

Высокомолекулярные соединения (серия А), 2020, T. 62, № 6, стр. 444-452
ОПТИМИЗАЦИЯ УСЛОВИЙ РАЗДЕЛЕНИЯ ПОЛИМЕРОВ МЕТОДОМ ГИДРОДИНАМИЧЕСКОЙ ХРОМАТОГРАФИИ
А. А. Курганов a, *, А. А. Королев a, Е. Н. Викторова a
a Институт нефтехимического синтеза им. А.В. Топчиева Российской академии наук
119991 Москва, Ленинский пр., 29, Россия
* E-mail: kurganov@ips.ac.ru
Поступила в редакцию 02.03.2020
После доработки 04.06.2020
Принята к публикации 19.06.2020
Аннотация
Для известной теоретической модели гидродинамической хроматографии, связывающей относительное удерживание аналита и эффективность разделения с характеристическим параметром системы, предложены выражения, позволяющие найти оптимальные условия проведения анализа. Теоретическая модель хорошо предсказывает относительное удерживание аналитов, но сильно переоценивает эффективность разделения, особенно для полимеров высокой молекулярной массы. Наблюдаемое расхождение обусловлено невозможностью в пределах одного разделения создать оптимальные условия для всех компонентов пробы и(или) влиянием полидисперсности исследуемых полимерных образцов на эффективность разделения.
Молекулярная масса полимера и его молекулярно-массовое распределение являются важнейшими характеристиками высокомолекулярных соединений. В хроматографии разработаны различные методы для определения этих характеристик [1]. Наиболее распространенный на сегодняшний день хроматографический метод анализа полимеров – это гель-проникающая хроматография, известная также как эксклюзионная хроматография [2]. Однако при анализе методом ГПХ полимеров с ММ выше нескольких миллионов дальтон в литературе были отмечены случаи расщепления пика полимера и появление зависимости результатов анализа от скорости потока подвижной фазы [3–5]. Такой эффект авторы работ связывали с конформационным переходом макромолекулы из клубка в вытянутую конформацию, равновесие между которыми смещается в сторону последней при увеличении скорости потока. Вытянутая цепочка в процессе движения по колонке будто “цепляется” на разветвлении транспортных каналов сорбента [6].
Чтобы избежать подобных осложнений, анализ очень высокомолекулярных полимеров предпочтительнее выполнять на колонках, не заполненных каким-либо адсорбентом. Одним из возможных хроматографических методов, потенциально способным реализовать подобное разделение, является гидродинамическая хроматография (ГДХ). Теоретические основы метода были заложены работой E.A. DiMarzio и C.M. Guttman [7], в которой они в приближении классической модели Дебая определили среднюю скорость движения полимерной молекулы, выступавшей в качестве жесткой, но проницаемой сферы в потоке жидкости, движущейся по каналу постоянного сечения. Параболический профиль потока жидкости рассматривался как постоянный и не подверженный влиянию частиц, перемещающихся потоком жидкости. По данным авторов, это ограничивает применение модели значениями характеристического параметра λ = rp /rо меньше 0.5 (rp и rо – радиус клубка полимера и капиллярного канала соответственно). Полученные в работе [7] уравнения для каналов различного сечения (здесь показано выражение для канала круглого сечения):
(1)
${{{v}}_{p}} = {{u}_{o}}\left[ {1 - \frac{{{{{(1 - {{r}_{p}}{\text{/}}{{r}_{o}})}}^{2}}}}{2} - {\gamma }{{{\left( {\frac{{{{r}_{p}}}}{{{{r}_{o}}}}} \right)}}^{2}}} \right]$Таблица 1.
Молекулярная масса, полидисперсность и размер клубка полистирольных стандартов, использованных в работе
| Мр × 10–3 | Ð | r0, нм | Dm × 10–5, мм2/c | Мр × 10–3 | Ð | r0, нм | Dm × 10–5, мм2/c |
|---|---|---|---|---|---|---|---|
| 0.078 | – | 0.16 | 296 | 500 | 1.07 | 27.6 | 2.11 |
| 11 | 1.08 | 2.93 | 18.1 | 675 | 1.07 | 32.93 | 1.78 |
| 39 | 1.07 | 6.16 | 8.88 | 730 | 1.01 | 34.48 | 1.70 |
| 68 | 1.05 | 8.54 | 6.49 | 770 | 1.01 | 35.58 | 1.65 |
| 76 | 1.04 | 9.12 | 6.10 | 1200 | 1.05 | 46.18 | 1.29 |
| 127 | 1.05 | 12.33 | 4.56 | 1440 | 1.09 | 51.41 | 1.16 |
| 180 | 1.07 | 15.14 | 3.74 | 3200 | 1.04 | 82.21 | 0.74 |
| 390 | 1.06 | 23.85 | 2.42 | 4400 | 1.10 | 99.14 | 0.62 |
| 411 | 1.06 | 24.59 | 2.35 | 9000 | 1.10 | 151.01 | 0.41 |
Примечание. Радиус клубка полимера рассчитан по уравнению r0= 1.23 × 10–2 $M_{p}^{{0.588}}$ нм, коэффициент диффузии полимера – по уравнению Dm = 1.23 × 10–2 $M_{p}^{{ - 0.564}}$ мм2/с [9]; приведенные уравнения получены для растворов в ТГФ. В работе использовали метилен хлорид; оба растворителя являются хорошими растворителями для полистирола, вносимой при этом неточностью пренебрегали.
Рис. 1.
Схема разделения полимерных молекул в ламинарном потоке жидкости в цилиндрическом капиллярном канале.
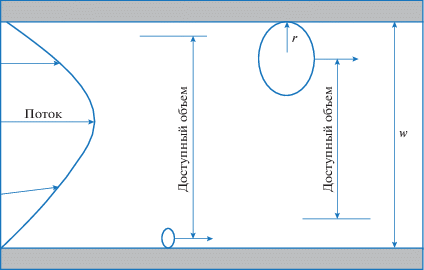
В дальнейшем уравнение (1) было приведено к более простому виду, где фигурировали только относительные величины: характеристический параметр λ и относительное удерживание полимера R = to/tp (to и tp – время элюирования из колонки низкомолекулярного маркера и полимера соответственно) [8]:
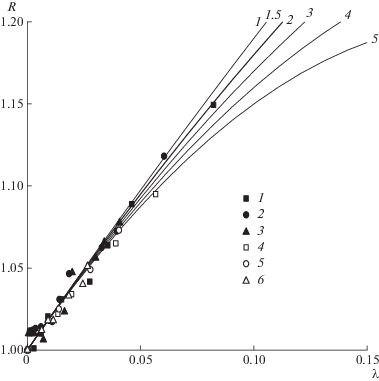
Вопрос о величине параметра С, заменившего параметр γ, вновь привлек внимание целого ряда исследователей [8]. В разных моделях величина параметра С варьируется в пределах от 0 до ~5. На рис. 2 представлена зависимость R от λ для различных величин параметра С. Большинство исследователей [9–12] считает, что параметр С имеет значение близкое к двум. При C = 2 максимальная величина R составляет ~1.5 и достигается при λ = 0.5, это соответствует тому, что диаметр клубка макромолекулы равен половине диаметра капилляра. Так, для капилляра радиусом 1 мкм максимальный радиус клубка равен 0.5 мкм, а ММ такого полимера составляет ~6.8 × 107. Теоретически такой капилляр позволяет анализировать полимеры с очень большой ММ. Однако максимальный временной диапазон, в котором будет происходить это разделение (т.е. интервал между временем выхода полимера максимально допустимой массы и временем выхода низкомолекулярного маркера), очень ограничен – всего 0.5 от времени выхода полимера tp. При таких условиях разделение полимеров возможно, только если колонка обладает достаточной эффективностью.
Рис. 2.
Зависимость теоретическая (кривые) и экспериментальная (точки) относительного удерживания полимеров R от характеристического параметра λ. Теоретические кривые приведены для серии значений параметра С, экспериментальные точки измерены на капиллярах диаметра d0 = 2.0 (1), 5.0 (2), 10.0 (3), 1.26 (4), 1.76 (5) и 2.72 мкм (6).
Эффективность разделения определяется процессами уширения первоначальной зоны введенной пробы, при движении пробы по каналу, и характеризуется высотой приведенной теоретической тарелки h. Экспериментально определяемая высота тарелки hap складывается из многих факторов, среди которых в случае анализа полимеров выделяют уширение, связанное с диффузионными процессами, в том числе с не оптимальностью условий разделения hD, и уширение, связанное с полидисперсностью анализируемых образцов hD. Роль диффузионных процессов в ГДХ, рассмотренная еще авторами работы [7], получила свое развитие [13–15] в качестве модели, предложенной для частиц нулевого размера. С учетом конечного размера макромолекул и поперечной диффузии в капилляре было предложено выражение [16] для зависимости приведенной высоты тарелки hD от приведенной скорости потока v, которое справедливо для 0 < λ < 0.2 [9]:
(3)
${{h}_{D}} = \frac{{{{{(1 - {\lambda })}}^{{ - 2}}}}}{{(1 + 2{\lambda } - 2{{{\lambda }}^{2}})}}\left( {\frac{2}{v}{{f}_{1}} + \frac{v}{{96}}{{f}_{2}}} \right)$Здесь hD – приведенная высота тарелки, обусловленная диффузионными процессами в капилляре,
и $v$ – приведенная скорость потока где ${{{v}}_{0}}$ = L/to – линейная скорость потока элюента; L – длина капиллярного канала; Dm – коэффициент диффузии сорбата; H = L/N – высота эквивалентной теоретической тарелки, находимая для деления длины колонки L на число теоретических тарелок N.Функции f1 и f2 зависят только от λ и задаются следующими выражениями:
(6)
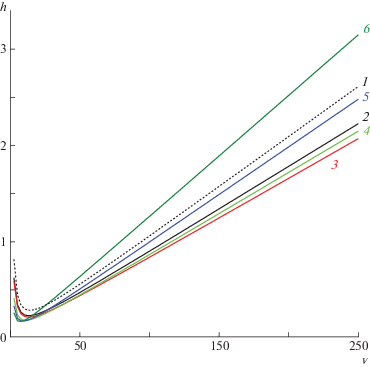
$\begin{gathered} {{f}_{1}} = 1 - 1.125{\lambda }\ln \left( {\frac{1}{{\lambda }}} \right) - 1.539{\lambda } \\ {{f}_{2}} = 1 - 3.862{\lambda } + 14.40{{{\lambda }}^{2}} \\ \end{gathered} $Как следует из уравнения (3), высота теоретической тарелки hD зависит только от приведенной скорости потока ${v}$ и характеристического параметра λ. Типичные профили зависимости, которая аналогична зависимости приведенной в работе [9], показаны на рис. 3 для серии значений λ < < 0.25. Обращает на себя внимание очень высокая потенциальная эффективность разделения (hD < 1 при ${v}$ < 100) и, вместе с тем, крутая правая ветвь представленной зависимости. На практике, однако, эффективность колонки стремительно падает с ростом ММ полимера [6, 8]. Вероятно, что представленная модель не вполне точно описывает процесс разделения высокомолекулярных полимеров в ГДХ, и здесь необходимы дополнительные экспериментальные исследования, которые должны пролить свет на поведение полимеров высокой ММ в процессе ГДХ. Результаты таких экспериментальных исследований и составляют основу данной публикации.
Рис. 3.
Зависимость высоты приведенной теоретической тарелки h от приведенной скорости потока v для серии значений характеристического параметра λ = 0 (1), 0.05 (2), 0.10 (3), 0.15 (4), 0.20 (5) и 0.25 (6).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В качестве разделительной колонки использовали немодифицированные кварцевые капилляры (длина и диаметр капилляров приведены в табл. 2) фирмы “Polymicro” (США). Со стороны выходного конца капилляра на расстоянии 0.5 м от конца удаляли полиимидное покрытие, чтобы создать “окошко” для измерения оптической плотности протекающего элюента. Длина “окошка” составляла ~5 мм. Подготовленный таким образом капилляр закрепляли в оптической ячейке УФ-детектора системы капиллярного электрофореза “Капель” (“Люмекс”, Россия). Выходной сигнал УФ-детектора брали непосредственно с выхода блока предусилителя, смонтированного в оптической ячейке “Капели”, и подавали на аналого-цифровой преобразователь системы обработки хроматографических данных фирмы “Бойсофт” (Россия). На выходной конец капиллярной колонки натягивали кусочек тефлонового капилляра и погружали его в емкость с элюентом, чтобы избежать испарения из него растворителя и засорения полимерными пробами, а также для визуального контроля потока элюента.
Таблица 2.
Параметры капиллярных колонок и условия разделения полимеров
| №, п/п | Параметры колонки | M × 10–3 | λ = rp/ro × × 10–3 | R | tp, c | σ, c | hap | hD | hp | ${{{v}}_{{ap}}}$ | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| экспери-ментальные данные | теоретиче-ские данные | ||||||||||
| 1 | L = 0.72 (0.85) м | 0.092 | 0.139 | 1.0 | 1.0 | 1572 | 13.0 | 7.06 | 9.31 | 0 | 0.21 |
| ro = 0.63 × 10–6 м | 68 | 6.78 | 1.023 | 1.027 | 1538 | 6.1 | 1.70 | 0.29 | 1.08 | 8.89 | |
| Δp = 3.8 мПа | 127 | 9.79 | 1.034 | 1.038 | 1521 | 7.3 | 2.37 | 0.26 | 2.15 | 12.60 | |
| uo = 0.46 мм/с | 411 | 19.52 | 1.077 | 1.075 | 1460 | 8.8 | 3.71 | 0.29 | 10.90 | 24.50 | |
| (по лит. данным [8]) | 775 | 28.34 | 1.105 | 1.107 | 1423 | 13.5 | 9.12 | 0.34 | 0.47 | 35.10 | |
| 2 | L = 4.5 (5.0) м | 0.078 | 0.159 | 1.0 | 1.0 | 4273 | 15.4 | 5.54 | 2.81 | 0 | 0.71 |
| ro = 1 × 10–6 м | 39 | 6.16 | 1.012 | 1.012 | 4222 | 10.7 | 2.05 | 0.32 | 9.83 | 23.70 | |
| Δp = 3.1 мПа | 76 | 9.12 | 1.018 | 1.018 | 4197 | 10.4 | 2.35 | 0.40 | 10.80 | 34.50 | |
| uo = 1.05 мм/c | 180 | 15.14 | 1.032 | 1.030 | 4141 | 10.3 | 2.59 | 0.58 | 55.30 | 56.20 | |
| (настоящая работа) | 390 | 23.85 | 1.051 | 1.047 | 4067 | 7.2 | 1.50 | 0.85 | 66.10 | 86.90 | |
| 730 | 34.48 | 1.067 | 1.067 | 4006 | 20.4 | 15.0 | 1.15 | 29.0 | 124.0 | ||
| 1440 | 51.41 | 1.103 | 1.097 | 3874 | 45.0 | 55.10 | 1.60 | 248.0 | 181.0 | ||
| 3200 | 82.21 | 1.154 | 1.151 | 3708 | 64.8 | 128.0 | 2.34 | 500.0 | 285.0 | ||
| 3 | L = 3.23 (3.30) м | 0.092 | 0.132 | 1.0 | 1.0 | 1386 | 4.7 | 3.61 | 0.88 | 0 | 2.34 |
| ro = 1.342 × 10–6 м | 68 | 6.36 | 1.009 | 1.013 | 1365 | 3.7 | 2.07 | 1.0 | 3.70 | 96.60 | |
| Δp =16.7 мПа | 127 | 9.19 | 1.017 | 1.018 | 1361 | 3.5 | 2.86 | 1.40 | 7.60 | 137.0 | |
| uo = 2.34 мм/с | 186 | 11.5 | 1.020 | 1.023 | 1353 | 8.9 | 13.60 | 1.71 | 17.60 | 170.0 | |
| (по лит. данным [8]) | 411 | 18.33 | 1.033 | 1.036 | 1339 | 6.0 | 6.03 | 2.60 | 35.0 | 266.0 | |
| 675 | 24.53 | 1.042 | 1.048 | 1322 | 9.2 | 12.0 | 3.36 | 72.20 | 352.0 | ||
| 775 | 26.61 | 1.050 | 1.052 | 1318 | 10.7 | 15.90 | 3.61 | 9.90 | 381.0 | ||
| 4 | L = 4.5 (5.0) м | 0.078 | 0.064 | 1.0 | 1.0 | 3004 | 6.8 | 0.83 | 0.59 | 0 | 3.59 |
| ro = 2.5 × 10–6 м | 11 | 1.17 | 1.009 | 1.002 | 2974 | 8.5 | 1.32 | 0.64 | 0.17 | 58.50 | |
| Δp = 5.6 мПа | 39 | 2.46 | 1.015 | 1.005 | 2958 | 10.2 | 1.94 | 1.25 | 0.65 | 119.0 | |
| uo = 2.12 мм/с | 76 | 3.65 | 1.019 | 1.007 | 2947 | 7.5 | 1.04 | 1.80 | 0.72 | 174.0 | |
| (настоящая работа) | 180 | 6.05 | 1.026 | 1.012 | 2928 | 10.0 | 1.91 | 2.89 | 3.80 | 283.0 | |
| 500 | 11.04 | 1.039 | 1.022 | 2890 | 18.7 | 6.82 | 5.04 | 12.20 | 504.0 | ||
| 770 | 14.23 | 1.047 | 1.028 | 2867 | 18.7 | 6.93 | 6.35 | 2.32 | 642.0 | ||
| 1200 | 18.47 | 1.052 | 1.036 | 2854 | 15.3 | 4.68 | 8.03 | 21.40 | 825.0 | ||
| 3300 | 33.49 | 1.073 | 1.065 | 2797 | 22.1 | 10.20 | 13.50 | 48.30 | 1460.0 | ||
| 4400 | 39.66 | 1.084 | 1.076 | 2769 | 25.5 | 13.80 | 15.60 | 199.0 | 1717.0 | ||
| 9000 | 60.40 | 1.114 | 1.113 | 2695 | 38.3 | 32.80 | 22.0 | 393.0 | 2571.0 | ||
| 5 | L = 2.8 (3.3) м | 0.092 | 0.20 | 1.0 | 1.0 | 2588 | 9.6 | 3.91 | 2.82 | 0 | 0.71 |
| ro = 0.882 × 10–6 м | 68 | 9.68 | 1.018 | 1.019 | 2545 | 5.5 | 1.32 | 0.36 | 11.10 | 29.40 | |
| Δp = 16.7 мПа | 127 | 13.98 | 1.027 | 1.028 | 2522 | 6.8 | 2.10 | 0.46 | 22.40 | 41.8 | |
| uo = 1.08 мм/c | 411 | 27.89 | 1.049 | 1.054 | 2469 | 9.6 | 4.30 | 0.78 | 99.10 | 81.1 | |
| (по лит. данным [8]) | 775 | 40.49 | 1.073 | 1.078 | 2412 | 15.0 | 11.10 | 1.06 | 26.90 | 116 | |
| 6 | L = 3.23 (3.30) м | 0.092 | 0.13 | 1.0 | 1.0 | 1123 | 5.1 | 4.53 | 0.73 | 0 | 2.87 |
| ro =1.342 × 10–6 м | 68 | 6.36 | 1.014 | 1.013 | 1108 | 5.7 | 5.73 | 1.22 | 3.70 | 119 | |
| Δp = 20.6 мПа | 127 | 9.19 | 1.019 | 1.018 | 1102 | 6.1 | 6.76 | 1.71 | 7.60 | 169 | |
| uo = 2.87 мм/с | 411 | 18.33 | 1.034 | 1.036 | 1086 | 6.7 | 8.16 | 3.20 | 35.0 | 328 | |
| (по лит. данным [8]) | 775 | 26.61 | 1.053 | 1.052 | 1067 | 11.7 | 26.10 | 4.44 | 9.80 | 469 | |
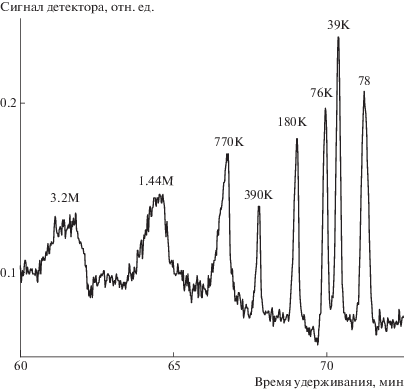
Входной конец капилляра через тройник, служащий делителем потока с минимизированными “мертвыми” объемами, присоединяли к крану-дозатору (“Reodyne”, США), снабженному петлей на 0.5 мкл. Подачу растворителя осуществляли жидкостным насосом LC-10AD фирмы “Shimadzu” (Япония). Для деления потока тройник снабжали рестриктором (капилляром) 1 м × 0.02 мм. Объем пробы, попадающей в капилляр, устанавливали объемом петли крана-дозатора (0.5 мкл) и коэффициентом деления делителя потока. Деление потока выбирали таким образом, чтобы вводимая проба была минимальна, но обеспечивала надежный сигнал детектора. Для хроматограммы, представленной на рис. 4, объемная скорость потока 3.3 × 103 мкм3/с (3.3 × 10–3 нл/с) и коэффициент деления потока 4.5 × 104. В этих условиях объем вводимой пробы равен 11.1 × 103 мкм3 (0.011 нл) и время ввода пробы составляет 3.4 с. Учитывая, что ширина самого эффективного пика на рис. 4 (пик 390 × 10–3) составляет ~28 с, вклад объема пробы в общую дисперсию не должен был превышать 10%.
Рис. 4.
Разделение методом ГДХ смеси полистирольных стандартов на капиллярной колонке диаметром 2 мкм; элюент – метилен хлорид, давление 3.1 МПа. ММ стандартов указаны на рисунке.
Кажущуюся эффективность колонки N рассчитывали по времени выхода пика to (или tp) и ширине пика на полувысоте σ, согласно известному выражению N = 5.54(tp/σ)2.
Пробы полимеров готовили растворением навески полимера в хлористом метилене. Концентрация проб составляла 0.1 мас. %.
Реагентами служили полистирольные стандарты с узким ММР производства “PolymerLabs” (Англия), “MZ Analysentechnik” и “PSS” (Германия). Характеристики стандартов приведены в табл. 1. Хлористый метилен, выступавший в качестве элюента при проведении разделений, имел квалификацию “для высокоэффективной жидкостной хроматографии (ВЭЖХ)” и его применяли без дополнительной очистки.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
На рис. 2 приведены экспериментальные величины относительного удерживания R высокомолекулярных ПС как функции характеристического размера λ. Несмотря на то что в эксперименте были использованы полистиролы с молекулярной массой до 9, все экспериментальные точки лежали в области λ < 0.1. В то же время, в области малых значений λ все модели, несмотря на различные значения параметра С, были очень близки друг к другу. Лишь для самых больших экспериментальных величин λ (близких к 0.1) можно отметить некоторое преимущество в аппроксимации экспериментальных точек уравнением (1) с параметром С = 2. Увеличить значение параметра λ можно либо повысив молекулярную массу ПС, либо уменьшив диаметр канала, в котором проводится ГДХ. Однако применение капилляров субмикронного диаметра представляет большие сложности с технической стороны, поскольку сильно снижается длина оптического пути УФ-детектора и, соответственно, его чувствительность. Понижение чувствительности детектора заставляет поднимать концентрацию вводимой пробы, что влечет повышение вязкости пробы и появление концентрационных эффектов (взаимное проникновение клубков). Чтобы избежать этих осложнений, рекомендуется работать с концентрацией полимера не выше 0.1% [8, 17–19], которые плохо совместимы с чувствительностью современных УФ-детекторов.
Авторы работы [8] использовали капилляры диаметром 1.24, 1.76 и 2.68 мкм. В настоящей работе эти данные были включены в зависимость, представленную на рис. 2, но это не привело к расширению диапазона характеристического размера λ, который остался ниже значения 0.1. Таким образом, расширение диапазона λ при увеличении ММ полимеров и(или) уменьшении диаметра капилляра пока не удается реализовать из-за технических сложностей. Для полимеров, исследованных в работе, характеристический параметр λ оставался ниже 0.1 (табл. 2). Относительное удерживание полимеров R, установленное экспериментально, удовлетворительно согласуется с величинами, найденными по уравнению (2) с параметром С = 2. Вместе с тем, величина λ < 0.1 означает, что относительное удерживание полимеров R, рассчитанное по уравнению (2) (см. рис. 2), не будет превышать 1.18 и что фракционирование образца полимера методом ГДХ пройдет в узком интервале времен элюирования.
Такое разделение потребует достаточно высокой эффективности колонки. Высокая (до 3 × 105 теоретических тарелок) эффективность капиллярной колонки в ГДХ полимерных сорбатов впервые отмечена в работе [8]. В последующих публикациях эффективность ГДХ с наполненными и монолитными колонками превышала 106 теоретических тарелок [9, 20], но при этом авторы работ отмечали быстрое снижение эффективности колонки и искажение формы пика полимеров с ростом его ММ.
Как отмечалось выше, дисперсия зоны при ее движении по каналу формируется из нескольких факторов. Диффузионная составляющая дисперсии hD в ГДХ складывается из обычной продольной диффузии и неравновесной диффузии в направлении, перпендикулярном к направлению потока. Эти два вклада как раз и описываются двумя слагаемыми в уравнении (3). Продифференцировав уравнение (3) по λ и приравняв производную к нулю, находим выражение для оптимальной скорости потока ${{v}_{{opt}}}$, при которой вклад диффузионного размывания пика минимален hDmin:
(7)
$\begin{gathered} {{{v}}_{{opt}}} = 8 \times \left( {\sqrt {\frac{{3{{f}_{1}}}}{{{{f}_{2}}}}} } \right)\quad {\text{и}} \\ {{h}_{{Dmin}}} = \frac{{\sqrt {{{f}_{1}}{{f}_{2}}} }}{{2\sqrt 3 {{{(1 - {\lambda })}}^{2}}(1 + 2{\lambda } - 2{{{\lambda }}^{2}})}} \\ \end{gathered} $Как следует из уравнений (7), оптимальная скорость потока ${{v}_{{opt}}}$ и минимальная высота теоретической тарелки hDmin зависят только от характеристического параметра λ. С увеличением λ от 0 до ~0.2 минимальная высота теоретической тарелки hDmin изменяется в довольно узком интервале значений, монотонно снижаясь с 0.28 до 0.16. Оптимальная скорость потока ${{v}_{{opt}}}$ для отмеченного интервала значений параметра 0 < λ < 0.2 также меняется в узком коридоре от 13.8 до 8.9. Таким образом, при оптимальной скорости потока колонка должна обладать высокой разделяющей способностью (hDmin < 1) и, более того, эффективность колонки должна возрастать с ростом ММ полимера. На практике, однако, наблюдается обратная картина, особенно для высокомолекулярных ПС. Так, экспериментально измеренные высоты приведенной теоретической тарелки hар (табл. 2) в большинстве случаев оказываются в несколько раз больше (т.е. хуже), чем hDmin, и само разделение происходит у большинства полимеров при неоптимальной скорости потока (табл. 2).
Неоптимальная скорость потока всегда приводит к уменьшению эффективности колонки, но иногда она применяется сознательно, например, чтобы сократить время анализа. В случае анализа полимеров неоптимальная скорость потока – это неотъемлемое следствие зависимости приведенной скорости потока (уравнение (5)) от коэффициента диффузии сорбата Dm, а следовательно, от его ММ. Соответственно, при выбранной скорости потока ${{v}_{o}}$ некоторые ММ элюирутся с приведенной скоростью $v$, близкой к оптимальной скорости ${{v}_{{opt}}}$, тогда как остальные полимеры будут элюироваться со значениями скорости либо выше, либо ниже оптимальной. Как правило, экспериментальная приведенная скорость элюирования ν ниже оптимальной (${{v}_{{opt}}}$ = 13.9–12.2 для 0 < < λ < 0.1) оказывается у низкомолекулярных сорбатов, тогда как у полимеров она обычно выше оптимальной, иногда очень значительно (табл. 2).
Значения приведенной высоты теоретической тарелки hD при неоптимальной скорости потока, которые были найдены при подстановке в уравнение (3) соответствующих значений λ и величин экспериментальной приведенной скорости потока ${{v}_{{ap}}}$, также представлены в табл. 2. В зависимости от того, насколько различны реальная и оптимальная приведенные скорости элюирования, приведенная высота теоретической тарелки hD может больше, чем на порядок, превосходить минимальную высоту теоретической тарелки (hDmin = 0.29–0.20 для 0 < λ < 0.1). В то же время, рассчитанные для неоптимальных потоков величины приведенной высоты теоретической тарелки hD в большинстве случаев оказывались меньше, чем экспериментальные величины приведенной высоты теоретической тарелки hap (табл. 2). Только в случае самых высокомолекулярных ПС, у которых наблюдаются большие отклонения указанной скорости потока от оптимального значения, величины приведенных высот теоретических тарелок сопоставимы друг с другом. Таким образом, экспериментальная дисперсия пика hap практически во всем диапазоне ММ оказывается выше предсказываемой теорией (уравнение (3)), и дополнительная дисперсия пика, вызванная неоптимальной скоростью его элюирования, оказывается недостаточной, чтобы объяснить этот эффект.
Фактором, специфическим для анализа именно полимерных объектов, является наличие у полимерной пробы ММР [2]. Ширина ММР характеризует полидисперсность пробы (P = Ð), ее влияние на величину дисперсии пика было исследовано в работе J.H. Knox и F. McLennan [21]. Предложенное ими уравнение было адаптировано позднее G. Stegeman [22] к условиям ГДХ полимеров на наполненных колонках:
(8)
${{h}_{p}} = \left( {\frac{L}{{{{d}_{c}}}}} \right)\left( {P - 1} \right)\left( {1 + {\alpha }} \right){{\left( {\frac{S}{{{{V}_{p}}}}} \right)}^{2}}$Здесь α = (11/4)(Р – 1) + (137/12) (Р – 1)2, S = = –d(Vр)/d (ln MМ) – наклон калибровочной зависимости, Vp – объем элюирования полимера. Учитывая, что R = tm/tp = Vm/Vp, а зависимость R от λ задается уравнением (2), то выражение S/Vp несложно преобразовать так, чтобы использовать его с универсальной для ГДХ калибровочной зависимостью dlnR/dlnλ:
где b = 0.588 – показатель степени в уравнении Марка–Хаувинка (табл. 2).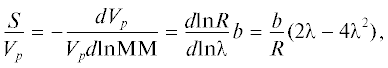
Подставив выражение (9) в уравнение (8), можно найти вклад полидисперсности образца ПС в дисперсию пика в ГДХ:
(10)
${{h}_{p}} = \left( {\frac{L}{{{{d}_{c}}}}} \right)\left( {P - 1} \right)\left( {1 + {\alpha }} \right)\frac{{{{b}^{2}}{{{(2{\lambda } - 4{{{\lambda }}^{2}})}}^{2}}}}{{{{{(1 + 2{\lambda } - 2{{{\lambda }}^{2}})}}^{2}}}}$Необычным в этом выражении является то, что дисперсия пика hp, вызванная полидисперсностью пробы, не зависит от скорости потока. В то же время, она зависит от геометрических параметров системы, увеличиваясь с ростом длины капилляра и(или) уменьшением его диаметра. Высота приведенной теоретической тарелки hp, обусловленная полидисперсностью пробы Р, быстро уменьшается, по мере того как Р приближается к единице. Тем не менее, этот вклад обычно считают доминирующим в размывании пиков при хроматографии полимеров [21, 22], а полидисперсность пробы Р = 1.1 считают предельно допустимой для определения эффективности колонок с полимерными пробами [21].
Найденные по уравнению (10) высоты приведенной теоретической тарелки hp для полистиролов, исследованных в этой работе, действительно оказались заметно больше, чем высоты приведенных теоретических тарелок hD, обусловленные диффузионными процессами. К сожалению, во многих случаях значения hp превосходили также и экспериментально найденные высоты теоретических тарелок hap (табл. 2), что уже трудно поддается объяснению. Аналогичное превышение hp над hap отмечено в работе [22], где авторы объяснили его неточностью значений полидисперсности образцов ПС, сообщаемых производителями. Вместе с тем, такое объяснение выглядит не очень обоснованным, если учесть, что для согласования величин hp и hap авторам [22] пришлось изменить полидисперсность образца ПС с Р = 1.01 до нереальных значений (1.0001 и ниже).
Таким образом, экспериментально обнаруженное уширение полос высокомолекулярных ПС в ГДХ может иметь в своей основе как уширение вследствие полидиспрености образцов полимеров, так и диффузионное уширение, связанное с невозможностью обеспечить всем компонентам пробы элюирование с оптимальной скоростью потока. Уширение пика, вызванное диффузионными процессами и неоптимальными условиями элюирования hD, оказывается, как правило, меньше, чем экспериментально измеряемая высота тарелки hap. Расхождение между этими величинами предполагает наличие дополнительных факторов, вызывающих повышенную дисперсию. Таким фактором, специфическим для полимерных объектов, может быть их полидисперсность. Однако дополнительный вклад в дисперсию, рассчитываемый для этого процесса по уравнению (10), иногда в разы превосходит экспериментальные величины hap, что не позволяет на данный момент сделать окончательный вывод о значении полидисперсности пробы для размывания пика в ГДХ.
Работа выполнена в соответствии с планом научных исследований ИНХС РАН и грантом Президиума РАН № 14.
Список литературы
Hameed B.S., Bhatt C.S., Nagaraj B., Suresh A.K. // Chromatography as an Efficient Technique for the Separation of Diversified Nanoparticles in Nanomaterials in Chromatography. Current Trends in Chromatographic Research Technology and Techniques / Ed. by C.M. Hussain. Amsterdam: Elsevier, 2018. P. 503.
Striegel A.M. // Size-Exclusion Chromatography in Liquid Chromatography / Ed. by S. Fanali. Amsterdam: Elsevier, 2017. P. 245.
Liu Y., Radke W., Pasch H. // Macromolecules. 2005. V. 38. P. 7476.
Liu Y., Radke W., Pasch H. // Macromolecules. 2006. V. 39. P. 2004.
Kurganov A.A., Svec F., Kanateva A.Yu. // Polymer. 2015. V. 60. P. A1.
Viktorova E.N., Korolev A.A., Orekhov V.A., Kanateva A.Yu., Kurganov A.A. // Rus. J. Phys. Chem. A. 2012. V. 87. P. 308.
DiMarzio E.A., Guttman C.M. // Macromolecules. 1970. V. 3. P. 131.
Tijssen R., Bos J., van Kreveld M.E. // Anal. Chem. 1986. V. 58. P. 3036.
Stegeman G., van Asten A.C., Kraak J.C., Poppe H., Tijssen R. // Anal. Chem. 1994. V. 66. P. 1147.
Advances in Polymer Science. New Developments in Polymer Analytics I / Ed. by M. Schmidt. Berlin; Heidelberg: Springer, 2000. V. 150.
Shendruk T.N., Slater G.W. // J. Chromatogr. A. 2012. V. 1233. P. 100.
Proulx K., Wilkinson K.J. // Environ. Chem. 2014. V. 11. P. 392.
Taylor G.I. // Proc. Roy. Soc. A. 1953. V. 219. Is. 1137. P. 186.
Aris R. // Proc. Roy. Soc. A. 1956. V. 235. Is. 1200. P. 67.
Wang Y., Jiang W., Miller S., Eckstein E. // J. Chromatogr. A. 2008. V. 1198. P. 140.
Brenner H., Gaydos L.J. // J. Colloid Interface. Sci. 1977. V. 58. P. 312.
Lipatov Yu.S. Selected Plenary Lectures of the IUPAC-Conference on Colloid and Surface Science in Budapest / Eds by F.H. Müller, A. Weiss, E. Wolfram., D. Steinkopff. Darmstadt: Verlag GmbH & Co. KG, 1976.
Martin M., Feuillebois F. // J. Sep. Sci. 2003. V. 26. P. 471.
Mandema W., Zeldenrust H. // Polymer. 1977. V. 18. P. 835.
Korolev E., Victorova E., Ibragimov T., Kanatyeva A., Kurganov A. // J. Sep. Sci. 2012. V. 35. P. 957
Knox J.H., McLennan F. // Chromatographia. 1977. V. 10. P. 75.
Stegeman G., Kraak J.C., Poppe H. // J. Chromatogr. 1993. V. 634. P. 149.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия А)