

Высокомолекулярные соединения (серия Б), 2019, T. 61, № 1, стр. 10-21
МИКРОВОЛНОВЫЙ СИНТЕЗ ПОЛИФЛУОРЕНОВ И СОПОЛИФЛУОРЕНОВ И ИХ ОПТИЧЕСКИЕ СВОЙСТВА
Г. И. Носова a, *, Л. С. Литвинова a, И. А. Березин a, Е. В. Жукова a, Р. Ю. Смыслов a, А. В. Якиманский a
a Институт высокомолекулярных соединений Российской академии наук
199004 Санкт-Петербург, Большой пр., 31, Россия
* E-mail: klengi@yandex.ru
Поступила в редакцию 23.05.2018
После доработки 12.10.2018
Принята к публикации 26.10.2018
Аннотация
Изучены закономерности синтеза сополифлуоренов по методу Сузуки под воздействием микроволнового нагрева в условиях, которые использовали ранее при конвекционном нагреве в открытой системе. С помощью метода эксклюзионной жидкостной хроматографии рассмотрены стадии поликонденсации: образование олигомеров, рост цепи и закрытие концевых групп. Проанализировано влияние температуры, растворителя и типа катализатора Pd(PPh3)4, PdCl2(dppf), полимер-связанного Pd(PPh3)4 и SillaCatDPP-Pd на молекулярно-массовые характеристики полимеров. Сопоставлены оптические свойства новых сополифлуоренов, полученных разными методами.
ВВЕДЕНИЕ
Сопряженные полимеры, к которым относятся полифлуорены, представляют интерес для создания опто-электронных устройств, таких как светоизлучающие диоды, полевые транзисторы, полимерные фотовольтаические ячейки, и для изготовления хемо- и биосенсоров [1–5]. Научные поиски в области полифлуоренов направлены на разработку химических структур с определенными оптическими, электронными, пленкообразующими свойствами (в комбинации с химической, термической, оптической стабильностью), и снижение затрат на синтез полимеров и совершенствование технологических аспектов изготовления материалов.
Сдерживающим моментом синтеза сополи-флуоренов (СПФ) является длительность их поликонденсационного синтеза в реакциях Сузуки и Ямамото (48 ч и более). Значительного сокращения времени синтеза сопряженных полимеров до 10–60 мин можно достигнуть путем проведения поликонденсации в микроволновом реакторе [6, 7]. С помощью микроволн можно не только нагревать реакционные смеси, но и значительно увеличивать скорость реакций. В жидких системах молекулы, имеющие постоянный дипольный момент (вода), могут вращаться в присутствии быстро меняющегося электрического поля микроволнового излучения, a ионы подвергаются быстрым поступательным движениям. В результате такого типа движения возникающее “внутреннее трение” приводит к нагреву полярной среды. При воздействии микроволнового излучения на реакционную систему происходит разупорядочивание структуры раствора и, как следствие снижение его вязкости, что приводит к увеличению эффективной скорости реакции по сравнению со скоростью в условиях термического нагрева. В итоге значительно быстрее доставляется энергия к реакционным частицам и легче преодолевается активационный барьер реакции. За счет молекулярного перемешивания создаются гомогенные условия протекания поликонденсации во всем объеме реакционной смеси без предварительного нагрева сосуда.
Успешное протекание синтеза полимеров в микроволновом реакторе зависит от полярности растворителя, температуры, давления в закрытой системе, мощности излучения, условий перемешивания, выбора температурного датчика и режима излучения: импульсного или непрерывного (как правило, используются устройства с импульсным излучением на частоте 2.45 ГГц) [8].
Определенные успехи достигнуты при применении микроволнового излучения для синтеза ароматических сопряженных полимеров прямым арилированием связей C–H ароматических мономеров с дибромариленами с Pd(OAc)2 [9], синтеза полифлуорена на основе 9,9-дигексилфлуорена в условиях окислительной полимеризации с FeCl3 [10], реакций кросс-сочетаний по механизмам Соногаширы и Стилле [8, 11–14], а также для проведения реакций в цепях сопряженных полимеров [15, 16]. В условиях как конвекционного, так и микроволнового нагрева выбор растворителей связан с их сольватирующими свойствами, способностью растворять исходные мономеры и образующиеся полимеры, что обеспечивает эффективный и гомогенный нагрев полимеризующейся смеси. Но, кроме того, скорость микроволнового синтеза существенно зависит от способности растворителей поглощать микроволновое излучение и преобразовывать сохраненную электромагнитную энергию в тепловую энергию при данной температуре и частоте, что определяется значением их диэлектрической проницаемости и коэффициентом диэлектрических потерь (толуол: е' = 2.26; е'' = 0.072; (Т = = 70°С; 2.45 ГГц); вода: е' = 63.4; е'' = 4.311; ТГФ: е' = 7.6; ДМФА: е' = 36.7 (25°С), N-метилпирролидон: е' = 32.1) [17–19]).
Использование полярных растворителей позволяет обеспечить гомогенный нагрев реакционной смеси, особенно на завершающих стадиях процесса, когда большая часть молекул мономера уже включена в состав макромолекул и ограничена в своей подвижности [8]. Полярный растворитель с высокой температурой кипения позволяет проводить реакцию при более высоких температурах. Тем не менее, неполярные растворители также используются в микроволновом синтезе. Их свойства могут быть улучшены за счет различных добавок (других растворителей, солей, ионных жидкостей), причем диэлектрические свойства растворителей по-разному изменяются с температурой, а их смешанные водные растворы часто имеют специфические свойства. ММ получаемого продукта зависит от выбора мощности излучения [17].
Ранее под действием микроволнового излучения в закрытом сосуде были получены поли(9,9-дигексилфлуорен) и поли(9,9-диоктилфлуорен) поликонденсацией по механизму реакции Ямамото с Мw = (43–50) × 103 (230°С; 10–60 мин; ДМФА–толуол = 1 : 2; 150 Вт) [7, 14, 18–22]. Как правило, с помощью этой реакции синтезируют полимеры, мономерами для которых служат, дибромиды или, в отдельных случаях, и менее реакционные дихлориды ароматических соединений. Так, в работах [14, 21] на основе дихлорароматического соединения в присутствии комплекса платины был синтезирован сополимер, содержащий 2–8 мол. % фосфоресцирующего сомономера, что позволило увеличить яркость светодиода на его основе до 3–6 кд /A.
Наше исследование синтеза СПФ, полученных по методу Ямамото в условиях термического нагрева, показало, что из-за наличия двух мономеров образуются блочные структуры, в результате чего оптические свойства полимеров значительно отличаются от СПФ, синтезированных в условиях кросс-сочетания по реакции Сузуки [23].
Основной реакцией синтеза СПФ с разнообразными оптическими свойствами является реакция Сузуки–Мияура кросс-сочетания дибром-ароматических соединений с производными диароматических бороновых кислот [1]. Таким способом в условиях микроволнового нагрева в закрытой системе (10 мл ампула) получены поли(9,9-диоктилфлуорен) и поли(9,9-дигексилфлуорен) с использованием катализаторов Pd(PPh3)4 [13, 24–28], PdCl2(PPh3)2 [12, 14] и PdCl2(dppf) [22, 25]. Проведение синтеза полифлуоренов под действием микроволнового излучения привело к многократному сокращению времени реакции при сохранении выхода полимера. При этом достигнуты значения молекулярных масс и индексов полидисперсности полифлуоренов, необходимые и для их практического применения. Поли(9,9-дигексилфлуорен) был синтезирован с Mw = 28 × 103 (Mw/Mп = 2.1) с катализатором Pd(PPh3)4 и 37.2 × 103 (2.0) с PdCl2(dppf) в ТГФ : вода (3 : 2) за 11–20 мин при 130°С и мощности 150 Вт [22, 25]. Однако с увеличением мощности излучения, температуры реакции или продолжительности синтеза ММ и выход полимера понижались. В ряде случаев также отмечено гелеобразование в ходе синтеза и потеря растворимости полимера при мощности 180 Вт [14, 16, 22, 25], что связывают с образованием сшивок полимерных цепей в присутствии водного основания [22]. Указанные процессы свидетельствует о недостаточной стабильности синтеза в найденных условиях микроволнового излучения. Наши исследования показали, что при проведении синтеза в закрытом сосуде (катализатор Pd(PPh3)4, ТГФ–вода) уже при 130–150°C (150 Вт) через 20 мин происходит желирование поли(9,9-диоктилфлуорена), и полученные в этих условиях полимеры уступают по оптическим свойствам тем, которые были синтезированы нами ранее при термическом нагреве [29].
Определенная методическая трудность синтеза полифлуоренов в микроволновом реакторе связана также с необходимостью закрытия концевых реакционных групп, присутствие которых приводит к снижению квантового выхода излучения и эффективности электролюминесценции, вызывает электрохимическое окисление полимера и разрушение электродов во время эксплуатации изделий [30]. В случае проведения процесса в закрытой системе для завершения синтеза необходимо дважды останавливать реакцию для введения монофункциональных соединений.
В целом следует отметить, что синтез полифлуоренов в условиях конвекционного нагрева значительно уступает микроволновому по длительности, но в то же время позволяет получать более высокомолекулярные полимеры, что важно для ряда практических приложений. В литературе недостаточно представлены оптические свойства гомополифлуоренов и практически отсутствуют данные как по синтезу, так и по свойствам СПФ, полученных в микроволновом реакторе.
Настоящая работа посвящена изучению реакции по методу Сузуки, на примере синтеза поли(9,9-дигексилфлуорена), поли(9,9-диоктилфлуорена) и СПФ при микроволновом нагреве в тех же условиях, что и при термическом нагреве: в открытой системе под аргоном при 80–100°С и с водно-щелочным агентом K2CO3. Задача работы – оценка и оптимизация условий стабильного микроволнового синтеза полифлуоренов с целью получения наиболее высокомолекулярных полимеров с определенными оптическими свойствами.
Рассмотрено влияние растворителя, температуры, типа катализатора и времени синтеза. Методом ЭЖХ исследован процесс поликонденсации, а именно образование олигомеров, роста цепи и закрытия концевых групп. Применяли катализаторы Pd(PPh3)4, PdCl2(dppf), а также полимер-связанный Pd(PPh3)4 и SillaCatDPP-Pd, последний из которых успешно используется в органическом синтезе [31]. Представляло интерес сопоставить молекулярно-массовые характеристики и оптические свойства поли(9,9-диоктилфлуорена) и сополифлуоренов более сложного состава, полученных разными методами.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Молекулярно-массовые характеристики полимеров определяли методом эксклюзионной жидкостной хроматографии при 40°С на установке “Agilent Technologies” 1260 Infinity c рефрактометрическим (DRI, Wavelength 660 nm) детектором. В установку входил набор последовательно соединенных хроматографических колонок: предколонка PLgel 5 μm Guard 50 × 7.5 mm и две стирогелевые колонки “Agilent Technologies PLgel” 5 μm MIXED-C, 300 × 7.5 mm. ТГФ использовали в качестве подвижной фазы, скорость которой составляла 1.0 мл/мин. Концентрация пробы 2 мг/мл, в колонку вводили 100 μL. Молекулярно-массовые характеристики рассчитывали по ПС-стандартам (Agilent Technologies). ТГФ перегоняли над КОН и использовали стабилизированный растворитель (0.02% 2,6-ди-трет-бутилгидрокситолуола).
Для изучения спектров фотолюминесценции и поглощения пленочные покрытия полифлуоренов и СПФ наносили на стекло в центробежном поле из растворов в толуоле (15 мг/мл), а в ряде случаев поливом СПФ в хлороформе. Концентрация растворов СПФ составляла 10 мг/мл. Электронные спектры поглощения полимерных пленок регистрировали на спектрофотометре UV-3100 СФ-256 УВИ (ЛОМО фотоника, Россия). Спектры фотолюминесценции получали на люминесцентном спектрофотометре LS-100 (фирма PTI®, Канада) в диапазоне 360–720 нм. Спектральная ширина щелей монохроматоров на входе и выходе отделения для образцов составляла 4 нм. Усиление ФЭУ варьировали от 500 до 700. Люминесценцию полимерных пленок снимали со стороны падения возбуждающего луча в диапазоне 400–700 нм при длине возбуждении λв = 380 нм.
Синтез полимеров проводили в одномодульном микроволновом реакторе “Discover LabMate” (“СЕМ Corporation”, USA), частота излучения 2.45 ГГц, максимальная мощность генератора 300 Вт). Температуру синтеза контролировали инфракрасным датчиком, расположенным под реакционным сосудом. Параметры реакции (температура, мощность, время, скорость перемешивания) устанавливали вручную.
Толуол и ТГФ перегоняли под аргоном над Na и бензофеноном. Мономерами служили 2,7-ди-бром-(9,9-диоктил)флуорен (М1), 2,7-дибром(9,9-дигексил)флуорен (М2), 9,9-дигексил-2,7-бис-(три-метиленборат)флуорен, 9,9-диоктил-2,7-бис-(три-метиленборат)флуорен, Aliquat® 336 и катализаторы Pd(PPh3)4, полимер-связанный Pd(PPh3)4, PdCl2(dppf) фирмы “Sigma–Aldrich”, SillaCatDPP-Pd SiliCycle® Inc. Синтез производных 3,6 и 2,7- дибромкарбазолов, а также и СПФ в условиях термического нагрева описан в работе [29].
Микроволновый синтез поли(9,9-диоктилфлуорена) (ПФ-8) (табл. 1, серии I, II) проводили в 100 мл колбе, снабженной холодильником, двумя адаптерами с кранами для работы в системе аргон–вакуум и отводами для введения растворителей и отбора проб. В колбу загружали 0.1425 г (0.255 ммоля) 9,9-диоктил-2,7-бис-(триметиленборат)флуорена, 0.1371 г (0.250 ммоля) 9,9-диоктил-2,7-дибромфлуорена и 10 мг (0.03812 ммоля) трифенилфосфина. Из системы удаляли кислород и загружали катализатор реакции 6.2 мг (0.0054 ммоля) Pd(PPh3)4 и повторно вакуумировали, промывали аргоном. Затем в колбу через холодильник вводили 2.5 мл толуола, 1.0 мл 2 M раствора K2CO3 и 15 мг триоктилметиламмоний хлорида (Aliquat® 336) в 0.3 мл толуола. Установку помещали в микроволновую печь и реакционный раствор перемешивали при 98–102°С (облучение 80 Вт) в течение 1.5 ч. Затем в реакционную зону добавляли 9,9-диоктил-2,7-бис-(триметиленборат)флуорен (10 мг в 0.5 мл толуола), а после 40 мин нагрева 2-бром(9,9-диоктил)флуорен (30 мг в 0.5 мл толуола). Перемешивание и нагревание продолжали еще 40 мин и полимер высаживали в метанол, промывали водой, ацетоном и отфильтровывали, повторно растворяли в CHCl3 и осаждали в метанол. Низкомолекулярные фракции полимера удаляли экстракцией ацетоном в аппарате Сокслета. Выход СПФ после очистки составлял 80%. В ходе синтеза через определенные интервалы времени из реакционной смеси под аргоном отбирали пробы, которые высушивали и экстрагировали CНCl3 и после удаления растворителя выделенный состав продуктов реакции анализировали методом ЭЖХ. Аналогично получали полимер поли(9,9-дигексилфлуорен) (ПФ-6) серии III–VI.
Таблица 1.
Условия проведения реакции поликонденсации, значения Mw и Mw/Mn синтезированных полимеров
| Серия | Катализатор | Содержание катализатора, мол. % | Tр, °С | Mw × 10–3 (Mw/Mn) | ||
|---|---|---|---|---|---|---|
| в конце реакции | после осаждения | после экстракции | ||||
| I | Pd(PPh3)4 | 1 | 100 | 61.66 (3.2) | 66.80 (2.90) | 72.45 (2.75) |
| II | Pd(PPh3)4 | 1 | 80 | 28.45 (3.17) | 33.44 (2.64) | 33.00 (2.17) |
| III | Pd(PPh3)4 | 3 | 100 | 34.88 (3.36) | 25.36 (2.52) | 27.70 (1.97) |
| IV | Pd(PPh3)4 полимер-связанный | 1 | 100 | 52.80 (3.94) | 59.67 (2.78) | 61.49 (2.45) |
| V * | [Pd(PPh3)4 | 1 | 80 | 37.86 (4.34 ) | – | 37.45 (2.23) |
| VI | PdCl2(dppf) | 1 | 100 | 94.67 (2.96) | 99.50 (2.74) | 103.51 (2.62) |
| VII** | Pd(PPh3)4 | 1 | 100–140 | – | 69.82 (2.9) | 79.00 (2.14) |
| VIII*** | Pd(PPh3)4 | 1 | 100 | 24.42 (3.56) | – | 31.00 (1.98) |
| CP1 | SillaCatDPP | 1 | 100 | 132.00 (2.55) | 134.40 (2.39) | |
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Синтез полимеров проводили в микроволновом реакторе “Discover LabMate” в растворителе толуол–вода. Ниже в качестве примера приведена реакция синтеза полифлуорена ПФ-8

В синтезе по методу Сузуки для образования диарилов необходим щелочной агент в двух стадиях реакции – окислительного присоединения и трансметаллирования. Комплекс нуль-валентного Pd с трифенилфосфиновыми лигандами (Pd(PPh3)4) в растворе диссоциирует с образованием координационно ненасыщенного комплекса. В результате окислительного присоединения галогенида к палладию возникает металлоорганическое соединение (двухвалентный Pd), а при его взаимодействии с основанием – интермедиат. Последний в результате переметаллирования с реакционным боратным комплексом образует дизамещенное палладийорганическое соединение, способное к восстановительному элиминированию, приводящему к целевому продукту и нуль-валентному Pd (Pd(PPh3)4). Для получения реакционных боратов также необходимо основание (водный K2СО3). Обнаружено также участие OH– и в ускорении стадии трансметаллирования (транс-цис-изомеризация) и этапа восстановительного элиминирования каталитического цикла в реакции Сузуки [32]. Реакция в выбранных условиях протекает в растворителе (толуол или ТГФ). Третичные аммонийные соли, в частности тетрабутиламмоний бромид, облегчают сольватацию органических субстратов в растворителях невысокой полярности и служат в качестве агентов фазового переноса гидроксил-аниона. Предполагается, что они участвуют в образовании реакционного комплекса бороната аниона [ARB (ОН)3]–[R4N]+ в смеси водной щелочи и растворителя [33]. В полимерном синтезе в реакции кросс-сочетания в основном вместо арил-дибороновых кислот используются их эфиры, очистка которых позволяет получить соединения более высокого качества с хорошей растворимостью в толуоле, ТГФ, ДМФА, хлороформе. Эфиры защищают бор-углеродную связь, предотвращая образование циклического ангидрида бораксина.
В синтезах ПФ-8 (табл. 1, серии I, II) и ПФ-6 (серия III–VI) а также СПФ в качестве агента транспортного переноса использовали триоктилметиламмоний хлорид (Aliquat® 336), который при 100°C и магнитном перемешивании в качестве поверхностно активного вещества дополнительно вспенивает реакционный раствор. Это способствует повышению массобмена и улучшению контакта реакционных центров активированных мономеров, олигомеров и макромолекул в ходе поликонденсации. Реакцию осуществляли при воздействии фосфиновых катализаторов Pd(PPh3)4, (табл. 1, серии I–III, V), PdCl2(dppf) (серия VI), и полимер-связанного Pd(PPh3)4 (серия IV), а также гетерогенного катализатора SillaCatDPP-Pd, (СП1).
При проведении реакции варьировали температуру (100 или 80°C), растворитель (толуол, ТГФ), мощность излучения (80–100 Вт) и концентрацию (1 или 3 мол. %) катализатора Pd(PPh3)4 (табл. 1). В ходе синтеза отбирали пробы и после их обработки выделенный состав продуктов реакции анализировали методом ЭЖХ (рис. 1).
Рис. 1.
ММР продуктов реакции, соответствующих времени отбора проб из реакционной смеси в серии синтезов I (a), IV (б), II (в) и V (д). а: 15 (1), 30 (2), 45 (3), 60 (4), 75 (5), 90 (6), 105 (7), 135 (8), 180 мин (9); б: 15 (1), 30 (2), 45 (3), 60 (4), 75 (5), 90 (6), 105 (7), 120 (8), 135 (9), 180 мин (10); в: 15 (1), 30 (2), 45 (3), 60 (4), 75 (5), 90 (6), 115 (7), 180 мин (8); г: 15 (1), 30 (2), 45 (3), 60 (4), 75 (5), 105 (6), 120 (7), 165 (8), 210 мин (9).

На рис. 1 видно, что синтез полимера на начальной стадии в толуоле протекает через характерный для поликонденсации этап образования димера (A–В) и тримера (В–А–В), хорошо различимые при 80°С (рис. 1в, 80°С, серия II) и при 100°С в случае использования полимер-связанного катализатора (рис. 1б, 100°С, серия IV). В более полярном растворителе ТГФ при 80°С (рис. 1г, серия V), а в толуоле при 100°С (Pd(PPh3)4) (рис. 1а, серия I) уже через 15 мин можно выделить четыре олигомерные зоны (Mw = 840, 1.3 × 103, 1.9 × 103 и 2.4 × 103), но затем, в силу уменьшения подвижности и реакционной способности олигомеров, молекулярная масса полимера в толуоле увеличивается значительно быстрее, чем в ТГФ и температурный фактор, а именно повышение температуры до 100°С, играет решающую роль (рис. 2).
Рис. 2.
ЭЖХ-хроматограмма продуктов реакции образцов серии V (а), кинетические кривые расхода мономеров (б) и выхода полимеров (в); а: 1 – мономер M2, 2 – реакционная смесь через 15 мин синтеза; б: 1, 4, 5 – М2 серий IV–VI, 2, 3 – М1 серий I и II; в: 1, 4, 5 – ПФ-6 серий IV, III, V; 2, 3 – ПФ-8 серий I и II.

Хроматографическая зона дибром-мономеров М1 и М2 не перекрывается с зонами других реакционных продуктов (для М2, димера и тримера RT = 18.5, 17.4 и 16.8 мин, рис. 2а), что позволяет определить конверсию мономера и выход образующихся полимерных продуктов в ходе поликонденсации (рис. 2б и 2в), а также анализировать стадию закрытия концевых реакционных групп. В первом случае расчет произведен на основании анализа площадей соответствующих хроматографических зон. В анализируемых пробах сигнал, отвечающий диборолану, отсутствует ввиду низкой чувствительности к этому соединению рефрактометрического детектора (отличие в чувствительности концентрации мономеров более чем на порядок).
В основном реакция образования диарилов по Сузуки лимитируется стадией окислительного присоединения галогенида к палладию. В данном случае конверсия дибромфлуорена (рис. 2а и 2б) в основном отражает скорость реакции на начальном этапе поликонденсации, т.е. образования олигомеров. Из рис. 2б, 2в и 3 следует, что дибром-мономеры M1 в сериях I, II и М2 в серии VI практически расходуется в течение 1 ч, в то время как в серии IV (100°С, полимер-связанный[Pd(PPh3)4]) и V (80°С, ТГФ) остается еще 20% мономера М2. Кривые расхода мономера и выхода полимера (рис. 2б и 2в) показывают, что скорость реакции при 100°С (толуол : вода 2.5 : 1.0) зависит от используемого типа катализатора и растет в ряду IV > VI > I. При 80°С толуол предпочтительнее ТГФ, что, возможно, связано с низким содержанием растворителя в смеси ТГФ : вода (1.7 : 1.0). (Увеличение соотношения до 2.7 : 1.0 привело к снижению температуры реакции с 80 до 64–67°С и к получению низкомолекулярного полимера). Эта же зависимость в целом сохраняется и для более глубокого этапа поликонденсации, что отражают кривые роста ММ, представленные на рис. 3. Начальный медленный этап реакции (стадия А) связан с образованием олигомеров и для ПФ-8 серии I (табл. 1, 100 °С, Pd(PPh3)4) он практически совпадает с конверсией мономеров 90%. Затем в результате взаимодействия олигомеров наблюдается быстрый рост длины молекулярных цепей. На 90-й минуте синтеза (120 мин в ТГФ, серия V) для закрытия концевых групп в зону реакции вводили небольшую порцию 9,9-диоктил-2,7-бис-(триметиленборат)флуорена. Из кривых рис. 3 (стадия В) следует, что на этом этапе реакции не происходит остановки роста цепи, а вплоть до введения монопроизводного 2-бром-(9,9-диоктил)флуорена – стадия С (135 мин, а в серии V – 160 мин) ММ полимеров продолжает повышаться.
Рис. 3.
Изменение Mn полимеров ПФ-6 и ПФ-8 (табл. 1) в процессе реакции. 1–5 – серии I–V соответственно. Пояснения в тексте.

Наблюдаемый эффект может быть связан с нарушением эквивалентности функциональных групп в процессе синтеза и вызван протеканием побочной реакции, например частичным гидролизом бороновых эфиров с образованием менее реакционных, но более полярных соединений – бороновых кислот. Арилбороновые кислоты способны тримеризоваться с образованием неактивных бораксинов, но при наличии воды эта реакция маловероятна. В то же время в присутствии основания может протекать реакция гидролитического депротоборанирования бороновых кислот, что приводит к нарушению стехиометрического соотношения реакционных центров [34, 35]. Успешное осуществление поликонденсации любого типа требует строгой эквивалентности функциональных групп. Используемый шестичленный триметиленборонатфлуорена наряду с пинаколятами является соединением, наиболее устойчивым к гидролизу, который замедляется при использовании несмешиваемых сред. Преимущество проведения реакции в среде водного Na2CO3, K2CO3 связано также с замедлением гидролиза в 18 раз по сравнению с KOH (скорость реакции деборанирования возрастает при увеличении силы и количества основания) [36].
Заметим, что на полимер-связанном Pd-катализаторе (иммобилизованном на сшитом ПС) (рис. 3, серия IV, кривая 4) рост полимерной цепи при 100°С происходит значительно медленнее чем в случае других катализаторов. Данный катализатор находится в виде взвешенных частиц в толуольной фазе, и взаимодействие с ним реакционных групп более затруднено по сравнению с растворимыми Pd(PPh3)4 и PdCl2(dppf). Снижение температуры на 20°С замедляет рост цепи и снижает ММ полимера (серии II и V, кривые 2 и 5). В растворителе ТГФ : Н2О (1.7 : 1.0; серия V) уменьшение скорости роста ММ по сравнению с системой толуол : Н2О (2.5 : 1.0) обусловлено плохой растворимостью полимера в водном ТГФ. В результате реакционная смесь однородна только на начальной стадии реакции, а затем происходит высаливание полимера и разделение раствора на две фазы. Но благодаря увеличению продолжительности синтеза ММ полимера достигает тех же величин, что и в толуоле при 80°С (табл. 1). При использовании в качестве растворителя ксилола температуру синтеза можно увеличить до 140°С, но на примере сополимера поли(9,9-дигексилфлуорен-2,7-диил-алт-2,2'-битиофен-5,5'-диил)а показано, что в этом случае удается получить полимеры с Мw не более 30 × 103 [37].
Увеличение загрузки катализатора в три раза (серия III, кривая 3) вызвало быстрый рост ММ на начальном этапе реакции (образование олигомеров и их взаимодействие происходят практически одновременно), и при этом имеет место потеря текучести раствора полимера при невысоких значениях Mn = (10–12) × 103. Возможно в данных условиях в результате физического структурирования (гелеобразования), вызывающего повышение вязкости раствора, ухудшаются условия перемешивания и реакция поликонденсации практически останавливается. Аналогичный процесс наблюдается и при увеличении мощности излучения до 100 Вт. При этом, несмотря на дополнительный обдув воздухом, не удается поддерживать температурный режим в синтезе (98–102°С) и система перегревается до 140°С. В таких случаях синтез останавливали на стадии А (рис. 3). После осаждения и экстракции полимеры были растворимы в ТГФ и CHCl3 (табл. 1, серии III и VII).
Существует два способа закрытия концевых групп полимерных цепей: путем последовательного добавления к реакционной смеси моноэфира фенилбороновой кислоты, и бромбензола [38] или вместо фенилбороновой кислоты вводят диборанат (9,9-диоктил-2,7-бис-(триметиленборат)флуорена), а затем соответствующее монобром ароматическое соединение [39]. В настоящей работе, как и при термическом нагреве (через 15 ч от начала реакции) был выбран второй способ завершения синтеза (рис. 4). Это позволило вводить в полимерную цепь одинаковые концевые группы с обеих сторон в макроцепи, что в ряде случаев важно для практического использовании полимеров. Сравнение двух указанных методов (табл. 1, ПФ-8 серии I и VIII) показывает, что использование для закрытия концевых групп только монофункциональных соединений приводит к получению более низкомолекулярных полимеров.
Рис 4.
ЭЖХ-хроматограммы продуктов реакции полимера ПФ-8 серий I (а) и II (б). 1 – в конце реакции (180 мин), 2 – после осаждения, 3 – после экстракции.
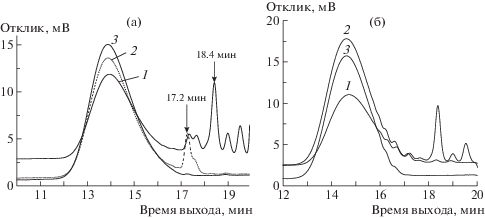
Характеристики полимеров после очистки представлены на рис. 4 и в табл. 1.
Из рис. 2б, 2в и рис. 3 следует, что к 180 мин синтеза олигомеры практически израсходованы как в серии I (100°С) так и в серии II (80°С), а значения их Mw и (Mw/Mn) соответственно равны 61660 (3.2) и 28450 (3.17), и эти полимеры содержат разное количество концевых групп. В реакционной среде более высокомолекулярного полимера серии I существует избыток неизрасходованного дибороланфлуорена, который при взаимодействии с избытком 2-бром-(9,9-диоктил)флуорена (RT = 18.4 мин на рис. 4а и 4б, кривая 1) образует новый олигомерный продукт (RT = 17.6–17.2 мин), в то время как в полимере с меньшей ММ (серия II, рис. 4б) значительно меньше как свободного диборалана, так и соответствующего олигомера. Осаждение полимера в метанол и промывка ацетоном позволяет удалить избыток 2-бромфлуорена. Олигомерные продукты (вплоть до Mw = 5 × 103) выводятся только при экстракции полимера ацетоном в сосуде Cокслета (рис. 4а и 4б кривые 3), результатом чего является снижение полидисперсности полимеров.
Катализаторы реакции Сузуки Pd(PPh3)4 и PdCl2(dppf) позволяют получать полимеры с высоким выходом и ММ, но они недостаточно устойчивы и изменяют активность при хранении. Использование полифлуоренов и СПФ в качестве эмиссионных слоев в светоизлучающих устройствах требует глубокой очистки полимера от катализатора, которой нельзя достичь даже при длительной экстракции полимера ацетоном [40]. В связи с этим исследован синтез полифлуоренов при микроволновом нагреве с полимерным гетерогенным катализатором (серия IV) и SillaCatDPP-Pd (CP1, табл. 1 и 2), силикатная матрица которого содержит лиганд, связанный с катализатором. Такие катализаторы могут быть удалены из реакционной смеси путем фильтрации, и при этом они позволяют получить полимеры с ММ, необходимыми для практического применения (табл. 1). Следует заметить, что полимер-связанный катализатор (иммобилизованный на сшитом ПС) при хранении (2 года) стал терять активность и оказался более чувствительным к кислородной среде, чем SillaCatDPP-Pd, что вызывало потемнение реакционного раствора в ходе синтеза в отличие от синтеза с катализатором SillaCatDPP-Pd.
Таблица 2.
Структура, Mw и Mw/Mn сополифлуоренов, полученных микроволновым синтезом и при конвекционном нагреве
| Сомономерные звенья сополимеров | Образец | Mw × 10–3 | Mw/Mn | Рисунок |
|---|---|---|---|---|
 |
ПФ-8* | 64.5 | 3.02 | 5а |
| ПФ-8 | 78.2 | 2.32 | ||
| СП-1 | 132.0 | 2.39 | ||
 |
СП-2* | 43.0 | 2.91 | 5б |
| СП-2 | 65.0 | 2.20 | ||
 |
СП-3 | 66.0 | 1.92 | 5в |
 |
CП-4 | 109.0 | 2.50 | 5в |
 |
СП-5* | 49.1 | 2.01 | 5г |
| СП-5-1 | 54.7 | 2.23 | ||
| СП-5-2 | 240.5 | 2.58 |
Найденные условия синтеза полимеров при микроволновом нагреве были опробованы для получения полифлуорена ПФ-8 и ряда СПФ, содержащих люминофорные группы, отличающиеся строением и количественным составом (табл. 2). Для функционализации СПФ использовали карбазол-2.7 (СП-1, СП-3, СП-5) или 3,6-диильные сомономерные звенья (CП-2 и CП-5), встроенные в основную цепь полифлуорена. С помощью этих звеньев через спейсерную гексильную или фенильную группы вводили люминофорные группировки нильского красного (НК, СП-5) и 4-пирролидинил-1,8-нафталимида (НИ-1 и НИ-2 в СП-2, СП-3 и СП-5), а также донорно-акцепторные карбазольные и диарил-1,3,4-оксадиазольные группы (транспортные допанты Т-1–Т-3) в боковые цепи СПФ. Введение 2,7- или 3,6-карбазолсомономерных звеньев в полифлуорены, обеспечивает достижение требуемой цветности излучения полимера, снижение уровня агрегирования в полимерной пленке, а также позволяет улучшить баланс электронных и дырочных носителей зарядов в полимере [30]. В табл. 2 представлены полимеры, в которых транспортные допанты также введены в полимер и через 9,9-фенилокси-флуореновые группы (CП-3–СП-5).
Звенья 4,7-бис-(2-тиенил)-2,1,3-бензотиадиазола (Т-БТ-Т) и Нильского красного являются красными люминофорами, а производное нафтилимида (НИ-1 и НИ-2) и сопряженный с флуореном фрагмент бензо [2, 3, 5]тиадиазола (БТ) – зелеными. Структура полимера CП-2 синтезирована с целью получения пленок зеленого свечения (рис. 5б), СП-3 (рис. 5в) – красного, а СП-4 и СП-5 (рис. 5г) – белого. Дополнительно проведен сопоставительный синтез ПФ-8, СП-2 и СП-5 в условиях термического и микроволнового нагрева. Оценка степени вхождения функциональных сомономерных фрагментов в полимерную цепь методом ЯМР затруднительна из-за их малых количеств. Наличие люминофоров фиксируется методом люминесцентного анализа пленок полимеров, а введение транспортных допантов приводит к изменению эффективности переноса энергии возбужденного состояния в пленках.
Рис. 5.
УФ-ФЛ-спектры пленочных покрытий полимеров ПФ-8 и CП-1-5; а: СП-1(1, 1'), ПФ-8* (2, 2'), ПФ-8**(3, 3'), ПФ-8 (4, 4'); б: СП-2 (1, 1'), СП-2*(2, 2'); в: СП-3(1), СП-4(2); г: СП-5* (1, 1'), CП-5-1 (2, 2') и CП-5-2(3'). Полимеры и сополимеры, отмеченные одной и двумя звездочками, получены при термическом нагреве по реакции Сузуки и Ямамото соответственно.

Спектральные характеристики полифлуорена ПФ-8 (рис. 5а), полученные разными методами (в том числе и по протоколу Ямамота при термическом нагреве), были индентичны. В полосе поглощения 340–450 нм с максимумом 390 нм дополнительно наблюдается плечо в диапазоне 410–440 нм. Это плечо характерно для поли-(9,9-диоктил)флуорена, содержащего β-фазу в полимере, стабилизированную взаимодействием ароматических циклов и октильных боковых групп [40]. Оказалось, что введение сопряженных с флуореном сомономерных 2,7-карбазольных звеньев (2.5 мол. %), содержащих транспортную оксадиазольную группу Т-1 (СП-1), привело к исчезновению плеча 410–440 нм в спектре поглощения СП-1 (рис. 5а, ср. кривые 1 и 2–4). Такой эффект можно интерпретировать как разрушение агрегатов β-фазы в пленках СП-1 данного состава. Благодаря механизму резонансного переноса энергии электронного возбуждения по Фёрстеру в полимере СП-2 происходит эффективный перенос энергии с донора (флуорена) на акцептор – нафталимидный фрагмент НИ-1 (1 мол. %), а в полимере СП-3 – последовательный перенос с флуорена на производное нафталимида (НИ-2), а затем на люминесцирующий красный дитиенилбензотиадиазол (1 мол. % Т-БТ-Т). В результате повышаются яркость и эффективность зеленой фотолюминесценции полимера СП-2, полученного двумя методами (рис. 5б, кривые 1 и 2), и красной для СП-3 (рис. 5в, кривая 1). Кроме того, при указанном соотношении мономеров в спектре отсутствует голубая составляющая спектра флуорена.
В результате направленного подбора стехиометрических соотношений люминофоров (сочетание голубого, зеленого и красного) были получены сополимеры СП-4 (рис. 5в, кривая 2) и СП-5 (рис. 5г). В силу высокой эффективности переноса энергии в пленках сомономерные звенья, ответственные за зеленое и красное излучение, взяты в малых количествах (0.03–0.15 мол. %). В спектрах флуоресценции (ФЛ) этих полимеров фиксируются три перекрывающиеся эмиссионные полосы в области 420–480 нм, голубое свечение от сопряженной цепи шести флуореновых звеньев, зеленое 475–575 нм для бензтиазола в основной цепи полимера и 475–530 для НИ-1 в боковой цепи, а также в широком диапазоне от зеленого до красного 575–730 нм от сопряженных фрагментов Т-ВТ-Т (СП-4) и 540–700 нм от НК (СП-5). Спектры этих полимеров CП-4 (рис. 5в, кривая 2) и CП-5 (рис. 5г) соответствуют белому свечению.
Спектры абсорбции и фотолюминесценции сополимера CП-5, полученного разными методами, имеют одинаковые профили поглощения и излучения, а небольшие различия в соотношении интенсивностей полос люминесценции, возможно, вызваны различиями их ММ.
ЗАКЛЮЧЕНИЕ
Полученные результаты показали, что проведение синтеза полифлуоренов и сополифлуоренов под действием микроволнового излучения в открытой системе при 100°С и 80 Вт по сравнению с закрытой (10 мл закрытый сосуд, 130–150°С, 130 Вт) протекает с увеличением времени синтеза с 20 до 90 мин, а с учетом закрытия концевых групп до 180 мин и значительно снижает время процесса по сравнению с конвекционным нагревом (25–45 ч) при сохранении условий синтеза. Преимущество используемой установки и рассмотренной схемы микроволнового синтеза состоит в том, что при 90–100°С достигается оптимально благоприятное соотношение как реакционной способности реагентов, так и уровня протекания побочных реакций, обеспечивается последовательность и непрерывность условий проведения стабильного синтеза полимеров. При этом растворитель толуол–вода имеет преимущество по сравнению с составом ТГФ–вода для получения более высокомолекулярных сополифлуоренов в диапазоне (30–25) × 103 и выше (в зависимости от типа и загрузки катализатора). Введение для закрытия концевых групп одного из мономеров 9,9-диоктил-2,7-бис-(триметиленборат)флуорена на определенной стадии реакции приводит к улучшению стехиометрического соотношения функциональных групп и, как следствие, к значительному росту ММ полимеров по сравнению с использованием моноборалана.
Сравнение свойств полифлуорена ПФ-8 и новых сополимеров сложного состава показало индентичность оптических параметров полимеров, полученных как при конвекционном, так и при микроволновом нагреве. Найденные условия микроволнового синтеза позволили ввести в полимерные цепи различные сомономерные звенья с целью снижения их агрегирования, а также получения сополифлуоренов, люминесценцирующих в зеленой или красной области спектра. Изученный микроволновый синтез позволяет также использовать сомономеры и в малых количествах (0.01–0.05 мол. %), необходимых для получения, например, белого излучения.
Список литературы
Polyfluorenes / Ed. by U. Scherf, D. Neher. Berlin: Springer, 2008.
Gunes S., Neugebauer Y., Sariciftci N.S. // Chem Rev. 2007. V. 107. № 4. P. 1324.
Курдукова И.В., Ищенко А.А. // Успехи химии. 2012. Т. 81. № 3. С. 258.
Bao B., Yuwen L., Zheng X., Weng L., Zhu X., Zhan X., Wang L. // J. Mater. Chem. 2010. V. 20. № 43. P. 9628.
Dong S., Ou D., Qin J., Li. Z. // J. Polym. Sci., Polym. Chem. 2011. V. 49. № 15. P. 3314.
Zhang W., Lu P., Wang Z., Ma Y. // J. Polym. Sci., Polym.Chem. 2013. V. 51. № 9. P. 1950.
Simon Y.C., Peterson J.J., Mangold C., Carter K.R., Coughlin E.B. // Macromolecules. 2009. V. 42. № 2. P. 512.
Komorowska-Durka M., Dimitrakis G., Bode D., Stankiewicza A.I., Stefanidis G.D. // Chem. Eng. J. 2015. V. 264. P. 633.
Choi S.J., Kuwabara J., Kanbara T. // ACS Sustainable Chem. Eng. 2013. V. 1. № 8. P. 878.
Zhang W., Wang Z., Zhang Y., Lu P., Liu L., Ma Y. // Polymer. 2014. V. 55. № 21. P. 5346.
Wu Z., Fan B., Li A., Xue F. Ouyang J. // Organic Electronics. 2011. V. 12. № 6. P. 993.
Nehls B.C., Asawapirom U., Fuldner S., Preis E., Farrell T., Scherf U. // Adv. Funct. Mater. 2004. V. 14. № 3. P. 352.
Jung I.H., Kim S.H., Jeong E., Yang R., K. Lee K., Woo H.Y., Shim H.-K. // J. Polym. Sci., Polym. Chem. 2011. V. 49. № 5. P. 1248.
Galbrecht F., Bünnagel T.W., Scherf U., Farrel T.l. // Macromol. Rapid Commun. 2007. V. 28. № 4. P. 387.
Li J., Song X., Feng Y., Wang Z., Zhang X., Shen F., Lu P. // Thin Solid Films. 2013. V. 545. № 1. P. 188
Xu W., Zhang W., Hu Q., Zhao L., Teng X., Xia R., Lai W.-Y., Nelson J., Huang W., Bradley D. D.C. // Organic Electronics 2014. V. 15. № 6. P. 1244.
Stenidis G.D., Munoz A.N., Sturm G.S.J., Stankiewicz A. // Rev. Chem. Eng. 2014. V. 30. № 3. P. 233.
Райхардт К. Растворители и эффекты среды в органической химии. М.: Мир, 1991.
Москва В.В. Растворители в органической химии // Соросовский образовательный журнал. 1996. № 12. С. 33.
McFarlane S.L., Piercey D.G., Coumont L.S., Tucker R.T., Fleischauer M.D., Brett M.J., Jonathan G.C., Veinot J.G.C. // Macromolecules. 2009. V. 42. № 3. P. 591.
Galbrecht F., Yang X.H., Nehls B.S., Neher D., Farrella T., Ullrich S. // Chem. Commun. 2005. № 18. P. 2378.
Zhang W.S., Lu P., Wang Z.M., Ma Y.G. // Sci . China Chem. 2012. V. 55. № 5. P. 844.
Yevlampieva N.P., Khurchak A.P., Nosova G.I., Smyslov R.Yu., Berezin I.A., Ilgachb D.M., Kopylova T.N., Gadirov R.M., Yakimansky A.V. // Chem. Phys. Lett. 2016. V. 645. № 1. P. 100.
Lai W.-Y., He Q.-Y., Zhu R., Chen Q.-Q., Huan W. // Adv. Funct. Mater. 2008. V. 18. № 2. P. 265.
Zhang W., Lu P., Wang Z., Ma Y. // J. Polym. Sci., Polym Chem. 2013. V. 51. № 9. P. 1950.
Fu Y., Cha H., Lee G.-Y., Moon B.J., Park C.E., Park T. // Macromolecules. 2012. V. 45. № 8. P. 3004.
Yong L.W., Dong L., We H. // Sci. China Chem. 2010. V. 53. № 12. P. 2472.
Lai W.-Y., Chen Q.-Q., He Q.-Y., Fan Q.-L., Huang W. // Chem. Commun. 2006. V. 42. № 18. P. 1959.
Nosova G.I., Lypenko D.A., Smyslov R.Yu., Berezin I.A., Zhukova E.V., Mal’tsev E.I., Dmitriev A.V., Litvinova L.S., Solovskaya N.A., Dobrokhotov O.V., Abra-mov I.G., Yakimanskii A.V. // Polymer Science B. 2014. V. 56. № 1. P. 59.
Wang Q., Zhang B., Liu L., Chen Y., Qu Y., Zhang X., Yang J., Xie Z., Geng Y., Wang L., Wang F. // J. Phys. Chem. C. 2012. V.116. P. 21733.
He P., Haswel S.J., Fletcher P.D.I., Kelly S.M. // Andrew Mansfield2 Beilstein J. Org. Chem. 2011. V. 76. № 4. P. 1150.
Wu X.F., Anbarasan P., Neumann H., Beller M. // Angew. Chem. Int. Ed. 2010. V. 49. № 39. P. 9047.
Arvela R.K., Leadbeater N.E., Sangi M.S., Williams V.A., Granado P., Singer R.D. // J. Org. Chem. 2005. V. 70. № 1. P. 161.
Murage J., Eddy J.W., Zimbalist J.R., McIntyre T.B., Wagner Z.R., Goodson F.E. // Macromolecules. 2008. V. 41. № 20. P. 7330.
Leadbeater N.E., Marco M. // Angew. Chem. Int. Ed. 2003, V. 42. № 12. P. 1407.
Hall D.G. // Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials / Ed. By D.G. Hall. Weinheim:Wiley-VCH Verlag GmbH & Co. KGaA, 2011. V. 1–2.
Dzhabarov V., Výprachtický D., Cimrová V. // Eur. Polym. J. 2018. V. 98. № 1. P. 420.
Wang E., Wang M., Wang M., Duan C., Zhang J., Cai W., He C., Wu H., Cao Y. // Macromolecules. 2009. V. 42. № 13. P. 4410.
Hou Q., Xu Y., Yang W., Yuan M., Peng J., Cao Y. // J. Mater. Chem. 2002. V. 12. № 10. P. 2887.
Sonar P., Grimsdale A.C., Heeney M., Shkunov M., McCulloch I., Müllen K. // Synth. Met. 2007. V. 157. № 22. P. 872.
Knaapila M., Bright D.l W., Nehls B.S., Garamus V.M., Almasy L., Schweins R., Scherfr U., Monkman A.P. // Macromolecules. 2011. V. 44. № 16. P. 6453.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)