

Высокомолекулярные соединения (серия Б), 2019, T. 61, № 2, стр. 83-98
СИНТЕЗ, СВОЙСТВА И ПРИМЕНЕНИЕ ПОЛИМЕРОВ, СОЧЕТАЮЩИХ АЗО- И АЗОМЕТИНОВЫЕ ГРУППЫ В СВОЕМ СОСТАВЕ
А. И. Ковальчук a, Я. Л. Кобзарь a, И. М. Ткаченко a, В. В. Шевченко a, *
a Институт химии высокомолекулярных соединений Национальной академии наук Украины
02160 Киев, Харьковское ш., 48, Украина
* E-mail: valpshevchenko@gmail.com
Поступила в редакцию 30.07.2018
После доработки 07.11.2018
Принята к публикации 22.11.2018
Аннотация
Рассмотрены методы синтеза, структура и свойства, а также области применения полимеров, сочетающих азо- и азометиновые группы в своем составе. Предложена классификация азо-азометинсодержащих полимеров, основанная на способах введения хромофорных групп в основную и боковую цепь полимеров, а также образования сетчатых структур. Проанализированы особенности азо-азометинсодержащих полимеров, вызванные сочетанием этих хромофорных групп в их составе.
ВВЕДЕНИЕ
Особое место среди высокомолекулярных соединений занимают органические полимеры с системой сопряженных связей, способных проводить электрический ток. Это определило широкий спектр практического применения таких полимеров, как полупроводниковые материалы [1]. Основными путями изменения их полупроводниковых свойств можно назвать регулирование общей протяженности системы сопряженных связей, а также варьирование их химической природы. Использование в качестве сопряженных фрагментов хромофорных групп, поглощающих видимый свет, наряду с сохранением традиционной электропроводности полимеров может придать им дополнительные свойства, не характерные для классических электропроводящих полимеров (полиацетилен, полифенилен, полианилин и другие) [2, 3].
В широкой гамме хромофоров, которые используются как сопряженные фрагменты, внимание привлекают азо- и азометиновые группы [4]. Помимо свойств, присущих обеим хромофорным группам (высокая оптическая чувствительность, близкий диапазон поглощения светового излучения, сольватохромные свойства), они имеют ряд отличий [5, 6]. Так, азобензольные фрагменты более склонны к фотоиндуцированным (УФ- и видимым излучением) обратимым транс-цис-транс-переходам, которые осуществляются с меньшими затратами энергии по сравнению с азометиновыми соединениями [7]. Поэтому азосоединения (в большей мере азосодержащие полимеры) являются ключевыми компонентами при создании фотоактивных реверсивных сред (“умных” поверхностей, поверхностно-рельефных решеток) [8, 9]. Способность азохромофоров к более эффективному (в сравнении с другими хромофорами) распределению электронной плотности по системе сопряжения в молекуле от электронодонорных к электроноакцепторным группам позволила создать на основе азополимеров материалы с нелинейно-оптическими свойствами [10–12].
Вместе с тем, благодаря повышенной отрицательной диэлектрической анизотропии азометиновых групп, а также их более высокой термостабильности в сравнение с азогруппами, азометиновая хромофорная система чаще используется в качестве мезогенных групп при конструировании полимерных ЖК-материалов [13]. Кроме того, способность азометиновых групп формировать комплексы (для этой цели, как правило, используют азометинсодержащие соединения на основе производных салицилового альдегида) с ионами различных металлов – эффективный путь придания им магнитных и регулируемых электрических свойств, позволяя тем самым конструировать на их основе сенсоры (на катионы металлов), катализаторы и биологически активные системы [14].
Имеется несколько обширных обзоров, посвященных синтезу, структуре и свойствам как азо- [10, 15–17], так и азометинсодержащих полимеров [13, 18, 19]. Однако информация о полимерах, сочетающих азо- и азометиновые группы в своем составе (ААП), в этих работах носит фрагментарный характер [18]. Учитывая упомянутые выше характеристики азо- и азометинсодержащих систем, интересной и перспективной представляется реализация их свойств при объединении в одном полимере, тем более что такой подход осуществлен для мономерных соединений.
В настоящем обзоре рассмотрены сведения о путях синтеза азо- и азометиновых полимеров, особенностях их химического строения и структуры, а также областях практического использования. Предложена классификация ААП, которая основывается на способах введения хромофорных групп в основную цепь линейных полимеров и их боковых ответвлений, а также образования сетчатых структур. В рамках предложенной классификации охарактеризованы линейные ААП с азо- и азометиновыми группами в основной цепи, с азометиновыми группами в основной цепи и азогруппами в боковой цепи, с азо- и азометиновыми группами в боковой цепи, а также ААП сетчатого строения.
ОБЩИЕ ПОДХОДЫ К СИНТЕЗУ АЗО- И АЗОМЕТИНОВЫХ ПОЛИМЕРОВ
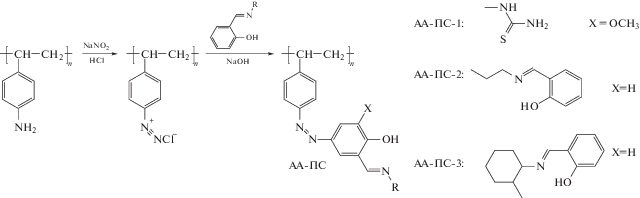
В настоящее время можно выделить два подхода к введению в состав полимеров сочетания азо- и азометиновых блоков. Первый из них строится на использовании непосредственно реакции образования хромофорных групп, как правило, азометиновой группы в реакциях формирования полимерной цепи ААП или полимераналогичных превращений [20–26], в основе которого лежит реакция азодиарилсодержащих аминов с альдегидной группой. Этот метод синтеза ААП позволяет рассматривать его в качестве азометинового подхода. Формирование же азогруппы при синтезе ААП отмечено только для поверхностной модификации аминосодержащих полистиролов азометинсодержащими соединениями за счет реакции азосочетания [27–29].
Второй подход к синтезу ААП базируется на применении мономеров, сочетающих в своем составе азо- и азометинсодержащие фрагменты (ААС) [30–35]. Этот подход применяют для синтеза как линейных, так и сетчатых, а также координационных азо- и азометиновых полимеров [30–32]. На сегодняшний момент описан целый ряд реакционноспособных ААС [31, 33–44]. Наибольший интерес среди них вызывают функционализированные соединения:
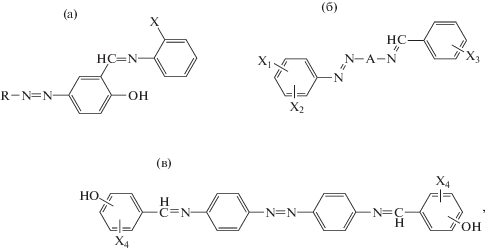
где R = Ph, X = H, OH, SH, COOH; X1, X2 = H, Cl, –OCH3, –NO2, X3 = H, Cl, –OH, –NO2, N(CH3)2; A = Ar или Het; X4 = H, CH3
Синтез таких соединений был направлен на создание хромогенных чувствительных к внешним воздействиям систем (stimuli-responsive systems) [45], способных проявлять термо- и фоторегулируемые ЖК-свойства, таутомерные переходы (под действием тепла, света, растворителя и pH среды) в сочетании со способностью к комплексообразованию с различными ионами [33–44]. Азо- и азометиновые группы в представленных соединениях могут находиться в единой системе сопряжения, либо выходить из нее в случае мета-изомеров или при введении в состав алифатических развязок. Использование некоторых ААС для синтеза ААП рассмотрено в соответствующей главе ниже.
Необходимо отметить, что большинство ААП, описанных в литературе и представленных в данном обзоре, имеют достаточно низкие значения молекулярной массы (Mn = (1.6–10.2) × 103). В некоторых случаях для синтезированных ААП вообще не приводятся величины ММ.
АЗОМЕТИНОВЫЙ ПОДХОД К СИНТЕЗУ АЗО- И АЗОМЕТИНОВЫХ ПОЛИМЕРОВ
Формирование полимерной цепи ААП за счет образования азометиновой связи является, по сути, единственным направлением синтеза линейных ААП. Этот путь позволяет синтезировать ААП, совмещающие обе группы в основной или боковой цепи полимера с сохранением цепи сопряжения между азо- и азометиновым фрагментами, либо разъединять их путем размещения одной из групп, как правило, азо-группы в боковой цепи. ААП линейного строения, сочетающие азо- и азометиновые группы в основной цепи полимера и полученные в рамках обоих подходов, представляют собой азосодержащие полиазометины (азо-ПАМ).
СИНТЕЗ АЗО-ПАМ С АЗОГРУППАМИ В ОСНОВНОЙ ЦЕПИ
Синтез полимеров данного типа основывается на взаимодействии азосодержащих диаминов с диальдегидами (табл. 1):
Таблица 1.
Свойства азо-ПАМ с азогруппами в основной цепи
| Азосодержащие полиазометины | х | R | Выход, % | λмакс, нм | [η], дл/г | Т10%, °С | σ, Ом–1 см–1 | ∆E*, eВ | Лите-ратура |
|---|---|---|---|---|---|---|---|---|---|
| азо-ПАМ-1 | 1 |  |
88 | 420 | 0.091 | 490 | >1.91 × 10–13 | – | [20] |
| азо-ПАМ-2 | 1 |  |
71 | 420 | 0.17 | 435 | 2.81 × 10–13 | – | [20] |
| азо-ПАМ-3 | 1 | 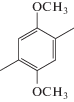 |
81 | 435 | 0.088 | 385 | 3.43 × 10–13 | – | [20] |
| азо-ПАМ-4 | 1 | 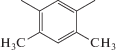 |
54 | 408 | 0.11 | 510 | <10–15 | – | [20] |
| азо-ПАМ-5 | 1 | 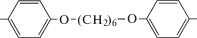 |
89 | 263, 402 | – | 310** | – | – | [21] |
| азо-ПАМ-6 | 2 |  |
– | – | – | – | 6.27 × 10–10 | 0.536 | [22] |
| азо-ПАМ-7 | 2 |  |
– | – | – | – | 3.46 × 10–7 | 0.504 | [22] |
| азо-ПАМ-8 | 4 |  |
– | – | – | – | 7.57 × 10–8 | 0.630 | [22] |
| азо-ПАМ-9 | 6 |  |
– | – | – | – | 3.05 × 10–9 | 0.78 | [22] |
Примечание. λмакс – Максимум поглощения УФ (для азо-ПАМ-1–азо-ПАМ-4 не приведен тип растворителя, для азо-ПАМ-5 в ДМСО); [η] – характеристическая вязкость (в муравьиной кислоте при 30°С); Т10% – температура 10% потери массы (инертная атмосфера); σ – удельная электропроводимость (комнатная температура, напряжение 1000 В).
* Значения энергии активации найдены по отношению log σ к абсолютной температуре [22].

Соединения азо-ПАМ-1–азо-ПАМ-4 получены взаимодействием 4,4'-(E)-диазен-1,2-диилдианилина с терефталевым, 2,5-диметил- и 2,5-диметокситерефталевым, а также 4,6-диметилизо-фталевым альдегидами соответственно (табл. 1) [20]. Эти полимеры (кроме азо-ПАМ-4) с достаточно высоким выходом продукта, а их величина [η] достигает значения 0.17 дл/г (табл. 1), тем самым практически не отличаясь от аналогичных ПАМ, не содержащих азогруппы [46]. Синтезированные азо-ПАМ имеют высокую термостабильность и растворимы в муравьиной, трифторуксусной, а также серной кислотах и не растворимы в органических растворителях. Все они способны проводить электрический ток, при этом величина их удельной электропроводимости σ зависит от характера заместителей в сопряженной цепи и ее электронно-конформационного строения. Так, включение электронодонорных метильных или метоксильных групп в состав терефталевого альдегида позволило получить азо-ПАМ-2 и азо-ПАМ-3 с повышенными значениями σ, в сравнение с азо-ПАМ-1. При этом азо-ПАМ-3 характеризуется и батохромным сдвигом максимума УФ-поглощения. Изменение подхода к дизайну азо-ПАМ путем использования 4,6-диметилизофталевого альдегида (азо-ПАМ-4) привело, с одной стороны, к повышению его термической стабильности и характеристической вязкости, а с другой – к понижению выхода и удельной электропроводимости (табл. 1).
Для улучшения растворимости и придания ЖК-свойств был синтезирован азо-ПАМ-5, содержащий гибкие алифатические фрагменты [21]. Этот полимер растворим в большинстве органических растворителей (хлороформ, ДМФА, ДМСО и других). Однако использование при синтезе азо-ПАМ-5 в качестве альдегидной компоненты 4,4'-[гексан-1,6-диил-бис-(окси)]дибенз-альдегида ведет к понижению его термической стабильности по сравнению с ароматическими азо-ПАМ-1–азо-ПАМ-4 (табл. 1).
Отметим, что регулирование длины цепи сопряжения азо-ПАМ-1–азо-ПАМ-5 позволяет изменять их максимум поглощения. Так, прерывание цепи сопряжения в азо-ПАМ-4 за счет использования изомерных мета-фрагментов ведет к гипсохромному сдвигу по сравнению с азо-ПАМ-1–азо-ПАМ-3, который еще больше возрастает в азо-ПАМ-5 при включении алифатических спейсеров в его состав. В то же время, в растворе ДМСО азо-ПАМ-5 имеет синее свечение с длиной волны максимума флуоресценции 459 нм, а его стоксов сдвиг составляет 52 нм. Для всех рассматриваемых азо-ПАМ-1–азо-ПАМ-5 наблюдается батохромный сдвиг по сравнению с аналогичными по строению азо-ПАМ, но не содержащих азогрупп [46]. При этом азо-ПАМ-5 формирует ЖК-фазу, близкую к нематическому типу [21]. Точно определить тип мезофазы и температурную область ее существования не удалось, так как температура перехода полимера в мезоморфное состояние совпадает с его температурой разложения.
Для улучшения оптических и электрооптических свойств, а также электронной проводимости азо-ПАМ, предложен подход, связанный с повышением концентрации азогрупп относительно азометиновых. Данный подход был реализован диазотированием 1,4-фенилендиамина и его азопроизводных с последующим азосочетанием полученных диазониевых солей с анилином. Таким способом получены азосодержащие диамины с двумя, четырьмя и шестью азогруппами в своем составе, на основе которых и были синтезированы полимеры азо-ПАМ-6–азо-ПАМ-9 соответственно (табл. 1) [22]. Такие полимеры имеют хорошую растворимость в ДМФА, из растворов которого были сформированы пленки на стекле методом “spin-coating”. Азо-ПАМ-6, полученный конденсацией 4,4'-{1,4-фенилен-бис-[(E)диазен-2,1-диил]}дианилина с глутаровым альдегидом, характеризируется значением температуры стеклования Tс = 75°С, а значение температуры его начальной потери массы Т0 = 198°С. Повышение ароматичности азо-ПАМ-7 путем замены глутарового альдегида (азо-ПАМ-6) на терефталевый позволило повысить значения Tс (186°С) и σ для указанного полимера. Однако дальнейшее увеличение концентрации азогрупп по отношению к азометиновым (азо-ПАМ-8 и азо-ПАМ-9), напротив, привело к понижению значений σ. По мнению авторов, это связано с понижением сегментальной подвижности цепей таких полимеров, поскольку энергия активации сегментальной подвижности ∆E для азо-ПАМ-7–азо-ПАМ-9 коррелирует с их значениями σ (табл. 1). При этом с повышением температуры наблюдается значительное увеличение электропроводности для азо-ПАМ-8 и азо-ПАМ-9. Последнее является следствием снижения ширины запрещенной зоны (вплоть до 1.5 еВ), а также увеличением сегментальной подвижности полимерных цепей для таких азо-ПАМ с ростом температуры [22, 47, 48].
Исследование фотоизомеризационных свойств для полимеров азо-ПАМ-6–азо-ПАМ-9 показало, что время достижения фотостационарного состояния для них наступает в интервале от 11 до 38 мин. Наименьшим временем перехода из транс- в цис-форму характеризуется азо-ПАМ-6, что связано с наличием гибких фрагментов в его составе [22]. Обратная термическая цис-транс- изомеризации для азо-ПАМ-6–азо-ПАМ-9 происходит в интервале от 16 до 68 мин. При этом с увеличением количества азобензольных фрагментов в элементарном звене для этой серии снижается время необходимое для обратного цис-транс-перехода [22].
В рамках азометинового подхода нами развивается направление создания фторсодержащих ААП [49]. Характерной особенностью является введение перфторароматических фрагментов в состав азо-азометиновой цепи сопряжения. С этой целью разработаны методы синтеза соответствующих азосодержащих бис-гидроксибензальдегидов, в которых в состав указанной цепи сопряжения входят изомерные тетрафторфениленовые или октафторбифениленовые фрагменты [50, 51]. Отметим также, что в отличие от синтеза описанных выше азо-ПАМ, разработанные мономеры содержат азогруппу в составе диальдегидной компоненты (структура (3а)) [49]:

Приведенный азо-ПАМ обладает аморфно-кристаллической структурой со степенью кристалличности ~50%, растворим в ионных жидкостях, не растворим в органических растворителях, стойкий к воздействию кислот и щелочей. Он характеризуется высокой термической стабильностью и отсутствием стеклования вплоть до 300°С [49].
Ввиду нерастворимости данного полимера в органических растворителях особенности фотооптического поведения перфторароматического азо-ПАМ изучены на примере модельного перфторсодержащего ААС, полностью отражающего особенности строения повторяющегося звена этого полимера и его дифильность (структура (3б)) [49]. В растворе ТГФ модельный ААС характеризуется интенсивным максимумом поглощения УФ при 365 нм, что характерно для π-π*-перехода азо-азометиновой хромофорной системы, и менее интенсивный максимум при 452 нм, который указывает на ее n-π*-переход. Установлено, что облучение ААС УФ-светом (365 нм) ведет к транс-цис-переходу последнего за счет азогрупп, фотостационарное состояние при этом наступает после 30 с облучения [49].
СИНТЕЗ АЗО-ПАМ С АЗОГРУППАМИ В БОКОВОЙ ЦЕПИ
Введение азо-групп в боковую цепь полимера осуществлено реакцией азосодержащих диаминов с диальдегидами:
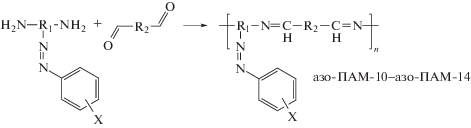
Видно, что азо- и азометиновые группы выведены из цепи сопряжения. Взаимодействием производных 4-[E-фенилдиазенил]бензол-1,3-диамина с пара-дибензальдегидами, содержащим оксиалифатические фрагменты различной длины, получена серия полимеров азо-ПАМ-10–азо-ПАМ-13 (табл. 2) [23]. Они характеризуются хорошей растворимостью в органических растворителях (ДМСО, ДМФА, N-метил-2-пирролидон (МП), 1,4-диоксан, дихлорметан, ацетон, толуол и другие). Очевидно, что хорошая растворимость полимеров связана как с наличием в их основной цепи алифатических фрагментов, так и является следствием невысоких ММ. В сравнении с азо-ПАМ, которые содержат азогруппы в основной цепи, полученные азо-ПАМ-10–азо-ПАМ-13 характеризуются относительно невысокой термической стабильностью. Синтезированные полимеры азо-ПАМ-10– азо-ПАМ-13 обладают высокой степенью кристалличности, при этом их значения Тс лежат в диапазоне 30–75°С (табл. 2).
Таблица 2.
Свойства азо-ПАМ с азогруппами в боковой цепи
| Азосодержа-щие полиазоме-тины | R1 | Х | R2 | Выход, % | λ*макс π → → π*-, n → π*-перехо-дов (нм) | Mn × 10–3 | Mw × 10–3 | Ð | Тс, °С | Т0, °С | Т10%, °С | Темпера-турные переходы ЖК-состоя-ния**, °С | Литера-тура |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| азо-ПАМ-10 | 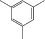 |
CH3-(p) |  |
69 | 360–365; 450–463 | 2.5 | 9.6 | 3.8 | 53 | 180 | 350 | 111 К, 138 Н, 205 И | [23] |
| азо-ПАМ-11 |  |
CH3-(o) |  |
99 | 363; 453 | 1.8 | 2.0 | 1.1 | 75 | 160 | 340 | 130 К, 158 Н, 206 И | [23, 52] |
| азо-ПАМ-12 | 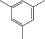 |
Cl-(p) |  |
97 | 360–365; 450–463 | 1.6 | 1.9 | 1.2 | 30 | 220 | 330 | 94 К, 106 Н, 215 И | [23] |
| азо-ПАМ-13 | 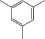 |
Cl-(p) | 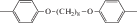 |
82 | 360–365; 450–463 | 4.3 | 4.6 | 1.1 | 59 | 230 | 295 | 120 К, 133 Н, 212 И | [23] |
| азо-ПАМ-14 | 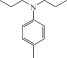 |
NO2-(p) | 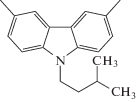 |
82 | – | 5.0 | 10.0 | 2.0 | 158 | 270 | – | – | [24] |
Введение азогруппы в боковую цепь линейных азо-ПАМ при сохранении цепи сопряжения оказалось эффективным инструментом для регулирования их нелинейно-оптических и ЖК-свойств (табл. 2) [23, 24]. Показано, что в интервале температуры 106–215°С в азо-ПАМ-10–азо-ПАМ-13 формируется термотропная мезофаза нематического типа. При этом самым широким температурным интервалом (109°С) существования мезофазы обладает азо-ПАМ-12, который содержит атомы хлора в пара-положении относительно азосвязи. Наряду с этим, указанный хлорсодержащий азо-ПАМ характеризуется и самыми низкими значениями ММ и Тс (сравнение азо-ПАМ-10, азо-ПАМ-11 и азо-ПАМ-13 в табл. 2). Замена 1,6-гексаметиленового фрагмента в азо-ПАМ-12 на 1,8-октаметиленовый спейсер приводит к росту как температуры начала образования мезоморфного состояния в азо-ПАМ-13, так и термостабильности. Однако при этом сужается температурный интервал существования мезоморфного состояния (сравнение азо-ПАМ-12 и азо-ПАМ-13). Введение же электронодонорной метильной группы в пара-положение к азогруппе вместо электроноакцепторного атома хлора (азо-ПАМ-10) привело к наиболее высокому значению Т10%. Среди полимеров азо-ПАМ-10–азо-ПАМ-13 самым низкомолекулярным считается азо-ПАМ-11, который содержит группу СН3 в орто-положении относительно азосвязи.
Изучение оптических свойств азо-ПАМ-10–азо-ПАМ-13 показало, что спектры поглощения в УФ-области для таких полимеров содержат два максимума. Первый соответствует π→π*-электронным переходам азо-азометиновой хромофорной системы, а второй проявляется в более длинноволновой области, что указывает на n→π*-переход (табл. 2). Однако по сравнению с исходными азосодержащими диаминами, в УФ-спектрах азо-ПАМ-10– азо-ПАМ-13 наблюдается батохромный сдвиг на 20 нм. Скорее всего, это является следствием расширения цепи сопряжения между азо-фенил-азометиновыми мезогенами [23, 52].
Азо-ПАМ с азогруппами в боковой цепи помимо применения в качестве ЖК-материалов могут быть использованы и для создания систем с нелинейно-оптическими свойствами. С этой целью получен азо-ПАМ-14 на основе 9-изофенил-9H-карбазол-3,6-дикарбальдегида и аминозамещенного красителя дисперсного красного-19 (DR-19) [24]. Этот полимер характеризуется хорошей растворимостью в ДМАА, ДМФА, МП, ТГФ и ДХ, и имеет высокие значения Т0 и Тс, а также ММ по сравнению с азо-ПАМ-10–азо-ПАМ-13 (табл. 2.). Использование в составе диальдегидного компонента объемных карбазольных блоков обусловлено, прежде всего, их способностью минимизировать диполь-дипольные взаимодействия между хромофорами, которая ведет к повышению активной концентрации хромофоров, их степени упорядочения при полинг-процессе и дает возможность получить полимеры с высоким значением коэффициента генерации второй гармоники (d33) [10]. Исследование нелинейно-оптических свойств второго порядка (d33) для азо-ПАМ-14 показало, что коэффициент генерации второй гармоники для него составляет 82 пм/В (при длине волны лазерного излучения 532 нм) [24].
Отметим, что для упомянутых ААП характерна густая сетка водородных связей, вызванная взаимодействием азометиновых групп, энергия которой составляет 39.0 кДж/моль [18]. Следствием этого является повышение Tс и уменьшение диполь-дипольных взаимодействий хромофоров, тем самым увеличивая время эксплуатации материалов на основе таких полимеров, в частности в нелинейно-оптической области [10]. Напротив, наличие густой сетки водородных связей ведет к понижению растворимости (азо-ПАМ-1–азо-ПАМ-4). В этом случае повышение растворимости ААП достигается включением в их состав объемных заместителей (азо-ПАМ-14), алифатических развязок (азо-ПАМ-5, азо-ПАМ-6, азо-ПАМ-10–азо-ПАМ-14), а также вывода хромофорных фрагментов в боковое ответвление цепи (азо-ПАМ-10–азо-ПАМ-14). Однако включение алифатических фрагментов в полимерную цепь приводит к уменьшению длины сопряжения и понижению их термической стабильности ААП [22], наличие же объемных заместителей ухудшает их механические характеристики ААП [53]. Наличие же азогрупп в боковой цепи полимеров, вследствие сильных диполь-дипольных взаимодействий, может приводить к антипараллельной направленности хромофорных фрагментов, что снижает нелинейно-оптический эффект [54], а также негативно сказывается на термической стабильности полимеров (сравнение Азо-ПАМ-5 и азо-ПАМ-10–азо-ПАМ-14) [21, 23, 24].
СИНТЕЗ ААП С АЗО- И АЗОМЕТИНОВЫМИ ГРУППАМИ В БОКОВОЙ ЦЕПИ
Сочетание азо- и азометиновых фрагментов в боковой цепи полимера также осуществлялось за счет образования азометиновой группы в реакциях полимераналогичных превращений. Этот путь реализован реакцией аминосодержащего азобензола с линейными элементоорганическими полимерами – полисиланами и полифосфазенами, содержащими альдегидные группы (структуры (5) и (6) соответственно):
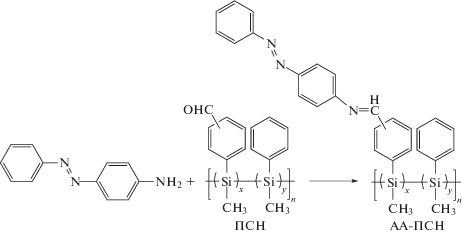

В случае полисиланов (ПСН) предварительно были синтезированы альдегидсодержащие полисиланы с 6 (ПСН-1) и 12 мол.% (ПСН-2) альдегидных групп в боковой цепи. Последующей конденсацией ПСН-1 и ПСН-2 с 4-аминоазобензолом получены соответствующие полимеры АА-ПСН-1 и АА-ПСН-2 [25]. Они растворимы в ТГФ и толуоле, их характеристики представлены в табл. 3. В электронных спектрах АА-ПСН-1 и АА-ПСН-2 присутствуют два максимума поглощения. Первый, более интенсивный, при 335 нм, свидетельствует о σ-σ*-переходах кремниевой цепи, а второй, менее интенсивный, при 360 нм, указывает на π-π*- и n-π*-переходы азо-азометиновой хромофорной системы (табл. 3). Величина мольного поглощения зависит от количества азо-азометиновых хромофорных фрагментов в боковой цепи полимера (сравнение АА-ПСН-1 и АА-ПСН-2 в табл. 3). Важно, что включение азо- и азометиновой групп в состав АА-ПСН-1 и АА-ПСН-2 влечет повышение их фотостабильности, однако при этом происходит гашение фото- и электролюминесценции [25].
Таблица 3.
Молекулярно-массовые характеристики и оптические свойства ПСН с азо- и азометиновыми группами в боковой цепи [25]
| Полисиланы | Mn × 10–3 | Mw × 10–3 | ε(σ-σ*), моль–1 см–1 | ε(π–π*,n–π*), моль1 см–1 | Выход, % |
|---|---|---|---|---|---|
| ПСН-1 | 8.2 | 33.0 | 7300 | – | – |
| ПСН-2 | 7.5 | 21.0 | 6500 | – | – |
| АА-ПСН-1 | 8.5 | 34.0 | 7800 | 1500 | 59 |
| АА-ПСН-2 | 7.8 | 22.5 | 7800 | 3000 | 66 |
Взаимодействием поли[бис-(п-формилфенокси)фосфазена] с избытком 4-аминоазобензола был получен азо-азометинсодержащий полифосфазен (ПФ) (структура (6)).
Этот полимер нерастворим в большинстве органических растворителей. Вследствие стерических препятствий степень функционализации полифосфазена азобензольными фрагментами достаточно низкая и составляет 34.3% (согласно спектроскопии ЯМР 1Н) [26]. По данным анализа ДСК, значение Тс для АА-ПФ составляет ~88°С, что существенно выше (на 32 оС) нефункционализированного полифоcфазена и является следствием повышения микрокристаличности в боковой цепи полимера, а также увеличения интенсивности полярных взаимодействий азометиновых групп. Синтезированный полимер представляет интерес для создания различных ЖК-материалов с возможностью регулирования их мезоморфного поведения с помощью фотоиндуцированных транс-цис-переходов [26].
Единственный пример введения фрагмента, сочетающего азо- и азометиновые группы в своем составе, за счет реакции образования азосвязи описан в работах [27–29]. Он связан с модификацией поверхности полиаминостирола путем его диазотирования с последующим азосочетанием диазониевой соли с азометисодержащим компонентом (производные салицилового альдегида):
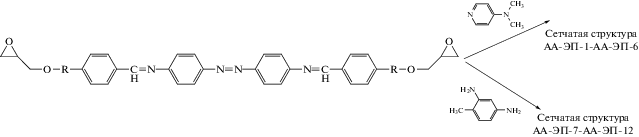
Для указанных полимераналогичных превращений были выбраны коммерчески доступные ПС (в виде гранул) марок XAD®-2 (АА-П-1) и XAD®-4 (АА-ПС-2, АА-ПС-3), характеризующиеся мезопористой структурой со средним диаметром пор ~5 и 9 нм соответственно. Выбор указанных выше марок ПС обусловлен, главным образом, их высокой сорбционной способностью. Полученные полимеры являются эффективными комплексообразователями, что позволяет использовать их для создания сорбционных колонок и аналитического определения (можно определить до 7.5 мкг) катионов таких металлов, как Cu2+, Zn2+, Pb2+, Ni2+ и Bi3+ [27–29].
ПОЛУЧЕНИЕ ААП НА ОСНОВЕ МОНОМЕРОВ, СОДЕРЖАЩИХ АЗО- И АЗОМЕТИНОВЫЕ ГРУППЫ В СВОЕМ СОСТАВЕ
Второй подход к синтезу ААП базируется на применении мономеров, сочетающих в своем составе азо- и азометинсодержащие фрагменты [30–35]. Использование реакционноспособных ААС позволило создать линейные, сетчатые и координационные ААП [30–35]. В этом случае формирование полимерной цепи и сшивки осуществляется за счет образования амидной, простой эфирной, аминной или координационной связей. В рамках данного подхода, ААС были применены также и для модификации поверхности целлюлозного волокна.
СИНТЕЗ ЛИНЕЙНЫХ ААП С АЗО- И АЗОМЕТИНОВЫМИ ГРУППАМИ В ОСНОВНОЙ ЦЕПИ
С целью получения полимерных материалов для очистки воды от ионов различных металлов поликонденсацией азо-азометинсодержащей дибензойной кислоты с ароматическим диамином получен азо-азометинсодержащий полиамид (АА-ПА) [30]:

Синтезированный с высоким выходом (95%) полимер хорошо растворим в сильнополярных апротонных растворителях (ДМАА, ДМФА, ДМСО, МП), а значения его [η], Mn и Mw составляют 0.72 дл/г (ДМФА, 25°С), 10.2 × 103 и 25.5 × 103 (в ДМАА, ГПХ) соответственно. По данным рентгенографического анализа, АА-ПА характеризуется аморфной структурой. Наличие в составе полимера фениленовых фрагментов, азометиновой и амидной связи позволило придать ему достаточно высокую термическую стабильность (Т0 = 283°С) [30].
В УФ-спектрах синтезированного полимера присутствуют максимумы поглощения при 316 и 376 нм, которые соответствуют π-π*- и n-π*-переходам азо-азометиновой хромофорной системы, что типично для ААП. При этом УФ-облучение (320 нм) раствора АА-ПА ведет к его фотолюминесценции, максимум эмиссии которой наблюдается при 467 нм.
Благодаря наличию в составе АА-ПА азометиновой и относительно ее в орто-положении гидроксильной группы, последняя является эффективным сорбентом тяжелых металлов и может быть применима для создания материалов для очистки воды от катионов Pb2+ [30].
СИНТЕЗ СЕТЧАТЫХ АЗО- И АЗОМЕТИНОВЫХ ПОЛИМЕРОВ
С целью получения ЖК-структуры АПП, мезоморфное состояние которых можно сохранить длительный период времени путем “замораживания” в полимерной сетке, были получены две серии азо-азометинсодержащих эпоксидных полимеров (ЭП) сетчатого строения [31]. В качестве исходного мономера были использованы α,ω-диэпоксидные соединения, содержащие жесткий азо-азометиновый блок в сочетании с метиленовой и гибкой оксиалкиленовой развязкой разной длины. Формирование сетчатой структуры проводилось по механизмам полимеризации и поликонденсации:
где АА-ЭП-1, АА-ЭП-7: R = –CH2–; АА-ЭП-2, АА-ЭП-8: R = 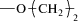 ; АА-ЭП-3, АА-ЭП-9: R = =
; АА-ЭП-3, АА-ЭП-9: R = = 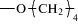 ; АА-ЭП-4, АА-ЭП-10: R =
; АА-ЭП-4, АА-ЭП-10: R =  ; АА-ЭП-5, АА-ЭП-11: R =
; АА-ЭП-5, АА-ЭП-11: R =  ; АА-ЭП-6, АА-ЭП-12: R =
; АА-ЭП-6, АА-ЭП-12: R =  .
.
Первая серия АА-ЭП-1–АА-ЭП-6 получена путем отверждения в присутствии 4-(N,N-диметиламино)пиридина. Вторая серия АА-ЭП-7– АА-ЭП-12 – с применением 2,4-диаминотолуола в стехиометрическом соотношении с исходными мономерами [31, 55]. Способ отверждения АА-ЭП определяет химическую природу сетчатой структуры и соответственно отражается на ее свойствах. Так, сетчатая структура для АА-ЭП-1–АА-ЭП-6 формируются за счет образования простой эфирной связи, тогда как сшивание полимерных цепей в АА-ЭП-7–АА-ЭП-12 обеспечивается посредством аминной связи [31].
Во время реакции сшивки первые два представителя полученных полимеров (АА-ЭП-1–АА-ЭП-2) образовывали термотропную мезофазу нематического типа, которая в конечном итоге была “заморожена” в результате образования сетчатой структуры. В то же время конечные материалы, созданные на основе остальных представителей данной серии полимеров (АА-ЭП-3–АА-ЭП-6) и содержащие от 4 до 10 алкильных фрагментов, характеризовались более упорядоченными мезофазами смектического типа (табл. 4).
Таблица 4.
Термические и ЖК-свойства азо-азометинсодержащих эпоксидных полимеров [31]
| Азо-азометинсодержащие эпоксидные полимеры | R | T10%, °C | Коксовый остаток полимера, % | Тип мезофазы* |
|---|---|---|---|---|
| АА-ЭП-1 | –CH2– | 328 | 60 | Н |
| АА-ЭП-2 | –O–(CH2)2– | 312 | 48 | Н |
| АА-ЭП-3 | –O–(CH2)4– | 313 | 36 | С |
| АА-ЭП-4 | –O–(CH2)6– | 318 | 30 | С |
| АА-ЭП-5 | –O–(CH2)8– | 297 | 33 | С |
| АА-ЭП-6 | –O–(CH2)10– | 331 | 24 | С |
| АА-ЭП-7 | –CH2– | 336 | 66 | Н |
| АА-ЭП-8 | –O–(CH2)2– | 390 | 46 | H |
| АА-ЭП-9 | –O–(CH2)4– | 328 | 37 | H |
| АА-ЭП-10 | –O–(CH2)6– | 317 | 32 | H |
| АА-ЭП-11 | –O–(CH2)8– | 313 | 24 | H |
| АА-ЭП-12 | –O–(CH2)10– | 341 | 27 | Н |
Отверждение по механизму поликонденсации позволило получить АА-ЭП при более низких значениях температуры и меньших временных затратах в сравнении с полимерами первой серии. Полимерные сетки АА-ЭП-7–АА-ЭП-12 независимо от длины спейсеров обладали мезофазами нематического типа (табл. 4). Авторы работы [31] полагают, что это связано с относительно быстрым процессом формирования сетчатых структур, а также с включением отвердителя в структуру сетки. Температурные диапазоны существования мезофаз для ЭП-1–АА-ЭП-12 не указаны [31]. Следует отметить, что для сетчатых азометинсодержащих полиэпоксидов подобного химического строения (но без наличия азогруппы в своем составе) наблюдаются аналогичные типы термотропных мезофаз [56]. Включение азогрупп в состав АА-ЭП открывает возможность дополнительного регулирования их термотропного мезоморфного состояния с помощью фотоиндуцированных транс-цис-переходов [57].
Синтезированные АА-ЭП обладают достаточно высокой термостабильностью (T10% > 312°С), которая несколько выше для полимеров второй серии. Отмечено, что зависимости между длиной спейсера и термостабильностью полимеров в пределах каждой из серий не наблюдается [31].
СИНТЕЗ КООРДИНАЦИОННЫХ АЗО- И АЗОМЕТИНОВЫХ ПОЛИМЕРОВ
Наличие азометиновой группы и относительно нее в орто-положении гидроксильной группы является эффективным инструментом для построения полимерной цепи посредством координационных связей. Функциональные характеристики таких полимеров регулируются типом катиона металла, входящего в координационную сферу [32].
С целью придания магнитных свойств ААП, получена серия линейных металлоорганических координационных полимеров МОП-1–МОП-5 на основе (1E,1'E)-5,5'-((1E,1'E)-[1,1'-бифенил]-4,4'-диил-бис-(диазен-2,1-диил))-бис-(2-гидроксибензальдегид)диоксима и хлоридов двухвалентных металлов Cu, Ni, Co, Mn и Zn соответственно (табл. 5) [32]:
Таблица 5.
Магнитные свойства металлоорганических координационных ААП [32]
| Металлоорганические координационные полимеры | Металл | Магнитный момент, μБ |
|---|---|---|
| МОП-1 | Cu | 1.83 |
| МОП-2 | Ni | 2.81 |
| МОП-3 | Co | 5.20 |
| МОП-4 | Mn | 5.58 |
| МОП-5 | Zn | – |

Полученные металлоорганические координационные полимеры (МОП) характеризуются наличием кристаллогидратной составляющей. Благодаря хорошей стабилизации низкого окислительного состояния катионов металлов азо-азометиновой системой сопряжения, а также наличию фенольных групп, позволяющих формировать ковалентную связь с ионами металлов, эти полимеры характеризуются хорошей стабильностью [32, 36, 58].
Полимеры из серии МОП-1–МОП-5 имеют октаэдрическое строение (по данным эмпирических расчетов на основе отражательной спектроскопии в видимой области); они не растворимы в большинстве органических растворителей. Температура разложения МОП-1–МОП-5 лежит в пределах 270–430°С, наибольшей термической стабильностью обладает Zn-содержащий МОП-5, а наименьшей – Cu-содержащий МОП-1. Все полимеры обладают магнитными свойствами, за исключением Zn-содержащего МОП-5, который является диамагнетиком. При этом максимальным значением магнитного момента характеризуется Mn-содержащий МОП-4. Таким образом, изменяя химическую природу катиона металла, входящего в состав полимера, можно регулировать его магнитные свойства. Данные координационные полимеры представляют интерес в качестве катализаторов [32].
ПРИВИВКА АЗО- И АЗОМЕТИНСОДЕРЖАЩИХ ФРАГМЕНТОВ К ЦЕЛЛЮЛОЗНОМУ ВОЛОКНУ
Азо- и азометинсодержащие мономеры, содержащие симметрично расположенные два азо-азометиновых фрагмента и концевые сульфатные группы, были использованы для придания новых свойств целлюлозным волокнам путем их поверхностной обработки [33–35]. В качестве таковых выбраны ААС N,N-бис-{p-[(2-сульфатоэтил)сульфонилфенилазо]сали-цилиден}-1,2-этилендиамин (ААС-1) и 3,5-бис-{2-гидрокифенил-5-[(2-сульфат-4-сульфоэтиленсульфонил-азобензол)метиленамино]}бензойная кислота (ААС-2). С их использованием в присутствии бикарбоната натрия получены две серии привитых образцов АА-Ц-1 и АА-Ц-2 с содержанием ААС в пределах 1.52 × × 10–2–5.08 × 10–2 ммоль/г (степень прививки определена по данным УФ-спектров ААС):
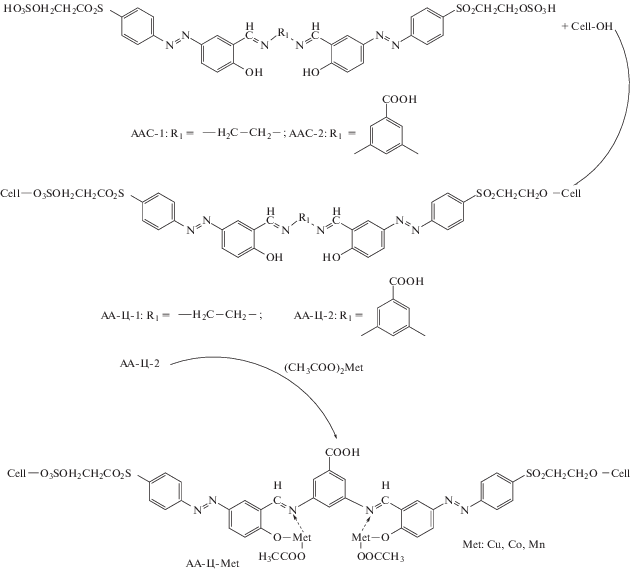
Цель прививки – придание комплексообразующей способности целлюлозному волокну и создания на его основе тканей с высокой степенью защиты от УФ-излучения и бактерицидными свойствами.
Проведенная прививка практически не сказывается на прочностных характеристиках волокна. Все полученные образцы привитой целлюлозы характеризуются высокой степенью защиты от УФ-излучения. Стоит отметить, что увеличение количества, привитого ААС, как в первой, так и во второй серии ведет к повышению степени защиты от УФ-излучения. При этом наилучшей степенью защиты обладает серия привитой целлюлозы на основе ААС-2, содержащая максимальное количество привитого ААС (5.08 × × 10–2 ммоль/г) [34–36]. Установлено, что образцы целлюлозы АА-Ц-2 обладают бактерицидными свойствами по отношению к штамму бактерий S. aureus, которые усиливаются по мере увеличения степени прививки [34].
Такие модифицированные с помощью ААС целлюлозные волокна были использованы для последующей модификации путем образования на поверхности хелатных соединений. С этой целью, волокна АА-Ц-2 (5.08 × 10–2 ммоль/г привитого ААС) выдерживались при кипячении в спиртовом растворе ацетатов (II) Cu, Co и Mn (см. схему (11) ) [34]. Cинтезированные металлокомплексы отмечаются наличием воды во внутренней координационной сфере, а температура начала их деструкции превышает 285оС. Однако немодифицированный образец целлюлозы характеризуется более высокой термической стойкостью (Т0 = 344°С). Такой тип модификации поверхности целлюлозного волокна также почти не влияет на его механические свойства [34, 35]. Модифицированные данным способом целлюлозные волокна проявляют высокую каталитическую эффективность при разложении пероксида водорода, которая максимальна в случае Mn-cодержащего соединения [34].
ПРИМЕНЕНИЕ АЗО- И АЗОМЕТИНОВЫХ ПОЛИМЕРОВ
Азо- и азометиновые полимеры, обладая комплексом уникальных физико-химических свойств, предмет все большего внимания современного материаловедения. Азо-ПАМ с азогруппами в основной цепи (азо-ПАМ-1– азо-ПАМ-9) перспективны в качестве сенсоров оптических переключателей [21, 22]. Они характеризуются полупроводниковыми и фотолюминисцентными свойствами, способностью подвергаться фотоиндуцированным транс-цис-транс-переходам, а также обладают высокой термической стабильностью [20–22].
Особое внимание привлекают полиазометины с азогруппами в боковой цепи (азо-ПАМ-10–азо-ПАМ-13), азо-азометинсодержащий полифосфазен (АА-ПФ), а также азо-азометинсодержащие полиэпоксиды (АА-ЭП-1– АА-ЭП-12) в качестве ЖК-среды, мезоморфное поведение которых можно изменять посредством изомеризации фотохромных групп, а также полимерных ЖК-матриц для допирования различных наночастиц (фуллерен, графен, нанотрубки) [23, 26, 31, 52]. Азо-ПАМ-14 можно применять для создания материалов с нелинейно-оптическими свойствами (d33 = 82 пм/В) [24]. Металлоорганические координационные полимеры (МОП-1–МОП-5) перспективны для получения катализаторов, материалов с регулированными магнитными свойствами [32]. Так, благодаря хорошей сорбционной способности, азо-азометинсодержащий полиамид (АА-ПА) является активным компонентом для очистки воды от катионов тяжелых металлов [30].
Отдельный интерес вызывают азо-азометинсодержащие поверхностно-модифицированные полимеры. Азо-азометинсодержащие полистиролы (АА-ПС-1–АА-ПС-3) используют как активные компоненты для сорбционных колонок при аналитическом определении катионов металлов [27–29]. Модифицированная с помощью ААС целлюлоза (АА-Ц-1 и АА-Ц-2) перспективна для изготовления тканей с высокой степенью защиты от УФ-излучения, а также катализаторов [33–35].
ЗАКЛЮЧЕНИЕ
Представленные в данном обзоре исследования свидетельствуют о существовании двух подходов к получению ААП. Первый, наиболее распространенный, так называемый азометиновый подход, основан на формировании полимерной цепи за счет образования азометиновой связи. Второй подход предполагает применение мономеров ААС, содержащих азо-азометиновый фрагмент в своем составе, для синтеза линейных, сетчатых, а также координационных полимеров. Полимерная цепь при этом формируется за счет образования амидной, простой эфирной, аминной или координационной связи. В рамках обоих подходов осуществлена прививка и поверхностная обработка полимерных материалов.
Синтезированные полимеры характеризуются следующими особенностями. Наличие азо- и азометиновой группы в одной системе сопряжения снижает энергию активации π → π*-перехода такой хромофорной системы и приводит к батохромному сдвигу, увеличивает стабильность цис-формы азогруппы и фотостабильность, а вместе с тем низкое окислительное состояние ионов металлов. Последнее используется для создания различных полихелатов на основе ААП, свойства которых в значительной степени могут быть изменены типом вводимого катиона металла. Для достижения комплексообразующих свойств ААП преимущественно получают на основе различных азосодержащих производных салицилового альдегида.
Наличие системы сопряжения отражается и на ЖК-свойствах, проявляющихся в увеличении температурного диапазона мезофазы и возможности регулировать мезоморфное поведение под воздействием света, а также в повышении термической стабильности ААП. В свою очередь, вывод из системы сопряжения азо- и азометиновой групп ведет к повышению температурно-временной стабильности нелинейно-оптических характеристик полимеров вследствие увеличения их температуры стеклования.
Дальнейшее развитие данного направления в синтетическом плане, очевидно, благодаря своей простоте, будет связано с применением азометинового подхода. Однако при этом требуются новые приемы регулирования электронной конфигурации ААП, придания способности к самоорганизации, повышения растворимости.
Следует ожидать также расширение спектра ААП за счет использования новых ААС, способных как к реакциям поликонденсации, к которым относятся большинство описанных к настоящему времени ААС, так и к реакциям полимеризации. Кроме того, в рамках азометинового подхода перспективным представляется поиск других типов азосодержащих диальдегидов с дополнительными функциональными возможностями, способствующих реализации свойств при сочетании азо- и азометиновых хромофоров. В этом плане нами предложен синтез и исследованы фотохимические и фотооптические свойства изомерных перфторароматических полифункциональных азосодержащих диальдегидов для получения на их основе новых типов ААП, а также других азосодержащих полимеров [50, 51].
Список литературы
Moliton A., Hiorns. R.C. // Polym. Int. 2004. V. 53. № 10. P. 1397.
Beaujuge P.M., Reynolds J.R. // Chem. Rev. 2010. V. 110. № 1. P. 268.
Cheng Y.-J., Yang S.-H., Hsu C.-S. // Chem. Rev. 2009. V. 109. № 11. P. 5868.
Liu C.-L., Tsai F.-C., Chang C.-C., Hsieh K.-H., Lin J.-L., Chen W.-C. // Polymer. 2005. V. 46. № 13. P. 4950.
Sek D., Siwy M., Bijak K., Grucela-Zajac M., Malecki G., Smolarek K., Bujak L., Mackowski S., Schab-Balcerzak E. // J. Phys. Chem. A. 2013. V. 117. № 40. P. 10320.
Sıdır Y.G., Sıdır İ., Berber H., Taşal E. // J. Mol. Liq. 2011. V. 162. № 3. P. 148.
Bandara H.M.D., Burdette S.C. // Chem. Soc. Rev. 2012. V. 41. P. 1809.
Xue X., Zhu J., Zhang Z., Zhou N., Tu Y., Zhu X. // Macromolecules. 2010. V. 43. № 6. P. 2704.
Mahimwalla Z., Yager K.G., Mamiya J., Shishido A., Priimagi A., Barrett C.J. // Polym. Bull. 2012. V. 69. № 8. P. 967.
Shevchenko V.V., Sidorenko A.V., Bliznyuk V.N., Tka-chenko I.M., Shekera O.V. // Polymer Science A. 2013. V. 55. № 1. P. 3.
Li Z., Wu W., Yu G., Liu Y., Ye C., Qin J., Li Z. // ACS Appl. Mater. Int. 2009. V. 1. № 4. P. 856.
Tang R., Zhou S., Xiang W., Xie Y., Chen H., Peng Q., Yu G., Liu B., Zeng H., Li Q., Li Z. // J. Mater. Chem. C. 2015. V. 3. № 17. P. 4545.
Hussein M.A., Abdel-Rahman M.A., Asiri A.M., Alamry K.A., Aly K.I. // Des. Monomers Polym. 2012. V. 15. № 5. P. 431.
Menon S.K., Parikh V.B. // Rev. Inorg. Chem. 2008. V. 28. № 2. P. 89.
Viswanathan N.K., Kim D.Y., Bian S., Williams J., Liu W., Li L., Samuelson L., Kumar J., Tripathy S.K. // J. Mater. Chem. 1999. № 9. P. 1941.
Mahimwalla Z., Yager K.G., Mamiya J., Shishido A., Priimagi A., Barrett C.J. // Polym. Bull. 2012. V. 69. № 8. P. 967.
Xie S., Natansohn A., Rochon P. // Chem. Mater. 1993. V. 5. № 4. P. 403.
Iwan A., Sek D. // Prog. Polym. Sci. 2008. V. 33. № 3. P. 289.
Pron A., Rannou P. // Prog. Polym. Sci. 2002. V. 27. № 1. P. 135.
Gopal J., Srinivasan M. // J. Polym. Sci. A. 1986. V. 24 № 11. P. 2789.
Zabulica A., Perju E., Bruma M., Marin L. // Liq. Cryst, 2014. V. 41. № 2. P. 252.
Khattab A.F., Abbas M.F. // IJERSTE. 2015. V. 4. № 8. P. 1.
Cozan V., Iftime M., Sava I., Bronnikov S. // High Perform. Polym. 2015. V. 27. № 5. P. 661.
Suh S.C., Shim S.C. // Synth. Met. 2000. V. 114. P. 91.
Výprachtický D., Cimrová V. // Macromolecules. 2002. V. 35. № 9. P. 3463.
Lee K.H., Lee D.C. // Polym. Bull. 1999. V. 42. P. 543.
Jain V.K., Sait S.S., Shrivastav P., Agrawal Y.K. // Talanta. 1997. V. 45. № 2. P. 397.
Kim Y.-S., In G., Han C.-W., Choi J.-M. // Microchem. J. 2005. V. 80. № 2. P. 151.
Topuz B., Macit M. // Environ. Monit. Assess. 2011. V. 173. № 1–4. P. 709.
Hajibeygi M., Shafiei-Navid S., Shabanian M., Vahabi H. // Appl. Clay Sci. 2018. V. 157. P. 165.
Castell P., Galià M., Serra A. // Macromol. Chem. Phys. 2001. V. 202. № 9. P. 1649.
Rana A.K., Shah N.R., Patil M.S., Karampurwala A.M., Shah J.R. // Die Makromol. Chemie. 1981. V. 182. № 12. P. 3387.
Hou A., Zhang C., Wang Y. // Carbohydr. Polym. 2012. V. 87. № 1. P. 284.
Gao A., Zhang C., Song K., Hou A. // Carbohydr. Polym. 2014. V. 114. P. 392.
Zhang K., Gao A., Zhang C., Xie K. // Desalin. Water Treat. 2016. V. 57. № 41. P. 19190.
İkiz M., İspir E., Aytar E., Ulusoy M., Karabuğa Ş., Aslantaş M., Çelik Ö. // New J. Chem. 2015. V. 39. № 10. P. 7786.
Kurtoglu N. // J. Serbian Chem. Soc. 2009. V. 74. № 8–9. P. 917.
Ghasemian M., Kakanejadifard A., Azarbani F., Zabardasti A., Kakanejadifard S. // J. Mol. Liq. 2014. V. 195. P. 35.
Mobinikhaledi A., Zendehdel M., Safari P., Hamta A., Shariatzadeh S.M. // Met. Nano-Metal Chem. 2012. V. 42. № 2. P. 165.
Ghasemian M., Kakanejadifard A., Azarbani F., Zabardasti A., Shirali S., Saki Z., Kakanejadifard S. // Spectrochim. Acta, Mol. Biomol. Spectrosc. 2015. V. 138. P. 643.
Refat M.S., El-Deen I.M., Ibrahim H.K., El-Ghool S. // Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2006. V. 65. № 5. P. 1208.
Yen M.S. // Pigment Resin Technol. 2012. V. 41. № 4. P. 216.
Karaer H., Gümrükçüoǧlu I.E. // Turkish J. Chem. 1999. V. 23. P. 67.
Naeimi H., Safari J., Heidarnezhad A. // Dye. Pigment. 2007. V. 73. № 2. P. 251.
Rezaeian K., Khanmohammadi H., Arab V. // Spectrochim. Acta, Mol. Biomol. Spectrosc. 2015. V. 151. P. 848.
Yang C.-J., Jenekhe S.A. // Macromolecules. 1995. V. 28. № 4. P. 1180.
Amundson K., Helfand E., Quan X., Smith S.D. // Macromolecules. 1993. V. 26. № 11. P. 2698.
Serin M., Sakar D., Cankurtaran O., Yilmaz F.K., Campus D. // J. Optoelectron. Adv. Mater. 2004. V. 6. № 1. P. 283.
Ковальчук А.И., Кобзарь Я.Л., Ткаченко И.М., Шекера О.В., Шевченко В.В. // Полімерний журн. 2018. Т. 40. № 3. С. 141.
Kovalchuk A.I., Kobzar Ya.L., Tkachenko I.M., Tolstov A.L., Shekera O.V., Shevchenko V.V. // Mendeleev Commun. 2017. V. 27. P. 599.
Kovalchuk A.I., Kobzar Ya.L., Tkachenko I.M., Tolstov A.L., Shekera O.V., Shevchenko V.V. // J. Mol. Struct. 2018. V. 1173. P. 671.
Bronnikov S., Kostromin S., Musteaţa V., Cozan V. // Liq. Cryst. 2015. V. 42. № 8. P. 1102.
Sethuraman S., Nair L.S., El-Amin S., Nguyen M.-T., Singh A., Krogman N., Greish Y.E., Allcock H.R., Brown P.W., Laurencin C.T. // Acta Biomater. 2010. V. 6. № 6. P. 1931.
Liang T., Cui Y., Yu J., Lin W., Yang Y., Qian G. // Thin Solid Films. 2013. V. 544. P. 407.
Mormann W., Bröcher M. // Macromol. Chem. Phys. 1996. V. 197. № 6. P. 1841.
Castell P., Serra A., Galià M. // J. Polym. Sci., Polym. Chem. 2004. V. 42. № 14. P. 3631.
Nardele C.G., Asha S.K. // J. Phys. Chem. B. 2014. V. 118. № 6. P. 1670.
Govindaswamy P., Sinha C., Kollipara M.R. // J. Organomet. Chem. 2005. V. 690. P. 3465.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)