

Высокомолекулярные соединения (серия А), 2019, T. 61, № 3, стр. 254-263
СОПОСТАВЛЕНИЕ ВЛИЯНИЯ НЕКОТОРЫХ МЕТОДОВ МОДИФИКАЦИИ НА ХАРАКТЕРИСТИКИ СВЕРХВЫСОКОМОЛЕКУЛЯРНОГО ПОЛИЭТИЛЕНА И КОМПОЗИТОВ НА ЕГО ОСНОВЕ
В. Г. Назаров a, *, В. П. Столяров a, Ф. А. Доронин a, А. Г. Евдокимов a, Г. О. Рытиков a, П. Н. Бревнов b, А. С. Заболотнов b, Л. А. Новокшонова b, А. А. Берлин b
a Московский политехнический университет
107023 Москва, Большая Семеновская ул., 38, Россия
b Институт химической физики им. Н.Н. Семенова Российской академии наук
119991 Москва, ул. Косыгина, 4, Россия
* E-mail: 110505n@gmail.com
Поступила в редакцию 18.10.2018
После доработки 21.11.2018
Принята к публикации 05.12.2018
Аннотация
Определена поверхностная энергия и рассчитана величина работы адгезии воды к поверхности сверхвысокомолекулярного полиэтилена и композитов на его основе, содержащих в качестве наполнителей нанопластины графита, дисульфид молибдена, шунгит и монтмориллонит. При получении композитов на основе сверхвысокомолекулярного полиэтилена методом полимеризационного наполнения in situ поверхностная энергия последних близка к характеристикам исходного полиэтилена и не превышает 35 мН/м. Рассмотренные в качестве вариантов модификации методы поверхностного фторирования, оксифторирования и обработки в кислородсодержащей плазме показали возможность существенного улучшения смачиваемости поверхности полимера вследствие увеличения поверхностной энергии. Наибольшее влияние на эту характеристику оказали оксифторирование и обработка в кислородсодержащей плазме. Эффективность обработки в кислородсодержащей плазме продемонстрирована на примере варьирования прочности при отрыве трафаретной краски 85000 Ondaflute от исходных и подвергнутых обработке образцов. При помощи сканирующей электронной микроскопии установлено сопровождающее поверхностную модификацию изменение морфологии поверхностных слоев образцов.
ВВЕДЕНИЕ
Физико-химические свойства поверхности полимеров, характеризуемые в основном свободной поверхностной энергией, имеют большое значение при разработке ряда технологий: формирование покрытий, получение биоматериалов для трансплантологии, изготовление приспособлений для решения задач микрофлюидики и другие [1–15].
В полной мере это относится к сверхвысокомолекулярному полиэтилену (СВМПЭ), находящему в последнее время все более широкое применение. СВМПЭ обладает рядом уникальных свойств, таких как высокое сопротивление удару и истиранию, низкое значение коэффициента трения скольжения, высокое удельное электрическое сопротивление, химическая стойкость и минимальное водопоглощение [16–20].
Сверхвысокомолекулярный полиэтилен является единственным полимером на сегодняшний момент, разрешенным для изготовления нагруженных частей имплантатов в связи с уникальным комплексом физико-механических свойств. Вследствие этого, изучение биостойкости и биосовместимости данного полимера представляется важной задачей [19].
Введение неорганических наполнителей позволяет повысить ряд важных эксплуатационных характеристик этого полимера, таких как износостойкость, жесткость и термостойкость, а также снизить газопроницаемость и горючесть при сохранении на высоком уровне остальных уникальных свойств.
Однако СВМПЭ, как и другие полиолефины, значится малополярным полимером с низкими показателями поверхностной энергии. Влияние наполнителей на поверхностные характеристики СВМПЭ, как правило, незначительно. В то же время, для практического применения необходимо увеличение поверхностной энергии СВМПЭ для улучшения межфазного взаимодействия [19, 20]. Эффективным способом достижения поставленной цели является модификация поверхности материалов и изделий. Наибольшее количество работ на эту тему связано с воздействием излучений различной природы на поверхность СВМПЭ: ионно-лучевая обработка [21–23], обработка потоком электронов [24], ультрафиолетовым лазером или γ-излучением [25, 26]. Более экономичным и простым в аппаратурном оформлении можно назвать метод плазмохимической обработки поверхности СВМПЭ [27–30], который позволяет увеличить адгезионную прочность, смачиваемость и поверхностную энергию полимера. Метод может быть видоизменен для целенаправленной химической модификации полимера с применением в качестве плазмообразующих газов кислорода, четырехфтористого углерода или других веществ [28, 29]. Из химических методов модификации поверхности СВМПЭ к настоящему времени известно лишь о поверхностном фторировании полимера газовой смесью фтора с инертными газами [11, 31–33]. Положительный эффект от поверхностной модификации заключается в увеличении адгезии волокон на основе СВМПЭ к олигомерным связующим, что дает возможность получать препреги улучшенного качества [32, 33].
Анализ литературных данных показывает, что существуют методы улучшения межфазного взаимодействия для СВМПЭ. Однако по сложности аппаратурного оформления, технологичности, экономической эффективности и другим показателям такие методы не равнозначны. По указанным характеристикам наиболее универсальными и перспективными, на наш взгляд, представляются методы плазмохимической и химической модификации полимера. Вместе с тем, вопросы сравнительной оценки этих методов с точки зрения достигаемых эффектов остаются еще малоисследованными, особенно для композитов на основе СВМПЭ. Кроме того, увеличение поверхностной энергии называется не единственным требованием, предъявляемым к изделиям из СВМПЭ. Так, например, этот полимер разрешен для изготовления нагруженных частей имплантатов, к качеству которых, как известно, предъявляются высокие требования по биостойкости, био- и гемосовместимости [19]. Эффективным методом улучшения указанных свойств может быть фторирование СВМПЭ, как показано на ряде примеров [11, 34].
Настоящая работа посвящена сравнительному исследованию влияния методов плазмохимической обработки, химической модификации газообразным фтором (фторирование) и смесью фтора с кислородом (оксифторирование) в буферном гелии на поверхностные свойства СВМПЭ и композитов на его основе. Выбор наполнителей для композиционных материалов обусловлен их высокой эффективностью для повышения механических, износостойких, электрофизических и барьерных характеристик СВМПЭ.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве объектов исследования использовали образцы ненаполненного сверхвысокомолекулярного полиэтилена (ТУ 2211-068-05796653-98; Открытое акционерное общество “Томский нефтехимический комбинат”) и композиты на основе СВМПЭ. Наполнителями служили нанопластины графита с удельной поверхностью 25.6 м2/г, толщиной 20 нм и продольными размерами 1–10 мкм, синтезированные путем термического расширения интеркалированного графита и последующего размола ультразвуком [35]; органомодифицированный монтмориллонит Сloisite 20А (ММТ), Sуд = 9.5 м2/г, исходный средний размер частиц 8 мкм; порошок дисульфида молибдена синтетический (МоS2) (ТУ 95-0106-024-92; Общество с ограниченной ответственностью “МетПрод”), средний размер частиц 1.6 мкм; шунгит (ТУ 5714-007-12862296-2016; Общество с ограниченной ответственностью Научно-производственный комплекс “Карбон-Шунгит”), природный минерал, содержащий углерод (30%) в виде фуллереноподобных структур, средний размер частиц 1.5 мкм.
В соответствии с методикой синтеза [35], композиты на основе СВМПЭ получали методом полимеризационного наполнения (полимеризации in situ) [35–37]. Синтез композиционных материалов на основе СВМПЭ и наполнителей (нанопластины графита, МоS2, шунгит) осуществляли путем закрепления (хемосорбции) катализатора (тетрахлорида ванадия) на поверхности частиц наполнителя и последующей полимеризации этилена на этих поверхностях.
Методика синтеза нанокомпозитов на основе СВМПЭ и ММТ описана в работе [38]. На первой стадии синтеза применяли интеркалирование катализатора в межслоевое пространство частиц. В результате последующей полимеризации достигали эксфолиации (расслоение) исходных частиц ММТ на отдельные нанослои толщиной ~1 нм, что было подтверждено методом рентгеноструктурного анализа [38].
Содержание наполнителя в композиционных материалах составляло: нанопластины графита 2.9 мас. % (образцы СВМПЭ + нанопластины графита), ММТ 5.3 мас. % (образцы СВМПЭ + + ММТ), МоS2 3.3 мас. % (образцы СВМПЭ + + МоS2) и шунгит 4.2 мас. % (образцы СВМПЭ + + шунгит). Средняя молекулярная масса полимера для всех типов композиционных материалов ~2.5 × 106 г/моль [35, 38]. Образцы материалов в виде пластин толщиной 2 мм для испытаний готовили методом горячего прессования при Т = = 160°С и давлении Р = 10 МПа.
Обработку поверхности образцов СВМПЭ и композитов на его основе в кислородсодержащей плазме проводили в вакуумной камере при давлении 13.3 Па. Образцы размером 12 × 12 см помещали внутрь цилиндрического активатора установки в зоне светящегося положительного столба низкотемпературного плазменного разряда и обрабатывали с одной стороны в течение 60 с при токе 60 мА.
Фторирование образцов осуществляли по разработанной методике [39] в реакторе из нержавеющей стали при 20°С фтором (15%) в смеси с гелием (содержащей в качестве технической примеси 0.5% кислорода) в течение 180 мин с предварительным и последующим вакуумированием реактора до остаточного давления 13.3 Па. Контроль результатов фторирования выполняли гравиметрическим методом по увеличению массы модифицированного образца, отнесенной к площади его поверхности (степень модификации $С _{A}^{F}$, кг/м2).
Оксифторирование образцов проводили в реакторе из нержавеющей стали объемом 300 мл с предварительным и последующим вакуумированием реактора. Использовали газовые смеси F2 7.5% и О2 10% в буферных газах (гелии и азоте с техническими примесями компонент воздуха ~0.5%). Содержание кислорода в газовой смеси 10 об. % задавали подачей в реактор расчетного количества осушенного воздуха, объем которого контролировали по вакуумметру с точностью 0.5 кПа (содержание кислорода в воздухе принимали 21 об. % [40]). Время обработки полимерных образцов составляло 180 мин. Контроль результатов оксифторирования также осуществляли гравиметрически (степень модификации $С _{A}^{M}$, кг/м2).
Для независимой верификации результатов гравиметрических измерений получали ИК-спектры с помощью ИК-фурье-спектрометра “СИМЕКС” ФТ-801 с использованием метода нарушенного полного внутреннего отражения на кристалле ZnSe.
Для определения величины краевого угла смачивания образцов СВМПЭ и композитов по дистиллированной воде и этиленгликолю до и после модификации применяли метод сидячей капли. Оптические изображения капли формировали путем извлечения отдельных кадров из потока видеоданных, формирующегося при скоростной микросъемке процесса растекания капли на специализированном лабораторном стенде. Для обеспечения одинаковых условий эксперимента капли жидкости объемом 6.1 мкл наносили на поверхность образца при 20°С микрометрическим шприцем, расположенным на фиксированной высоте от образца полимера. Относительная ошибка измерений составила ±8%. Расчет величины поверхностной энергии γ, ее полярной γP и дисперсионной γD составляющих проводили по уравнениям, приведенным в монографии [41].
Для определения адгезии образцов СВМПЭ и соответствующих композитов использовали трафаретную краску 85000 Ondaflute. Прочность при отрыве σ (ГОСТ 32299-2013 [42]) определяли на разрывной машине РМ-50. Нанесение красочного оттиска площадью 1 см2 на поверхность образцов осуществляли методом трафаретной печати на ручном станке карусельного типа Argon HT, путем продавливания краски сквозь незакрытые печатающие элементы (круг площадью 1 см2) полиэфирной печатной формы. Необходимый контакт между печатной формой и запечатываемой поверхностью и перенос краски достигали давлением упруго-эластичного ракеля на печатную форму. Исследуемый образец СВМПЭ с красочным оттиском размещали в устройстве фиксации образцов машины РМ-50. На красочном оттиске исследуемого образца клеем “Момент” (“Henkel”) фиксировали металлический цилиндр с площадью торца 1 см2, который посредством гибкой тяги крепили к верхнему зажиму машины РМ-50. Регистрацию прочности при отрыве краски от поверхности образца осуществляли в программе Stretch Test.
Для контроля морфологических изменений получали изображения поверхностей модифицированных полимеров, сформированные с помощью автоэмиссионного растрового электронного микроскопа JSM 7500-F (“JEOL”, Япония), функционировавшего при ускоряющем напряжении 10 кВ. Достигнутое пространственное разрешение составило 1 нм. Для снижения уровня статического заряда и предотвращения разрушения под действием электронного пучка непроводящих образцов проводили предварительное напыление на поверхность образцов слоя платины (~8 нм толщиной).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Первоначально были изучены поверхностные характеристики исходного СВМПЭ и композитов на его основе без модификации поверхности образцов.
Результаты измерений краевых углов смачивания по воде и этиленгликолю, а также результаты расчетов поверхностной энергии (ее полярной и дисперсионной составляющих) для исходных образцов СВМПЭ и композитов на его основе представлены в табл. 1. Кроме того, по уравнению Дюпре–Юнга [43]:
где Wa – работа адгезии, σжт – поверхностное натяжение жидкости, θ – краевой угол смачивания, была проведена оценка работы адгезии воды к поверхностям исследованных экспериментальных образцов (табл. 1).Таблица 1.
Значения краевого угла смачивания, поверхностной энергии, ее полярной и дисперсионной составляющих, а также работы адгезии воды к полимеру для СВМПЭ и композитов на его основе
| Материал | Краевой угол смачивания θ, град | $\gamma _{s}^{D}$, мН/м | $\gamma _{s}^{P}$, мН/м | γs, мН/м | Wa × 10–2, мДж/м2 |
|
|---|---|---|---|---|---|---|
| θвода | θэг | |||||
| СВМПЭ без наполнителя | 86 | 60 | 23 | 5.6 | 28 | 0.8 |
| СВМПЭ + ММТ | 89 | 57 | 33 | 2.2 | 35 | 0.8 |
| СВМПЭ + нанопластины графита | 71 | 54 | 12 | 21 | 32 | 0.8 |
| СВМПЭ + МоS2 | 80 | 58 | 18 | 10 | 28 | 0.9 |
| СВМПЭ + шунгит | 78 | 60 | 13 | 14 | 28 | 0.9 |
Из полученных результатов следует, что присутствие в композитах наполнителей мало сказывается на значениях краевых углов смачивания по воде и этиленгликолю. Как следствие, расчетные значения γs и Wa отличаются не более чем на 15–20%. Это связано с особенностями получения композитов методом полимеризационного наполнения, когда катализатор наносится на наполнитель и после полимеризации полимер обволакивает наполнитель. Такое предположение согласуется и с данными ИК-спектроскопии (рис. 1). Основные полосы поглощения, соответствующие колебаниям групп СН2 в полиэтилене, для всех образцов совпадают.
Рис. 1.
ИК-спектры исходного СВМПЭ (1) и композитов на его основе ММТ (2), нанопластины графита (3), МоS2 (4) и шунгит (5). Спектры 2–5 смещены на 10–40% вниз соответственно.
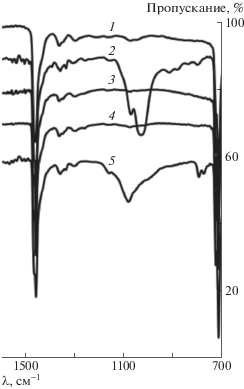
Новые слабые полосы поглощения в области 1200–1000 см–1 наблюдаются лишь для ММТ и шунгита. В случае ММТ эти полосы связаны, вероятно, с колебаниями связи Si–О, а для шунгита – с наличием в его составе еще и фуллереноподобных структур, о чем свидетельствует более широкая полоса поглощения.
Если введение наполнителя в СВМПЭ слабо влияет на поверхностную энергию композита на его основе, то дисперсионная и полярная составляющая поверхностной энергии существенно изменяются. Так, полярная составляющая $\gamma _{s}^{P}$ при введении в СВМПЭ нанопластин графита, МоS2 или шунгита увеличивается в 2–4 раза в зависимости от наполнителя. С одной стороны, это может быть связано с изменением аморфно-кристаллической структуры поверхностного слоя, так как, например, концентрация и структура кристаллитов на поверхности аморфно-кристаллических полимеров могут существенно влиять на величину $\gamma _{s}^{P}$ [43]. С другой стороны, при введении в сверхвысокомолекулярный полиэтилен ММТ наблюдается возрастание на 50% дисперсионной составляющей $\gamma _{s}^{D}$, что, вероятно, связано с увеличением плотности упаковки макромолекул в поверхностном слое и уменьшением свободного объема полимера [43].
Таким образом, значения поверхностной энергии СВМПЭ и композитов на его основе находятся в пределах 27–35 мН/м, в то время как для ряда задач практического применения этого пластика требуются более высокие значения поверхностной энергии. Например, для успешного запечатывания красками на полимер, по данным работы [44], теоретически допустимый уровень этой величины 42–44 мН/м, а следовательно, в случае СВМПЭ и композитов на его основе необходимо применение поверхностной модификации.
В оптимальных условиях обработки СВМПЭ и композитов на его основе кислородсодержащей плазмой (60 мА в течение 60 с), фторированием (F2 15%, 3 ч) и оксифторированием (F2 7.5%, О2 10%, 3 ч) были получены образцы с поверхностными характеристиками, представленными в табл. 2.
Таблица 2.
Значения краевых углов смачивания, поверхностной энергии, ее полярной и дисперсионной составляющих, а также работы адгезии воды к полимеру для различных способов модификации поверхности
| Образец | Способ обработки | Краевой угол смачивания, град | $\gamma _{s}^{D}$, мН/м | $\gamma _{s}^{P}$, мН/м | γs, мН/м | Wa × 10–2, мДж/м2 | |
|---|---|---|---|---|---|---|---|
| θ вода | θ эг | ||||||
| СВМПЭ без наполнителя | Кислородсодержащая плазма | 57 | 39 | 11 | 33 | 44 | 1.1 |
| Оксифторирование | 52 | 24 | 16 | 31 | 47 | 1.2 | |
| Фторирование | 60 | 33 | 18 | 24 | 42 | 1.1 | |
| СВМПЭ + ММТ | Кислородсодержащая плазма | 47 | 17 | 14 | 37 | 51 | 1.2 |
| Оксифторирование | 57 | 20 | 23 | 22 | 46 | 1.1 | |
| Фторирование | 62 | 31 | 23 | 19 | 42 | 1.1 | |
| СВМПЭ + нанопластины графита | Кислородсодержащая плазма | 45 | 20 | 12 | 42 | 53 | 1.2 |
| Оксифторирование | 50 | 17 | 17 | 33 | 50 | 1.2 | |
| Фторирование | 55 | 32 | 13 | 32 | 45 | 1.1 | |
| СВМПЭ + MoS2 | Кислородсодержащая плазма | 47 | 17 | 14 | 38 | 52 | 1.2 |
| Оксифторирование | 41 | 17 | 9.4 | 48 | 58 | 1.3 | |
| Фторирование | 59 | 37 | 14 | 28 | 42 | 1.1 | |
| СВМПЭ + шунгит | Кислородсодержащая плазма | 45 | 18 | 12 | 42 | 54 | 1.2 |
| Оксифторирование | 49 | 12 | 18 | 33 | 50 | 1.2 | |
| Фторирование | 58 | 46 | 6.5 | 38 | 44 | 1.1 | |
Из приведенных в табл. 2 данных видно, что исследованные методы модификации поверхности СВМПЭ и композитов на его основе позволяют существенно улучшить смачиваемость полимера и повысить поверхностную энергию на 20–80%, а работу адгезии воды к полимеру на ~40%. Наиболее высокие значения этих параметров достигаются при использовании для модификации поверхности полимера кислородсодержащей плазмы или оксифторирования, причем получаемые каждым из этих методов значения γs и Wa близки друг к другу и колеблются для первого показателя от 46 до 54 мН/м2, а для второго – (1.1–1.2) × 102 мДж/м2.
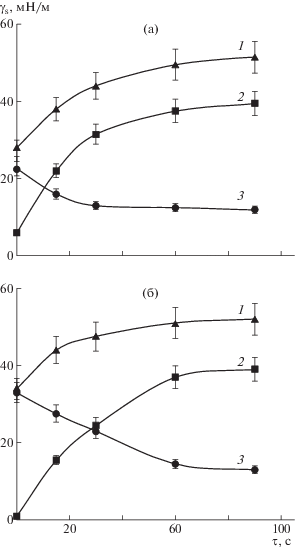
При модификации поверхности полимера кислородсодержащей плазмой большое значение имеет время обработки. На рис. 2 показана кинетика изменения поверхностной энергии и ее составляющих на примере исходного СВМПЭ и композита, содержащего ММТ. Видно, что существенное изменение поверхностной энергии наблюдается в первые 20 с и достигает практически максимальных значений к 60 с. Таким образом, оптимальной продолжительностью плазмохимической модификации полимера можно считать временной интервал в 60 с.
Рис. 2.
Кинетика изменения поверхностной энергии γs (1), ее полярной $\gamma _{s}^{P}$ (2) и дисперсионной $\gamma _{s}^{D}$ (3) составляющих для СВМПЭ (а) и композита СВМПЭ + + ММТ (б) при обработке в кислородсодержащей плазме.
При модификации поверхности полимера фторированием наблюдаются более низкие значения поверхностной энергии и работы адгезии воды к полимеру, что объясняется интенсивным фторированием макромолекул поверхностного слоя, уменьшающим его гидрофильность.
Очевидно, что для всех трех исследованных методов достигаемый при модификации эффект связан с образованием на поверхности полимера полярных кислородсодержащих групп. Об этом свидетельствует резкое увеличение полярной составляющей поверхностной энергии в 2–20 раз. Это подтверждают и данные ИК-спектроскопии в режиме НПВО, которые показаны на примере СВМПЭ и композита, содержащего шунгит (рис. 3 и 4); для остальных образцов получены аналогичные данные.
Рис. 3.
ИК-спектры СВМПЭ без наполнителя: 1 – исходный СВМПЭ, 2 – фторированный образец, 3 – оксифторированный образец, 4 – образец после обработки в кислородсодержащей плазме.
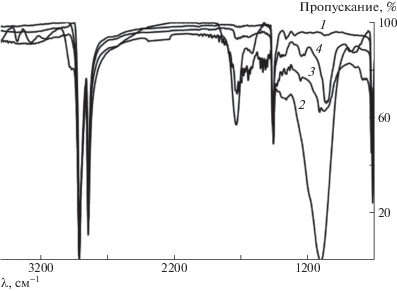
Рис. 4.
ИК-спектры композита СВМПЭ+шунгит: 1 – исходный СВМПЭ + шунгит, 2 – фторированный образец, 3 – оксифторированный образец, 4 – образец после обработки в кислородсодержащей плазме.
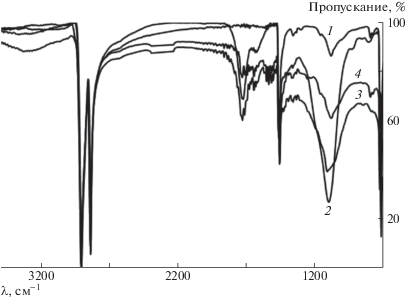
В ИК-спектрах исходного СВМПЭ и композита после модификации появляются полосы поглощения разной интенсивности в области 1600–1800 см–1, подтверждающие образование в полимерной цепи карбонильных групп разного строения за счет присутствующего в газообразном фторе и в низкотемпературной плазме кислорода [45], а также уширенная полоса поглощения в области 3200–3400 см–1, которую можно отнести к группе ОН, и сильная широкая полоса в области 950–1300 см–1 для фторированных и оксифторированных образцов, соответствующая полосам поглощения групп С–Fx (х = 1, 2 и 3).
Кроме того, для всех трех методов модификации образцов исходного СВМПЭ и композитов отмечается существенное снижение дисперсионной составляющей поверхностной энергии $\gamma _{s}^{D}$, в бóльшей степени при обработке кислородсодержащей плазмой. Это, по-видимому, связано с уменьшением плотности упаковки фрагментов макромолекул в поверхностном слое в процессе модификации, т.е. с его “разрыхлением” [46].
Существенное изменение морфологии поверхностного слоя, особенно при обработке в кислородсодержащей плазме, подтверждают фотографии поверхности СВМПЭ и композитов на примере СВМПЭ+ММТ, выполненные на автоэмиссионном растровом электронном микроскопе (рис. 5).
Рис. 5.
Фотографии поверхности композита СВМПЭ+ММТ: а – исходный СВМПЭ + ММТ, б – фторированный образец, в – образец после обработки в кислородсодержащей плазме, представленный в разном масштабе.

Видно, что после модификации поверхность образца приобретает сильную волнистость, т.е. увеличивается ее шероховатость (см. также работы [47, 48]). Этот эффект, очевидно, вносит свой вклад в увеличение гидрофильности модифицированной поверхности полимера согласно уравнению Венцеля–Дерягина [43].
Эффективность методов модификации для увеличения поверхностной энергии СВМПЭ и композитов на его основе изучена на примере изменения прочности при отрыве краски σ от образцов до и после их обработки в кислородсодержащей плазме (рис. 6).
Рис. 6.
Гистограмма значений прочности σ при отрыве краски от СВМПЭ и композитов на его основе: 1 – СВМПЭ без наполнителя, 2 – СВМПЭ + ММТ, 3 – СВМПЭ + нанопластины графита, 4 – СВМПЭ + MoS2, 5 – СВМПЭ + + шунгит; темные полосы – исходные образцы, серые полосы – образцы, модифицированные в кислородсодержащей плазме.
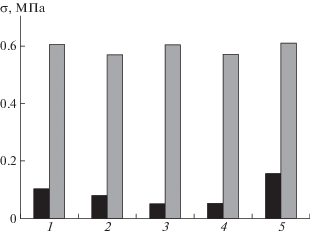
Из приведенной гистограммы следует, что такая обработка увеличивает прочность при отрыве краски от образцов полимера в 4–11 раз. Важно, если разрушение контакта “полимер–краска–приклеенный к ней металлический цилиндр” для исходных немодифицированных образцов происходит на границе раздела “полимер–краска”, то для модифицированных образцов наблюдается отрыв на границе раздела “краска–приклеенный к ней металлический цилиндр” (рис. 7).
Рис. 7.
Характер отслоения краски от композита СВМПЭ + шунгит: а – исходный образец, б – образец, модифицированный кислородсодержащей плазмой.

Таким образом, характер разрушения контакта “полимер–краска–приклеенный к ней металлический цилиндр” в случае немодифицированных образцов свидетельствует, что наблюдаемая при этом величина прочности при отрыве соответствует адгезии краски к полимеру. В случае модифицированных образцов разрушение контакта на границе клеевого соединения краски с металлическим цилиндром предполагает, что адгезия краски к полимеру больше фиксируемой в эксперименте величины прочности при отрыве σ.
ЗАКЛЮЧЕНИЕ
При получении композитов на основе СВМПЭ методом полимеризационного наполнения in situ поверхностные характеристики последних близки к характеристикам исходного СВМПЭ и по поверхностной энергии не превышают 35 мН/м. Для решения ряда задач практического применения рассмотренных материалов необходима модификация поверхности полимера. Рассмотренные в качестве вариантов модификации методы поверхностного фторирования, оксифторирования и обработки в кислородсодержащей плазме показали возможность существенного улучшения смачиваемости поверхности полимера за счет увеличения поверхностной энергии. Наибольшую эффективность воздействия на увеличение указанных характеристик демонстрируют методы оксифторирования и обработки в кислородсодержащей плазме, что, в частности, подтверждается значительным увеличением прочности при отрыве краски от образцов после их обработки в кислородсодержащей плазме.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (код проекта 18-29-05037 мк).
Список литературы
Ye Yuan, Xu Yongjun, Liu Yuping, Tu Weiping, Liao Junxu // Polym. Mater. Sci. Technol. Eng. 2013. V. 29. № 11. P. 187.
Nie Z., Kumacheva E. // Nature Mater. 2008. V. 7. № 4. P. 277.
Patankar N.A. // Langmuir. 2003. V. 19. P. 1249.
Barish J.A., Goddard J.M. // J. Appl. Polym. Sci. 2011. V. 120. № 5. P. 2863.
Extrand C.W. // Langmuir. 2005. V. 21. P. 10370.
Callies M., Que’re D. // Soft Matter. 2005. V. 1. P. 55.
Feng X., Jiang L. // Adv. Mater. 2006. V. 18. P. 3063.
Gao L., McCarthy T.J. // Langmuir. 2007. V. 23. P. 3762.
Changqing Yi, Cheuk-Wing Li, Shenglin Ji, Mengsu Yang // Anal. Chim. Acta. 2006. V. 560. № 1–2. P. 1.
Vinogradova O.I., Belyaev A.V. // J. Phys. Condens. Matter. 2011. V. 23. P. 184104.
Назаров В.Г. Поверхностная модификация полимеров. М.: Московский гос. ун-т печати, 2008.
Logtenberg H., Lopez-Martinez M.J., Feringa B.L., Browne W.R., Verpoorte E. // Lab. Chip. 2011. V. 11. № 12. P. 2030.
Чалых А.Е., Степаненко В.Ю., Щербина А.А., Стоянов О.В. // Вестн. технол. ун-та. 2015. Т. 18. № 18. С. 36.
Пахомов К.С., Антипов Ю.В., Чалых А.Е., Будылин Н.Ю., Стоянов О.В., Симонов-Емельянов И.Д. // Вестн. технол. ун-та. 2016. Т. 19. № 1. С. 100.
Чалых A.E., Бусыгин В.Б. // Высокомолек. соед. Б. 1996. Т. 38. № 11. С. 1917.
Андреева И.Н., Веселовская Е.В., Наливайко Е.И., Печенкин А.Д., Бухгалтер В.И., Поляков А.В. Сверхвысокомолекулярный полиэтилен высокой плотности. Л.: Химия, 1982.
Галицын В.П. Дис. … д-ра хим. наук. Тверь: Федеральное гос. унитарное предприятие “Научно-исследовательский институт синтетического волокна с экспериментальным заводом”, 2012.
Ю-Винг Май, Жонг-Жен Ю. Полимерные нанокомпозиты. М.: Техносфера, 2011.
Сенатов С.Ф. Дис. … канд. физ.-мат. наук. М.: Национальный исследовательский технологический университет “Московский государственный институт стали и сплавов”, 2013.
Maity J., Jacobs C., Das C.K., Alam S., Singh R.P. // Mater. Sci. Eng. A. 2008. V. 479. № 1–2. P. 125.
Valenza A., Visco A.M., Torrisi L., Campo N. // Polymer. 2004. V. 45. № 5. P. 1707.
Abdul-Kader A.M., Turos A., Jagielski J. // Vacuum. 2005. V. 78. № 2–4. P. 281.
Turos A., Abdul-Kader A.M., Ratajczak R., Stonert A. // Vacuum. 2009. V. 83. P. 54.
Visco A.M., Campo N. // Inc. Plasma Sci. Plasma Technol. 2003. V. 158. № 10. P. 621.
Kraus E., Baudrit B., Heidemeyer P., Bastian M., Stoyanov O., Starostina I. // J. Adhesion. 2017. V. 93. № 3. P. 204.
Панин С.В., Корниенко Л.А., Пувадин Т., Мержиевский Л.А., Шилько С.В., Полтаранин М.А., Иванова Л.Р., Коробейников М.В., Штарклев Е.А. // Трение и смазка в машинах и механизмах. 2011. № 12. С. 26.
Eun Hee Cho, Sang Gil Lee, Jeong Koo Kim // Curr. Appl. Phys. 2005. V. 5. № 5. P. 475.
Oosterom R., Ahmed T.J., Poulis J.A., Bersee H.E.N. // Med. Eng. Phys. 2006. V. 28. № 4. P. 323.
Teodoru S., Kusano Yu., Rozlosnik N., Michelsen P.K. // Plasma Proc. Polym. 2009. V. 6. № S1. P. 375.
Kraus E., Orf L., Baudrit B., Heidemeyer P., Bastian M., Bonenberger R., Starostina I., Stoyanov O. // App. Surf. Sci. 2016. V. 371. P. 365.
Kharitonov A.P., Simbirtseva G.V., Tressaud A., Durand E., Labrugere C., Dubois M. // J. Fluor. Chem. 2014. V. 165. P. 49.
Tressaud A., Durand E., Labrugere C., Kharitonov A.P., Simbirtseva G.V., Kharitonova L.N., Dubois M. // Acta Chim. Sloven. 2013. V. 60. № 3. P. 495.
Nazarov V.G., Stolyarov V.P., Evlampieva L.A., Fokin A.V. // Dokl. Phys. Chem. 1996. V. 350. P. 268.
Sevast'yanov V.I., Nemets E.A., Stolyarov V.P., Baranov V.A., Bozhko N.N., Nazarov V.G. // Inorg. Mater. Appl. Res. 2011. V. 2. № 2. P. 146.
Brevnov P.N., Kirsankina G.R., Zabolotnov A.S., Krasheninnikov V.G., Grinev V.G., Berezkina N.G., Sinevich E.A., Shcherbina M.A., Novokshonova L.A. // Polymer Science C. 2016. V. 58. № 1. P. 38.
Новокшонова Л.А., Мешкова И.Н. // Высокомолек. соед. А. 1994. Т. 36. № 4. С. 629.
Ivanyuk A.V., Gerasin V.A., Rebrov A.V., Pavelko R.G., Antipov E.M. // J. Eng. Phys. Thermophys. 2005. V. 78. № 5. P. 926.
Brevnov P.N., Zabolotnov A.S., Krasheninnikov V.G., Novokshonova L.A., Pokid’ko B.V., Bakirov A.V., Babkina O.N. // Kinet. Katal. 2016. V. 57. № 4. P. 482.
Nazarov V.G., Stolyarov V.P., Baranov V.A., Evlam-pieva L.A. // Russ. J. General Chem. 2009. V. 79. P. 565.
Кнунянц И.Л., Зефиров Н.С., Кулов Н.Н. Химическая энциклопедия. М.: Советская энциклопедия, 1990. Т. 2.
Ван Кревелен Д.В. Свойства и химическое строение полимеров / Пер. с англ. под ред. А.Я. Малкина. М.: Химия, 1976. Van Krevelen D.W., Te Nijenhuis K. Properties of Polymers. New York. Elsevier Pub. Co., 1972.
ГОСТ 32299-2013 (ISO 4624:2002). Материалы лакокрасочные. Определение адгезии методом отрыва.
Щукин Е.Д., Перцов А.В., Амелина Е.А. Коллоидная химия. М.: Высш. шк., 2006.
Ричард Сотвард Р. Материальные аспекты УФ-печати [Электронный ресурс] // PUBLISH/ДИЗАЙН. ВЕРСТКА. ПЕЧАТЬ. 2008. 23 июня. URL: https://www.publish.ru/articles/200806_5169221 (дата обращения: 04.06.2018)
Купцов А.Х., Жижин Г.Н. Фурье-КР и Фурье-ИК-спектры полимеров. М.: Техносфера, 2013.
Shcherbina A.A., Chalykh A.E. // Prot. Metals Phys. Chem. Surf. 2015. V. 51. № 3. C. 341.
Назаров В.Г., Нагорнова И.В., Столяров В.П., Доронин Ф.А., Евдокимов А.Г., Бревнов П.Н., Заболотнов А.С., Новокшонова Л.А. // Химическая физика. 2018. № 12. С. 63.
Nazarov V.G., Kondratov A.P., Stolyarov V.P., Evlampieva L.A., Baranov V.A., Gagarin M.V. // Polymer Science A. 2006. V. 48. № 11. P. 1164.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия А)