительно понижает термоокислительную стабильность полимера. Разрушение ПЛА и прежде всего начальная стадия процесса (до ~20% потери массы) ускоряется. На что указывает уменьшение температуры начала разрушения, асимметричное уширение пика на ДTГ- кривой со сдвигом к области 110–70°С с уменьшением максимальной скорости полураспада в области 380–210°С (рис. 6, кривые 4', 5').
Для модифицированного ПЛА обнаружено существование нескольких эффективных ET и A, характерных для ступенчатого механизма деструкции. Аномально низкие величины А и ЕT у модифицированного ПЛА, характеризующие термоокислительный распад полимера на начальной стадии в интервале температуры от 40 до 180°С, указывают, что ускорение разрушения полимера до 20% потери массы обусловлено разрушением слабых связей, скорее всего, отрывом сложноэфирных боковых групп (табл. 2). На основании формы пика в интервале температуры 210–380°C разрушение полимера в этом интервале можно объяснить деструкцией связей С–С в макромолекулах.
Важно отметить, что изменение формы пика, появление низкотемпературной области ускоренного разрушения полимера коррелируют с концентрацией порофора – модификатора ПЛА (рис. 6, кривые 4, 4' и 5, 5'). У образца, наполненного 3% углекислого аммония, потеря массы в области 210–380°С достигает ∼16, а у содержащего 10% (NH4)2CO3 – 26%, максимум пика 50% – потери массы с T1/2 = 360°C смещается к 330°C (кривые 4', 5'). На основании указанных выше данных можно сделать вывод, что наполнение полимера порофором способствует ускорению процесса термоокислительной деструкции.
Так как температурная область распада порофора в полимере перекрывает область начала потери массы полимера, то можно предположить существование инициирующего влияния продуктов распада порофора на начальную стадию радикального процесса термоокисления. Однако величины кинетических параметров термораспада порофора превышают параметры начальной стадии разрушения ПЛА. В этом случае распад порофора, скорее всего, будет тормозить радикальные процессы термоокисления. Более вероятен другой механизм воздействия порофора на реакционную способность ПЛА. Это модифицирующий эффект, способствующий увеличению сегментарной подвижности цепей полимера. Повышенная скорость деструкции ПЛА, соответствующая низкому значению энергии активации термоокислительной деструкции, обусловлена появлением высокоподвижных сегментов полимерной цепи, способных передавать кинетическую цепь окисления с макромолекулы на макромолекулу внутри- и межмолекулярно.
По-видимому, рост подвижности цепей ПЛА в большей степени будет увеличивать скорость термоокислительного разрушения связей С–С, особенно на глубокой стадии процесса. В пользу данной гипотезы свидетельствует тот факт, что 80% потери массы наполненного ПЛА приходится на интервал температуры (Tн–Tк) 380–210, в котором соответствующие этому интервалу величины ET и A характерны для термораспада макрорадикалов.
Анализ ТГ- и ДTГ-кривых пористых образцов ПЛА показал, во-первых, смещение этих кривых и пиков изменения скорости потери массы в низкотемпературную область на 200°С по сравнению с кривыми для чистого ПЛА; во-вторых, сложность формы пиков потери массы на ДTГ-кривых (рис. 6, кривые 6, 6' и 7, 7'). Можно предположить, что наблюдаемый характер кривых деструкции ПЛА в пористых образцах может быть следствием неоднородности процесса, наложением реакций термоокисления с разными константами скорости или отличными друг от друга механизмами разрушения макроцепей ПЛА. Отсутствие начальной ускоренной стадии потери веса на TГ-кривой, указывающей на разрушение полимера, в пористых образцах, возможно, обусловлено отсутствием модифицирующего и инициирующего действия порофоров, удаленных из матрицы. Ускорение термоокислительной деструкции, резкое понижение температуры начала протекания разрушения полимерной матрицы пористого ПЛА может быть связано с ростом содержания слабых связей. Такими могут быть карбонатные связи. Однако в этом случае, скорость падения массы полимера должна быть существенно меньшей по сравнению со скоростью, определяемой из интенсивности пика ДTГ-кривой.
Низкие значения величин Tн, энергии активации распада в пористых образцах по сравнению с исходным ПЛА при высоких величинах предэкспоненты, высоких значениях скорости потери массы, по-видимому, указывают на то, что в пористом ПЛА превалирует разрушение макроцепей по напряженным связям С–С. Важно отметить, что по литературным данным полученные в работе величины эффективной энергии активации (табл. 2) соответствуют ET распада напряженных углеводородных связей. Согласно кинетической концепции разрушения, роль внешнего напряжения сводится к уменьшению энергии активации разрыва связей и к увеличению вероятности их разрыва. В пористых полимерах напряженные связи могут образоваться в ходе порообразования под действием поверхностного натяжения. Наличие нескольких перекрывающихся пиков потери массы на ДTГ-кривой, скорее всего, указывает на неоднородное разрушение полимерных цепей вследствие гетерогенного распределения напряженных связей по пористой матрице ПЛА.
ЗАКЛЮЧЕНИЕ
Изменения в строении кристаллитов пористой матрицы могут быть вызваны действием, по крайней мере, трех факторов – это пластификация полимера в результате его наполнения порофорами, высокотемпературное воздействие и газообразование на стадии формирования пор. Из данных, приведенных в настоящей работе, можно заключить, что разрушение кристаллических структур ПЛА в большей степени обусловлено пластифицирующим действием порофоров и высокотемпературным воздействием. Относительно изменения структуры кристаллитов в процессе формирования пор можно предположить, что внутреннее давление, создающее поры, существенно разрушает порядок в молекулярной упаковке полимерных цепей, изменяет поверхностное натяжение и, как следствие, меняет закономерности и кинетику формирования кристаллических структур в процессе охлаждения полимера. Вклад этого эффекта зависит от метода порообразования, природы порообразователя и полимера.
Из полученных данных следует, что пористая структура в биодеградируемом полимере – полилактиде, применяемом в медицинских имплантатах, зависит от природы порофоров, методики порообразования и формы пор, определяется пластифицирующим действием порофора.
Установлено, что порообразование в полимере приводит к разрушению кристаллических областей и изменению структуры макромолекул аморфных областей. Структура пористого ПЛА может оказывать влияние на механизм и кинетику термоокислительной деструкции и, в результате, на термоокислительную стабильность полимера.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (код проекта 15-29-04862).
Список литературы
Штильман М.И. Полимеры медико-биологического назначения. М.: Академкнига, 2006.
Кирилова И.А., Подорожная В.Т., Легостаева Е.В., Шаркеев Ю.П., Уваркин П.В., Аронов А.М. // Хирургия позвоночника. 2010. № 1. С. 81.
Волова Т.Г. // Журн. Сибирского федерального ун-та. Сер. Биология. 2014. Т. 7. № 2. С. 103.
Бояндин А.Н., Николаева Е.Д., Шабанов А.В., Васильев А.Д. // Журн. Сибирского федерального ун-та. Сер. Биология. 2014. Т. 7. № 2. С. 174.
Misra S., Ansari T., Valappil S. // Biomaterials. 2010. № 31. C. 2806.
Иванов А.Е., Зубов В.П. // Успехи химии. 2016. Т. 85. № 6. С. 565.
Корж Н.А., Кладченко Л.А., Малышкина С.В., Тимченко И.Б. // Ортопедия, травматология и протезирование. 2005. № 4. С. 118.
Кирилова И.А. // Хирургия позвоночника. 2011. № 1. С. 68.
Тер-Асатуров Г.П., Лекишвили М.В., Бигваева А.Т., Аджиев К.С., Панкратов А.С., Рябов А.Ю., Юрасова Ю.Б. // Клеточная трансплантология и тканевая инженерия. 2012. № 1. С. 81.
Shumilova A.A., Nikolaeva E.D. // J. Siberian Federal Univ. Biology. 2016. V. 1. № 9. C. 53.
Khalyfa A., Vogt S., Weisser J., Grimm G., Rechtenbach A., Meyer W. // J. Mater. Sci.: Mater. Med. 2007. V. 18. № 5. P. 909.
Мураев А.А., Иванов С.Ю., Петров И.Ю., Ким Л.Е. Современные костезамещающие материалы в стоматологии и челюстно-лицевой хирургии. М.: Российский ун-т дружбы народов, 2015.
Бухарова Т.Б., Фатхудинов Т.Х., Цедик Л.В., Ильющенко А.Ф., Гольдштейн Д.В. // Клеточные технологии в биологии и медицине. 2009. Т. 147. № 1. С. 38.
Кокорев О.В., Аникеев С.Г., Дамбаев Г.Ц., Гюнтер В.Э., Ходоренко В.Н. // Сибирский медицинский журн. 2013. Т. 117. № 2. С. 117.
Копылов А.Н., Максимкин А.В., Анисимова Н.Ю., Сенатов Ф.С., Чуков Д.И., Чердынцев В.В., Лебединская О.В., Киселевский М.В. // Росс. биотерапевт. журн. 2013. Т. 12. № 4. С. 39.
Николаенко В.П., Астахов Ю.С. // Офтальмологические ведомости. 2008. Т. 1. № 2. С. 35.
Краснов А.П., Афоничева О.В., Соловьева В.А. Пат. 2327709 Россия. 2008.
Salasznyk R.M., Williams W.A., Boskey A., Batorsky A., Plopper G.E. // J. Biomed. Biotechnol. 2004. V. 2004. № 1. P. 24.
Ogura N., Kawada M., Chang W., Zhang Q., Lee S., Kondoh T., Abiko Y. // J. Oral Sci. 2004. V. 46. № 2. P. 207.
Никитин Л.Н., Николаев А.Ю., Саид'Галиев Э.Е., Гамзазаде А.И., Хохлов А.Р. // Сверхкритич. флюиды: теория и практика. 2006. Т. 1. № 1. С. 77.
Sarasua J.R., Lopez A.A., Balerdi P., Maiza I. // J. Mater. Sci. 2005. V. 40. № 7. P. 1855.
Garlotta D. // J. Polym. Environment. 2002. V. 9. № 2. P. 63.
Берштейн В.А., Егоров В.М. Дифференциальная сканирующая калориметрия в физикохимии полимеров. Л.: Химия, 1990.
Greco A., Maffezzoli A. // J. Therm. Anal. Cal. 2003. V. 72. № 3. P. 1167.
Wunderlich B. Macromolecular Physics. Crystal Melting. New York; London; Toronto; Sydney; San Francisco: Acad. Press, 1980. V. 3.
Скородумов В.Ф. // Физикохимия полимеров: синтез, свойства, применение. 1999. № 5. С. 40.
Chan J.H., Balke S.T. // Polym. Degrad. Stab. 1997. V. 57. № 1. P. 135.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия А)