

Высокомолекулярные соединения (серия Б), 2019, T. 61, № 2, стр. 99-115
Полимеризация виниловых мономеров в присутствии 4,6-ди-трет-бутил-N-(2,6-диизопропилфенил)-О-иминобензохинона
О. С. Лизякина a, Л. Б. Ваганова a, *, М. Г. Чегерев b, А. В. Пискунов b, Д. Ф. Гришин a
a Национальный исследовательский Нижегородский государственный университет им. Н.И. Лобачевского
603950 Нижний Новгород, пр. Гагарина, 23, Россия
b Институт металлоорганической химии им. Г.А. Разуваева Российской академии наук
603950 Нижний Новгород, ул. Тропинина, 49, Россия
* E-mail: vaganova_lb@mail.ru
Поступила в редакцию 28.06.2018
После доработки 17.10.2018
Принята к публикации 31.10.2018
Аннотация
Изучены особенности радикальной полимеризации ряда виниловых мономеров в присутствии 4,6-ди-трет-бутил-N-(2,6-диизопропилфенил)-о-иминобензохинона. Установлено влияние указанного соединения на кинетические закономерности синтеза полимеров при различных значениях температуры, а также их молекулярно-массовые характеристики. Определены оптимальные условия получения соответствующих полимеров и блок-сополимеров с участием 4,6-ди-трет-бутил-N-(2,6-диизопропилфенил)-о-иминобензохинона в широком диапазоне молекулярных масс и составов.
ВВЕДЕНИЕ
Одной из актуальных задач химии высокомолекулярных соединений является разработка эффективных методов направленного синтеза полимеров с заданным составом и свойствами в условиях радикального инициирования. Для ее решения в последние годы активно применяют концепцию контролируемой радикальной полимеризации, в том числе методы обратимого ингибирования. В качестве агентов, способных регулировать молекулярно-массовые характеристики полимеров по данному механизму, предложено задействовать ряд органических и металлоорганических соединений [1, 2]. В частности, весьма широкое распространение получили нитроксильные и пространственно-затрудненные углерод-центрированные радикалы как регуляторы молекулярно-массовых характеристик полимеров [3, 4].
Ингибиторы фенольного типа как источники феноксильных радикалов также можно рассматривать в качестве агентов, способных целенаправленно изменять кинетические параметры полимеризации и регулировать молекулярную массу и полидисперсность полимеров [5]. При этом, как правило, здесь имеются в виду бинарные системы на основе хинонов или комплексов металлов с хиноновыми лигандами [6, 7]. Примеров непосредственного использования хинонов и их производных в индивидуальном состоянии для проведения радикальной полимеризации в контролируемом режиме в литературе приведено мало [5].
В работах [8, 9] для целенаправленного изменения молекулярно-массовых характеристик полимеров впервые был апробирован ряд N-(арил)-о-иминохинонов различного строения. Указанные соединения являются структурными аналогами о-хинонов и активно применяются как редокс-активные лиганды в химии координационных соединений [10]. Показано, что N-(2,6-диалкилфенил)-о-иминобензохиноны способны проводить радикальную полимеризацию метилметакрилата в режиме обратимого ингибирования. При этом 4,6-ди-трет-бутил-N-(2,6-диизопропилфенил)-о-иминобензохинон можно назвать наиболее эффективным в плане получения ПММА c заданными молекулярно-массовыми характеристиками [8, 9]:

Цель настоящей работы – изучение и проведение сравнительного анализа процессов радикальной полимеризации виниловых мономеров различного строения с участием 4,6-ди-трет-бутил-N-(2,6-диизопропилфенил)-о-иминобензохинона (imQ), а также выявление факторов, влияющих на его эффективность в качестве регулятора молекулярно-массовых характеристик конкретных полимеров.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Объектами исследования в данной работе служили эфиры акриловой и метакриловой кислот, а также стирол, акрилонитрил и N-винилпирролидон. Инициатором выступал ДАК.
Мономеры, инициатор и применяемые растворители очищали общепринятыми способами [11–13]. о-Иминохинон получали по методике, описанной в работе [14]. Физико-химические константы всех использованных соединений соответствовали литературным данным.
Образцы для полимеризации готовили следующим образом: раствор мономера с ДАК и imQ в расчетном соотношении помещали в стеклянные ампулы, трижды дегазировали, перемораживая ампулы в жидком азоте. Остаточное давление составляло ~1.3 Па. Ампулу отпаивали и помещали в термостат на строго заданное время.
Контроль кинетики полимеризации осуществляли гравиметрическим методом. Для очистки полимеров от остатков мономера, инициатора и imQ образцы переосаждали из раствора в хлористом метилене (или ДМФА) в соответствующий осадитель, затем сушили в вакууме до постоянной массы.
Образцы полимеров для дальнейшего применения в качестве макроинициатора получали при различном соотношении мономер : ДАК : imQ при температуре 70°С. Навеску дважды переосажденного полимера помещали в ампулу и добавляли необходимое количество свежеперегнанного мономера. Концентрация макроинициатора в смеси составляла 10–20 и 20–25 мас. % для постполимеризации и блок-сополимеризации соответственно. После гомогенизации системы ампулу трижды дегазировали, перемораживая в жидком азоте, после чего отпаивали. Постполимеризацию и блок-сополимеризацию проводили при температуре синтеза макроинициатора.
Молекулярно-массовые характеристики полимеров определяли методом ГПХ [15]. Анализ образцов полиакрилатов, полиметакрилатов, полистирола и продуктов их сополимеризации осуществляли на установке “Knauer” с каскадом линейных колонок 103–105 Å (“Phenomenex”, США). В качестве детектора использовали дифференциальный рефрактометр “RI Detektor K-2301” и УФ-детектор “UV Detektor K-2501”. Элюентом служил ТГФ (25.0 ± 0.1°С). Для калибровки применяли узкодисперсные стандарты ПММА и ПС.
Анализ образцов ПАН, поли-N-винилпирролидона (ПВП) и продуктов сополимеризации ПММА с акрилонитрилом и N-винилпирролидоном проводили на установке “Knauer” с линейной колонкой 105 Å. В качестве детектора использовали дифференциальный рефрактометр “RI Detektor K-2300”. Элюентом служил раствор LiBr 0.01 моль/л в ДМФА (40.0 ± 0.1°С). Для калибровки применяли узкодисперсные стандарты ПММА.
Продукты взаимодействия imQ и инициатора получали таким образом: навески imQ (230 мг, 0.58 ммоля) и ДАК (в мольном соотношении imQ : ДАК = 1.0 : 1.0, 1.0 : 1.5 или 1.0 : 2.0) смешивали в ампуле, добавляли 3 мл растворителя (ацетонитрила, бензола или толуола). Ампулу с раствором трижды дегазировали, отпаивали и помещали в термостат при температуре 90 или 110°С. По истечении заданного времени ампулу вскрывали и удаляли растворитель. Продукты реакции анализировали методом времяпролетной масс-спектрометрии. Регистрацию масс-спектров проводили на приборе “Bruker Microflex LT” с использованием матрично-активированной лазерной десорбцией–ионизацией с матрицей транс-2-[3-(4-трет-бутилфенил)-2-метил-2-пропенил-иден]малонитрил (DCTB) в линейном режиме при положительных значениях потенциала. Исследуемый образец (~1 мг) растворяли в 100 мкл ТГФ. Полученный раствор (~1 мкл) наносили на подложку из нержавеющей стали и сушили на воздухе, после чего регистрировали спектр. Экспериментальные данные обрабатывали с помощью программного обеспечения “Bruker flexControl” и “flexAnalysis”.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Для оценки влияния imQ на кинетические особенности радикальной полимеризации и молекулярно-массовые характеристики были выбраны полимеры акриловых (метилакрилат, бутилакрилат) и метакриловых (метилметакрилат ММА, н-бутилметакрилат БМА, н-октилметакрилат ОМА) эфиров, а также стирол, акрилонитрил и N-винилпирролидон. Поведение данных мономеров при радикальном инициировании полимеризации существенно отличается [16]. Закономерности полимеризации указанных мономеров в условиях обратимого ингибирования, например с участием нитроксилов, также различные [3]. В частности, ТЕМПО и его аналоги успешно регулируют синтез ПС и неэффективны в синтезе полиметакрилатов. В этой связи, исследование особенностей полимеризации мономеров различного строения в присутствии imQ, который был успешно применен для синтеза ПММА с относительно низкой полидисперсностью, представляет несомненный интерес.
Полимеризация акриловых и метакриловых эфиров
Полимеризация акриловых мономеров, инициируемая ДАК, уже при 50–70°С проходит с очень высокой скоростью (рис. 1а, кривая 1). Получаемые при этом образцы полиакрилатов характеризуются высокими значениями молекулярной массы и полимодальностью кривых ММР (табл. 1).
Рис. 1.
Временные зависимости ln[m0]/[m] мономеров бутилакрилата (а – 1, 3; б – 2, 5) и БМА (а – 2, 4; б – 1, 3, 4). Т = 70 (а) и 90°С (б); инициатор 0.1 мол. % ДАК. Концентрация imQ, мол. %: а – 0 (1, 2) и 0.1 (3, 4); б – 0 (1), 0.1 (2, 3) и 0.2 (4, 5).

Таблица 1.
Влияние imQ на молекулярно-массовые характеристики полиметакрилатов (концентрация ДАК 0.1 мол. %)
| Мономер | Т, °С | Концентрация imQ, мол. % | Время, ч | Выход полимера, % | Мn × 10–3 | Đ |
|---|---|---|---|---|---|---|
| Метилакрилат | 50 | 0.1 | 40 | 0 | – | – |
| 70 | 0.1 | 8 | 2 | 132 | 22.89 | |
| Бутилакрилат | 50 | 0 | 0.25 | 25 | 1900 | 3.52 |
| 0.1 | 60 | 2 | 9.1 | 2.12 | ||
| 70 | 0 | 0.25 | 61 | 65.2 | 51.14 | |
| 0.1 | 10 | 9 | 34.0 | 5.92 | ||
| 90 | 0 | 0.05 | 50 | 47.8 | 13.89 | |
| 0.1 | 1 | 63 | 103 | 3.88 | ||
| 0.2 | 1 | 2 | 4.2 | 1.61 | ||
| ММА | 50 | 0 | 6 | 88 | 717 | 2.75 |
| 0.1 | 60 | 10 | 16.0 | 1.43 | ||
| 70 | 0 | 2 | 94 | 435 | 7.57 | |
| 0.1 | 10 | 61 | 71.4 | 1.45 | ||
| 0.2 | 10 | 24 | 28.4 | 1.51 | ||
| 90 | 0 | 0.75 | 95 | 119 | 4.99 | |
| 0.1 | 1 | 47 | 54.1 | 1.63 | ||
| 0.2 | 1 | 34 | 30.0 | 1.59 | ||
| БМА | 70 | 0 | 3 | 87 | 373 | 3.18 |
| 0.1 | 10 | 24 | 75.6 | 1.40 | ||
| 0.2 | 10 | 7 | 32.5 | 1.53 | ||
| 90 | 0 | 0.67 | 91 | 187 | 2.40 | |
| 0.1 | 1 | 35 | 117 | 1.34 | ||
| 0.2 | 1 | 28 | 59.4 | 1.54 | ||
| ОМА | 70 | 0 | 4 | 94 | 1089 | 3.96 |
| 0.1 | 15 | 67 | 163 | 1.63 | ||
| 0.2 | 15 | 39 | 104 | 1.71 |
Введение imQ в систему приводит к уменьшению конверсии мономера за заданный промежуток времени. Значения среднечисленных молекулярных масс Мn и коэффициентов полидисперсности Ð полиакрилатов также понижаются (табл. 1).
Рассмотрим подробнее закономерности полимеризации акриловых и метакриловых эфиров с участием imQ при различных значениях температуры. Так, введение imQ при 50°С уже в эквимольном соотношении с инициатором практически полностью ингибирует процесс полимеризации метилакрилата и бутилакрилата (табл. 1). При температуре 70°С использование imQ в равномольном соотношении с инициатором при полимеризации бутилакрилата влечет появление длительного периода индукции, гель-эффект несколько сглаживается (рис. 1а, кривые 1 и 3). В случае синтеза полибутилакрилата при 90°С (соотношение imQ : ДАК = 1 : 1) гель-эффект отсутствует (рис. 1б, кривая 2), конверсия бутилакрилата при этом достигает 90%. Однако при увеличении концентрации imQ появляется период индукции, а предельная степень превращения мономера понижается до ~50%.
При полимеризации метакриловых эфиров в аналогичных условиях скорость процесса также значительно понижается. Выход ПММА при температуре 50°С при эквимольном соотношении imQ : ДАК составляет ~10% за 60 ч (табл. 1). В интервале 70–90°С процессы полимеризации метакриловых эфиров с участием imQ отличаются от синтеза полиакрилатов (рис. 1). Так, imQ уже в эквимольном соотношении с инициатором полностью подавляет гель-эффект при полимеризации ММА [8, 9]. Скорость полимеризации и предельные степени превращения понижаются в соответствии с увеличением концентрации imQ как для ММА, так и для БМА и ОМА. Предельная степень превращения при 70°С БМА составляет ~90%, ОМА ~80% при соотношении imQ : ДАК = = 1 : 1. При температуре 90°С общая скорость полимеризации ММА уменьшается пропорционально росту концентрации imQ.
Влияние imQ на молекулярно-массовые характеристики полиакрилатов и полиметакрилатов различно (табл. 1; рис. 2, 3).
Рис. 2.
Зависимость Mn образцов ПБА от конверсии мономера Р. Т = 70 (1) и 90°С (2, 3); инициатор 0.1 мол. % ДАК. Концентрация imQ 0.1 (1, 2) и 0.2 мол. % (3).

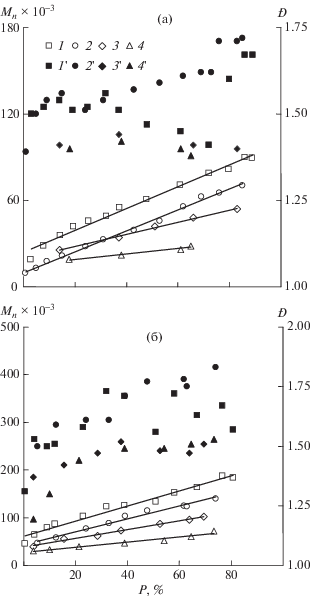
Рис. 3.
Зависимости Mn (1–4) и Ð (1'–4') ПММА (а) и ПОМА (б) от конверсии мономера. Т = 70°С; инициатор ДАК 0.1 (1, 1', 2, 2'), 0.2 (3, 3') и 0.4 мол. % (4, 4'). Концентрация imQ 0.1 (1, 1'), 0.2 (2, 2', 3, 3') и 0.4 мол. % (4, 4').
Зависимость среднечисленной ММ полибутил-акрилата от конверсии мономера в условиях эквимольного соотношения инициатор : добавка при 70°С имеет S-образный вид (рис. 2, кривая 1). При этом кривые ММР полибутилакрилата полимодальны вне зависимости от конверсии мономера, а значения Ð ~ 5–6.
При температуре 90°С характер изменения молекулярно-массовых характеристик полибутил-акрилата от конверсии зависит от соотношения imQ : ДАК. Так, при эквимольном количестве добавки и инициатора наблюдается линейный рост ММ полибутилакрилата с увеличением конверсии (рис. 2, кривая 2) в интервале (60–120) × 103. С увеличением концентрации imQ зависимость Мn от конверсии бутилакрилата приобретает S-образный вид (рис. 2, кривая 3), значения ММ составляют от нескольких тысяч на начальных и более 500 тыс. на предельных конверсиях. При этом на начальном этапе Ð образцов полибутилакрилата имеют значения ~1.7 и возрастают до 4–5 к предельным конверсиям.
В отличие от полиакрилатов, кривые ММР образцов полиметакрилатов, получаемых с участием imQ, унимодальны во всем интервале используемых значений температуры, соотношений добавка : инициатор и достигаемых конверсий мономеров.
При полимеризации ММА введение imQ уже в эквимольном соотношении с инициатором позволяет наблюдать пропорциональный рост ММ образцов ПММА с увеличением конверсии (рис. 3а). При повышении концентрации imQ линейный рост Мn сохраняется. Образцы ПММА вплоть до глубоких степеней превращения имеют значения Ð ~ 1.4–1.8 как при 70°С (рис. 3а, кривые 1'–4'), так и при 90°С. Среднечисленная ММ образцов ПБМА и полиоктилметакрилат (ПОМА) (рис. 3б) также пропорционально увеличивается с ростом конверсии мономера вне зависимости от используемой концентрации imQ (рис. 3б, кривые 1–4). Значения Ð для ПБМА составляют ~1.3–1.5, а для ПОМА ~ 1.3–1.8 (рис. 3б, кривые 1'–4').
Следует отметить, что при увеличении количества вводимой добавки с сохранением эквимольного соотношения imQ : инициатор наблюдается понижение темпов нарастания ММ полиметакрилатов (рис. 3, кривые 1, 3 и 4).
Появление периода индукции, а также скачкообразное увеличение Мn и полимодальность образцов при полимеризации акрилатов с участием imQ указывают на его необратимое взаимодействие с инициирующими или олигомерными радикалами. Вероятно, при полимеризации акриловых эфиров imQ проявляет свойства сильного ингибитора. Радикальная полимеризация метакриловых эфиров в аналогичных условиях с участием imQ протекает в режиме обратимого ингибирования.
Полимеризация стирола
Влияние imQ на кинетические закономерности полимеризации стирола и молекулярно-массовые характеристики ПС отличается как от синтеза полиакрилатов, так и полиметакрилатов. В частности, более существенными факторами оказываются температура процесса и концентрация инициатора в системе.
При инициируемой ДАК полимеризации стирола общая скорость процесса при 70°С с введением imQ понижается пропорционально его концентрации (рис. 4); предельная степень превращения стирола при эквимольном количестве imQ и инициатора составляет ~60%. При температуре 90°С наблюдаются аналогичные закономерности. Важно, что при двукратном увеличении концентрации imQ относительно ДАК только при 70°С появляется период индукции (рис. 4, кривая 3). При температуре 110°С imQ понижает скорость полимеризации как с участием ДАК, так и автополимеризации стирола. Предельная степень превращения стирола при этом остается ~80% для инициируемых процессов и уменьшается до ~60% для процессов без инициатора.
Рис. 4.
Зависимость ln[m0]/[m] стирола от времени его полимеризации при 70°С; инициатор 0.1 мол. % ДАК. Концентрация imQ 0 (1), 0.1 (2) и 0.2 мол. % (3).
Молекулярно-массовые характеристики ПС и их изменение по ходу полимеризации также определяются концентрацией imQ и инициатора (рис. 5). Так, при эквимольном соотношении imQ : ДАК во всем исследуемом интервале температуры (70–110°С) значение Мn ПС пропорционально возрастает с увеличением конверсии (кривые 2, 3). Интересно, что при повышении концентрации imQ значения ММ образцов ПС снижаются только при 110°С (кривые 3, 5). В интервале 70–90°С при увеличении количества imQ численные значения ММ образцов ПС оказываются выше, чем для образцов, полученных при их эквимольном соотношении, рост же ММ незначителен (кривые 2, 4), при этом значение Ð не превышает 2.0.
Рис. 5.
Зависимость Mn образцов ПС от конверсии мономера. Т = 110 (1, 3, 5) и 70°С (2, 4). Концентрация ДАК 0 (1) и 0.1 мол. % (2–5), imQ 0.1 (1–3) и 0.2 мол. % (4, 5).
Значения Ð образцов ПС, полученных в условиях инициирования ДАК, при равномольном соотношении imQ : инициатор при 70°С монотонно возрастают в пределах ~1.7–3.0. С повышением концентрации imQ или температуры процесса интервал изменения данных величин составляет ~1.6–2.0.
Молекулярная масса ПС, получаемого в результате автополимеризации стирола при 110°С, практически не зависит от конверсии и составляет порядка (230.0–250.0) × 103. Значения Ð монотонно возрастают в пределах ~1.8–2.9. При проведении полимеризации в присутствии imQ значения Мn ПС оказываются ниже, чем для образцов без его участия. Следует отметить, что до конверсии ~20% наблюдается рост ММ, однако с течением времени синтеза ММ выходит на постоянное значение (рис. 5, кривая 1). Значения же Ð остаются ~1.8–2.0.
Таким образом, в процессе радикальной полимеризации стирола imQ взаимодействует с растущими радикалами, однако в качестве регулятора молекулярно-массовых характеристик ПС он малоэффективен.
Полимеризация акрилонитрила и N-винилпирролидона
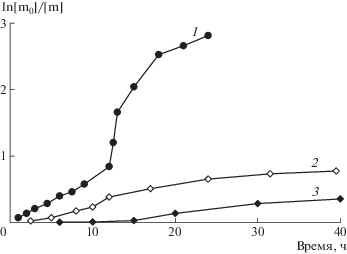
Полимеризация акрилонитрила, инициируемая 0.1 мол. % ДАК, в растворе ДМСО при 70°С менее, чем за 3 ч протекает до конверсии ~90% (рис. 6, кривая 1). При введении в систему imQ в эквимольном соотношении с инициатором наблюдается появление периода индукции. При этом предельная конверсия акрилонитрила понижается до ~55% (кривая 4).
Рис. 6.
Зависимость ln[m0]/[m] акрилонитрила от времени его полимеризации при 70°С. Концентрация ДАК 0.1 (1, 2, 4, 5) и 0.2 мол. % (3); imQ 0 (1, 2), 0.1 (4, 5) и 0.2 мол. % (3). Растворители ДМСО (1, 4) и ДМФА (2, 3, 5); соотношение акрилонитрил : растворитель = 1 : 2.

В растворе ДМФА при температуре 70°С и использовании imQ также появляется период индукции, а предельная степень превращения акрилонитрила не превышает ~35% (рис. 6, кривая 5). В случае двукратного увеличения концентрации ДАК и imQ закономерности, связанные с периодом индукции и уменьшением максимального выхода ПАН, сохраняются (кривая 3).
Скорость полимеризации акрилонитрила как с участием imQ, так и без него в ДМСО выше, чем в среде ДМФА (рис. 6, кривые 1, 2 и 4, 5). Как и следовало ожидать, увеличение соотношения растворитель : акрилонитрил приводит к увеличению времени синтеза и уменьшению предельных степеней превращения акрилонитрила.
Полимеризация винилпирролидона в массе при 70°С, инициируемая 0.1 мол. % ДАК, характеризуется высокой скоростью и наличием гель-эффекта (рис. 7, кривая 1). При осуществлении данного процесса в среде ДМФА (соотношение винилпирролидон : ДМФА = 1 : 2) предельная конверсия винилпирролидона понижается до ~80% (кривая 2).
Рис. 7.
Зависимость ln[m0]/[m] винилпирролидона от продолжительности его полимеризации при 70°С. Инициатор 0.1 мол. % ДАК. Концентрация imQ 0 (1, 2), 0.1 (3, 4) и 0.2 мол. % (5, 6). Полимеризация в массе (1, 3, 6) и в ДМФА (2, 4, 5). Объемное соотношение винилпирролидон : ДМФА = 1 : 2.
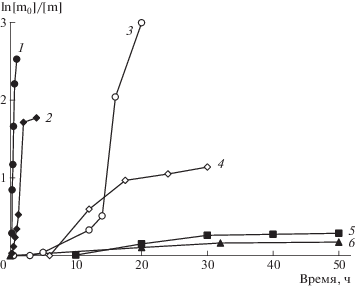
Использование imQ уже в эквимольном соотношении с инициатором в процессах полимеризации винилпирролидона и в массе, и в ДМФА приводит к возникновению периода индукции (рис. 7, кривые 3 и 4). Полимеризация в массе характеризуется ярко выраженным автоускорением (кривая 3). В случае же растворной полимеризации с участием imQ ярко выраженное автоускорение не наблюдается (кривая 4), при этом предельная степень превращения винилпирролидона не превышает 70%.
Увеличение концентрации imQ влечет понижение максимального выхода ПВП до ~20% вне зависимости от проведения процесса в массе или растворе (рис. 7, кривые 5 и 6).
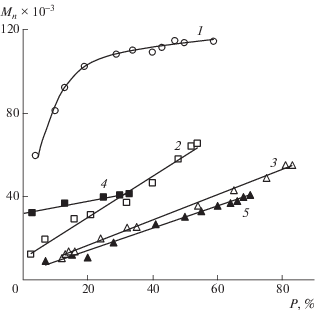
Молекулярная масса образцов ПАН, полученных в среде ДМСО с использованием 0.1 мол. % ДАК при 70°С, не зависит от конверсии мономера и составляет ~110.0 × 103. При введении imQ в эквимольном соотношении с инициатором значения ММ понижаются до ~70.0 × 103, при этом с ростом конверсии акрилонитрила данные величины также не изменяются. Значения Ð для образцов ПАН, синтезированных как в присутствии одного инициатора, так и с использованием imQ, остаются на уровне ~1.7–1.8.
ПАН, полученный с участием 0.1 мол. % ДАК в среде ДМФА при 70°С, характеризуется незначительным понижением Мn с ростом конверсии мономера (рис. 8, кривая 4). Такое изменение ММ типично для данного полимера, синтезированного в среде ДМФА в условиях радикального инициирования. Вероятно, это обусловлено частичным сшиванием полимера, а также может быть связано с реакцией передачи цепи на растворитель. Молекулярные массы образцов ПАН, синтезированного в присутствии imQ, практически не отличаются от аналогичных величин для полимеров, полученных при участии только одного инициатора (кривые 4 и 5), Ð остаются на уровне 1.6–1.7. Значение Мn полимеров, полученных в условиях кратного увеличения концентрации imQ и ДАК, имеет чуть меньшие показатели, при этом также наблюдается ее незначительное уменьшение в течение синтеза (кривая 6).
Рис. 8.
Зависимость Mn образцов ПВП (1–3) и ПАН (4–6) от конверсии мономеров; Т = 70°С. Концентрация ДАК 0.1 (1–5) и 0.2 мол. % (6), imQ 0 (2, 4), 0.1 (1, 3, 5) и 0.2 мол. % (6). Полимеризация в массе (1) и в ДМФА (2–6). Объемное соотношение мономер : : ДМФА = 1 : 2.
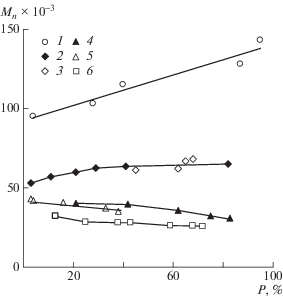
Предельная степень превращения акрилонитрила при радикальной полимеризации в ДМФА с использованием ДАК при температуре 90°С составляет ~40%, а при 110°С ~ 25%. Молекулярная масса образцов ПАН при 90°С незначительно увеличивается в интервале (12.0–15.0) × 103. При температуре 110°С значение Мn образцов ПАН не зависит от конверсии мономера и составляет ~10.0 × 103. Введение imQ в эквимольном соотношении с инициатором при 90–110°С не оказывает существенного влияния ни на общую скорость полимеризации акрилонитрила, ни на степень превращения мономера. Молекулярно-массовые характеристики ПАН при полимеризации с участием imQ также практически не изменяются.
Полученный в массе в присутствии ДАК при 70°С ПВП характеризуется высокими значениями ММ. В случае растворной полимеризации молекулярная масса ПВП остается практически постоянной (рис. 8, кривая 2), при этом наблюдается монотонное увеличение Ð в интервале 2.2–3.0. Образцы ПВП, синтезированного в присутствии imQ в ДМФА, характеризуются меньшими значениями ММ в сравнении с образцами, полученными в массе. При этом молекулярные массы образцов с участием imQ практически не отличаются от аналогичных характеристик полимеров, синтезированных с участием только одного инициатора (кривые 2 и 3). Вне зависимости от способа проведения полимеризации значения Ð образцов ПВП, полученных с участием imQ, остаются в пределах 2.0–2.5.
Таким образом, imQ при полимеризации акрилонитрила и N-винилпирролидона выступает в качестве ингибитора и не оказывает существенного влияния на молекулярно-массовые характеристики ПАН и ПВП. Численные значения ММ полимеров в значительной степени определяются природой используемых растворителей.
Взаимодействие 4,6-ди-трет-бутил-N-(2,6-диизопропилфенил)-о-иминобензохинона с инициирующими радикалами в немономерных средах
Согласно полученным результатам, imQ либо позволяет проводить полимеризацию виниловых мономеров в регулируемом режиме, либо ингибирует процесс. Понижение скорости процессов и в том, и в другом случае указывает на взаимодействие инициирующих или олигомерных радикалов с imQ.
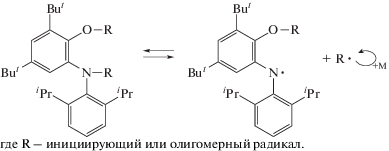
Используемый в работе imQ является структурным аналогом классического ингибитора 3,5-ди-трет-бутил-о-хинона [17]. Общую схему реакции imQ с радикалами можно представить как последовательное акцептирование с образованием эфира соответствующего о-аминофенола (APR2) (схема 1). Наличие о-иминосемихинонов (imSQR) в среде метилметакрилата зафиксировано ранее методом ЭПР [8].

При полимеризации метакриловых эфиров высокие значения конверсии мономеров, линейный рост ММ макромолекул и относительно низкие Ð свидетельствуют об обратимом взаимодействии производных imQ и растущих радикалов (схема 2).
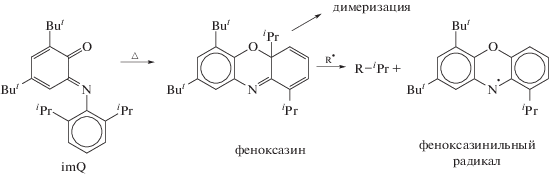

Известно из работ [14, 18, 19], что при нагревании о-иминохиноны склонны к внутримолекулярной циклизации с образованием феноксазинов (схема 3 ), которые необратимо димеризуются. Данная реакция наиболее вероятна в неполярных средах при высоких значениях температуры. Согласно полученным данным, даже при 90–110°С в ряде случаев ингибирующая способность imQ сохраняется.
Логично предположить, что взаимодействие растущих радикалов возможно и с феноксазиновой формой imQ. В частности, при наличии в системе радикалов возможен отрыв от циклической формы изопропильного заместителя (схема 3).
Феноксазинильный радикал может вступать и в дальнейшие реакции (схема 4).
Способность феноксазинильных радикалов влиять на скорость полимеризации и молекулярно-массовые характеристики ПММА показана в работе [9].
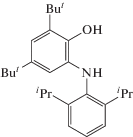
С помощью времяпролетной масс-спектрометрии были охарактеризованы продукты взаимодействия imQ с ДАК в различных средах. Так, после прогревания в толуоле при 90°С в течение 6 ч при эквимольном соотношении imQ : ДАК удалось зафиксировать лишь один продукт с m/z = 380.2. Данный сигнал относится, по-видимому, к 4,6-ди-трет-бутил-N-(2,6-диизопропилфенил)-о-аминофенолу (APH2):
Очевидно, что при использовании в качестве реакционной среды толуола последний выступает в качестве активного передатчика цепи и взаимодействует с imQ с образованием о-аминофенола. Замена растворителя на менее склонный к реакциям отрыва водорода бензол должна способствовать реакции imQ именно c цианизопропильными радикалами.
Действительно, при анализе продуктов реакции в среде бензола при 90°С (соотношение imQ : : ДАК = 1.0 : 1.5) в спектре MALDI-TOF обнаружены четыре основных пика (рис. 9). Первый продукт с m/z = 380.2 является о-аминофенолом APH2. Второй (449.3 m/z), третий (516.3 m/z) и четвертый (583.3 m/z) продукты являются производными imQ, содержащими один, два и три цианизопропильных радикала в своем составе. Аналогичное сочетание пиков от продуктов реакции зафиксировано при взаимодействии imQ с ДАК в среде бензола и при 110°С (мольное соотношение imQ : ДАК = 1 : 2, время синтеза 2 ч).
Рис. 9.
Спектр MALDI-TOF продуктов взаимодействия imQ и ДАК в бензоле. T = 90°C; время реакции 6 ч; эквимольное соотношение imQ : ДАК.
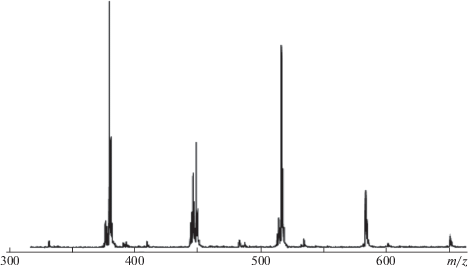
Продукты взаимодействия imQ с ДАК, вероятно, представляют собой производные о-аминофенола вида APRH, APR2 и APR3:
В ацетонитриле в результате реакции imQ с ДАК получен только один продукт с m/z = 449.5, т.е. производное с одним цианизопропильным радикалом (APRH).
Присутствие в качестве продуктов реакции производных аминофенола, содержащих несколько цианизопропильных радикалов, подтверждает их ступенчатое взаимодействие с imQ. Схему образования продукта, содержащего три инициирующих радикала, можно представить как последовательное присоединение их к различным резонансным формам imSQR (схема 5).
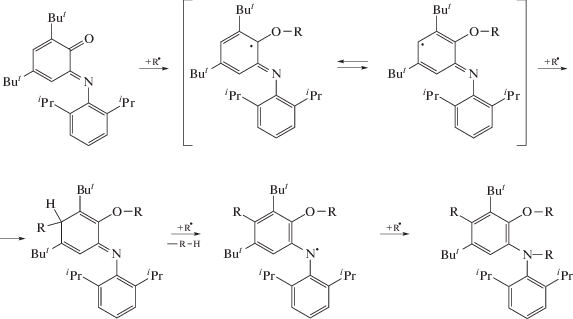
Возможность присоединения алкильных радикалов в положение 5 о-иминохинонового кольца была ранее продемонстрирована в реакциях комплексов непереходных металлов, содержащих восстановленную форму imQ [20–23].
Таким образом, состав продуктов взаимодействия imQ и ДАК зависит от реакционной среды. Влияние imQ на кинетические закономерности полимеризации различных виниловых мономеров и молекулярно-массовые характеристики полимеров, скорее всего, также связано как с побочными реакциями самого imQ, так и с различными продуктами взаимодействия imQ и инициатора.
Постполимеризация и блок-сополимеризация
Для подтверждения протекания полимеризации метакриловых мономеров с участием imQ по схеме 2, полученные полимеры были апробированы в роли макроинициаторов в процессах постполимеризации и блок-сополимеризации (табл. 2, 3; рис. 10–12).
Таблица 2.
Постполимеризация метакриловых мономеров (Т = 70°С, время 5 ч)
| Мономер | Характеристики макроинициатора | [Макро-инициатор], мас. % | Характеристики продуктов постполимеризации | |||||
|---|---|---|---|---|---|---|---|---|
| imQ/ДАК* | время, ч | Mn × 10–3 | Đ | конверсия мономера, % | Mn × 10–3 | Đ | ||
| ММА | 1 : 1 | 6 | 42.8 | 1.32 | 20 | 40 | 98.8 | 1.29 |
| 12 | 76.6 | 1.39 | 10 | 10 | 184 | 10.79** | ||
| 20 | 25 | 162 | 6.25** | |||||
| 2 : 2 | 12 | 41.7 | 1.36 | 10 | 26 | 132 | 1.59 | |
| 10*** | 35 | 199 | 1.96 | |||||
| 20 | 30 | 103 | 1.41 | |||||
| 20*** | 40 | 140 | 1.56 | |||||
| 4 : 4 | 12 | 28.7 | 1.38 | 10 | 25 | 106 | 1.55 | |
| 20 | 35 | 65.8 | 1.45 | |||||
| 8 : 8 | 12 | 16.9 | 1.50 | 10 | 31 | 84.9 | 1.47 | |
| 20 | 32 | 53.6 | 1.37 | |||||
| БМА | 1 : 1 | 12 | 86.9 | 1.29 | 10 | 5 | 150 | 14.0** |
| 2 : 2 | 12 | 55.9 | 1.36 | 10 | 16 | 92.2 | 2.72** | |
| 20 | 29 | 106 | 1.91** | |||||
| 4 : 4 | 12 | 32.1 | 1.46 | 10 | 20 | 72.0 | 2.54** | |
| 20 | 41 | 55.3 | 2.05** | |||||
| 8 : 8 | 12 | 22.6 | 1.48 | 10 | 35 | 64.5 | 2.06** | |
| 20 | 41 | 48.1 | 1.88 | |||||
| ОМА | 2 : 2 | 8 | 72.7 | 1.52 | 20 | 22 | 102 | 5.07** |
Таблица 3.
Блок-сополимеризация метакриловых мономеров (Т = 70°С, концентрация макроинициаторов 20 мас. %)
| Характеристики макроинициатора | Характеристики продуктов блок-сополимеризации | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| мономер 1 | imQ/ДАК, мол. %/мол. % | время, ч | Mn × 10–3 | Ð | мономер 2 | время, ч | конверсия мономера 2, % | Mn × 10–3 | Ð |
| ММА | 0.1 : 0.1 | 6 | 42.8 | 1.32 | ЭМА | 5 | 43 | 99.1 | 1.66 |
| БМА | 5 | 23 | 105.0 | 1.42 | |||||
| ОМА | 5 | 28 | 124.0 | 1.55 | |||||
| 0.2 : 0.2 | 12 | 41.7 | 1.36 | Метилакрилат | 15 | 17 | 56.6 | 2.02* | |
| Стирол | 15 | 5 | 51.8 | 1.80* | |||||
| ММА/стирол (аз.)** | 15 | 7 | 46.7 | 1.62 | |||||
| БМА | 2 | 14 | 45.9 | 1.54 | |||||
| 0.4 : 0.4 | 12 | 23.8 | 1.69 | Акрилонитрил*** | 15 | 2 | 24.8 | 2.07* | |
| Акрилонитрил в ДМФА*** | 15 | 12 | 23.4 | 5.73* | |||||
| Винилпирролидон*** | 16 | 13 | 50.1 | 3.85* | |||||
| 0.4 : 0.4 | 12 | 28.7 | 1.38 | Метилакрилат | 15 | 12 | 56.2 | 5.66* | |
| ЭМА | 5 | 46 | 72.9 | 1.69 | |||||
| БМА | 5 | 53 | 109.0 | 1.36 | |||||
| ОМА | 5 | 65 | 73.8 | 1.69 | |||||
| 0.8 : 0.8 | 12 | 15.8 | 1.56 | Акрилонитрил*** | 15 | 3 | 28.3 | 2.48* | |
| Акрилонитрил в ДМФА*** | 15 | 11 | 20.5 | 3.61* | |||||
| Винилпирролидон*** | 16 | 22 | 30.7 | 3.75* | |||||
| 0.8 : 0.8 | 12 | 16.9 | 1.50 | Метилакрилат | 15 | 5 | 36.0 | 4.15* | |
| Стирол | 15 | 14 | 34.7 | 2.41* | |||||
| ММА/стирол (аз.)** | 15 | 16 | 19.6 | 1.41 | |||||
| ЭМА | 5 | 50 | 52.6 | 1.60 | |||||
| БМА | 5 | 39 | 75.1 | 1.34 | |||||
| БМА | 0.8 : 0.8 | 12 | 22.6 | 1.48 | Стирол | 5 | 8 | 27.6 | 1.77 |
| ММА | 5 | 31 | 50.6 | 1.67 | |||||
| ЭМА | 5 | 58 | 85.7 | 2.01* | |||||
| ОМА | 5 | 30 | 87.6 | 1.40 | |||||
| ЦМА | 5 | 47 | 78.6 | 1.70 | |||||
| ОМА | 0.2 : 0.2 | 8 | 72.7 | 1.52 | ММА | 5 | 18 | 145.0 | 2.76* |
| БМА | 5 | 17 | 113.0 | 2.60* | |||||
| 0.4 : 0.4 | 12 | 60.0 | 1.51 | ММА | 5 | 23 | 96.4 | 3.11* | |
| БМА | 5 | 4 | 82.4 | 2.98* | |||||
| ЦМА | 5 | 4 | 72.4 | 1.85 | |||||
Рис. 10.
Кривые ММР образцов ПММА-инициатора (1) и продуктов его постполимеризации (2–4) при 70°С. ПММА получен в присутствии 0.4 мол. % imQ и 0.4 мол. % ДАК, Mn = 18.8 × 103, Ð = 1.4. Массовое соотношение ПММА : ММА = 0.2; время постполимеризации 1 (2), 2 (3) и 3 ч (4); конверсия ММА 30 (2), 38 (3) и 45 мас. % (4).
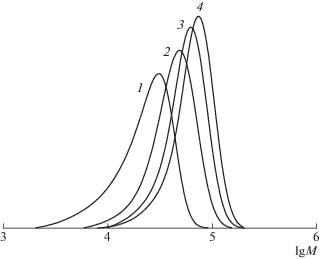
Рис. 11.
Кривые ММР образцов ПММА-инициатора (1) и продуктов его сополимеризации с мономером 2 – азеотропом ММА : стирол (2, 4) и стирол (3, 5) при 70°С. ПММА получен в присутствии 0.8 мол. % imQ и 0.8 мол. % ДАК, Mn = 16.9 × 103, Ð = 1.5. Массовое соотношение ПММА : мономер 2 = 0.25 (2, 4) и 0.2 (3, 5). Время синтеза 15 ч; конверсия мономера 2 – 16 (2, 4) и 14 мас. % (3, 5). Детекторы – рефрактометр (1–3) и УФ (4, 5).

Рис. 12.
Кривые ММР продуктов сополимеризации при 70°С макроинициаторов на основе ПММА (а) с мономерами ЭМА (2), БМА (3), ОМА (4) и ПОМА (б) с мономерами ММА (2), БМА (3), ЦМА (4). Кривые 1: а – ПMМА получен в присутствии 0.1 мол. % ДАК и 0.1 мол. % imQ, Mn = 42.8 × 103, Đ = 1.32; б – ПОМА получен в присутствии 0.4 мол. % ДАК и 0.4 мол. % imQ, Mn = 60 × 103, Đ = 1.51. Количество макроинициатора 20 мас. % от мономера 2; время сополимеризации 2 (а) и 5 ч (б); конверсия мономера 2: а – 43 (2), 23 (3) и 28 мас. % (4); б – 23 (2) и 4 мас. % (3, 4).
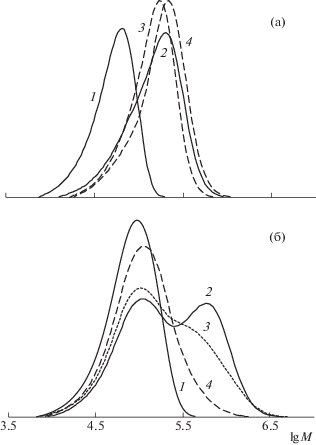
Как следует из табл. 2, образцы ПММА и ПБМА, синтезированные с участием imQ, при добавлении новой порции метилметакрилата и бутилметакрилата соответственно способны к реинициированию полимеризации.
Установлено, что за одинаковый промежуток времени конверсия добавленного мономера увеличивается с ростом концентрации макроинициатора и, как правило, снижается ММ продукта постполимеризации. Данный факт указывает на возобновление полимеризации мономера именно за счет используемых образцов ПММА и ПБМА. Отметим, что ММ получаемых продуктов постполимеризации увеличивается вне зависимости от ММ изначального макроинициатора.
В случае бимодальности кривых ММР продуктов постполимеризации мода с меньшей ММ либо точно соответствует ММР макроинициатора, либо незначительно смещена в область больших ММ, мода с большей ММ относится к образующимся постполимерам. Появление высокомолекулярной моды подтверждает линейное удлинение части макромолекул макроинициатора. Увеличение времени синтеза приводит к росту конверсии добавленного мономера и смещению высокомолекулярной моды в область больших ММ (табл. 2). Наличие пика, соответствующего кривой ММР изначального образца макроинициатора, указывает на образование “мертвого” полимера на стадии его получения или при дальнейшей постполимеризации.
Использование в роли макроинициатора образцов полиметакрилатов, синтезированных при кратном увеличении количества ДАК и imQ относительно мономера, позволило получить унимодальные продукты постполимеризации (рис. 10, кривые 2–4). Таким образом, эффективность реинициирования процесса зависит от характеристик используемого полиметакрилатного инициатора, условий его получения и времени синтеза. Путем варьирования ММ макроинициатора и условий синтеза можно добиться унимодальных узкодисперсных постполимеров, что позволяет наращивать ММ в широком диапазоне. Сохранение унимодальности кривых ММР и низких Ð указывает на высокую эффективность эфирных производных imQ, содержащих полиметакрилатный радикал, в роли макроинициатора для постполимеризации.
В случае же ПС-инициаторов продукты постполимеризации бимодальны вне зависимости от ММ используемых в качестве макроинициатора образцов и условий процесса. Получить унимодальные постполимеры не удалось даже при кратном увеличении количества imQ при синтезе макроинициатора. По-видимому, это связано с протеканием ряда побочных реакций при полимеризации стирола с участием imQ.
Интересные закономерности обнаружены при изучении реинициирующей способности образцов полиметакрилатов, синтезированных с использованием imQ, и в процессах блок-сополимеризации (табл. 3). В качестве макроинициатора при этом также апробировались образцы ПММА, ПБМА и ПОМА. Гомополимеризация соответствующих мономеров, согласно полученным результатам, с участием imQ проходит в режиме обратимого ингибирования (см. выше).
Как следует из данных табл. 3, образцы полиметакрилатов способны к реинициированию полимеризации различных мономеров. При концентрациях макроинициатора 5–10 мас. %, аналогично процессам постполимеризации, конверсии добавляемых мономеров невелики. Кривые ММР продуктов блок-сополимеризации бимодальны вне зависимости от природы макроинициатора и добавленного мономера. Мода с меньшей ММ, как правило, соответствует кривой ММР используемого макроинициатора; мода с большей ММ, предположительно, относится к образующимся блок-сополимерам. Увеличение концентрации макроинициатора приводит к увеличению скорости полимеризации и понижению значений ММ образующегося продукта. В дальнейшем будет использована наиболее оптимальная концентрация макроинициатора в мономере 20–25 мас. %.
Возможность реинициирования процесса полимеризации акриловых эфиров рассмотрена на примере метилакрилата (табл. 3). Согласно полученным результатам, конверсия добавляемого мономера невелика, а растворимые продукты сополимеризации характеризуются полимодальностью кривых ММР.
При реинициировании полимеризации акрилонитрила образцами ПММА в массе мономера значения конверсии акрилонитрила также крайне низкие, а кривые ММР продуктов сополимеризации бимодальны. Возможно, это связано с ограниченной растворимостью продукта сополимеризации в акрилонитриле. Проведение процесса в растворе ДМФА позволяет увеличить конверсию акрилонитрила, однако кривые ММР остаются бимодальными. Аналогичные закономерности можно наблюдать и в случае прививки винилпирролидона к ПММА.
Таким образом, получение блок-сополимеров с заданными молекулярно-массовыми характеристиками при прививке к полиметакрилатам метилакрилата, акрилонитрила и винилпирролидона затруднительно. Это связано с тем, что гомополимеризация рассматриваемых мономеров с участием imQ проходит в неконтролируемом режиме.
Несколько другие закономерности обнаруживаются при синтезе сополимеров со стиролом. При реинициировании полимеризации стирола его конверсия невысока, а кривые ММР продукта сополимеризации являются бимодальными вне зависимости от концентрации полиметакрилатного макроинициатора и его характеристик. При увеличении концентрации макроинициатора в мономере до 20–25 мас. % количество заполимеризовавшегося стирола растет, однако кривые ММР остаются бимодальными. Применение УФ-детектора при анализе продуктов сополимеризации полиметакрилатов со стиролом позволяет наблюдать появление полимера, содержащего звенья стирола. Кривая ММР унимодальна и совпадает с высокомолекулярной модой кривой ММР продукта сополимеризации, что однозначно указывает на получение именно блок-сополимеров ПММА–блок–ПС (рис. 11). Бимодальность кривых продуктов сополимеризации в случае прививки стирола к образцам ПММА является следствием того, что для него характерна худшая регулируемость процесса с участием imQ, чем для ММА (см. выше).
Согласно литературным данным [24], сополимеризация азеотропной смеси, один из мономеров которой полимеризуется в контролируемом режиме, подчиняется тем же закономерностям, что и гомополимеризация мономера, способного к регулируемой полимеризации. Использование образцов ПММА, полученных с участием imQ, для инициирования полимеризации азеотропной смеси стирола с метилметакрилатом приводит к синтезу продукта с унимодальными кривыми и относительно узким ММР (табл. 3). Анализ их молекулярно-массовых характеристик с использованием УФ-детектора подтвердил появление одной широкой моды, которая соответствует смещенной моде кривой блок-сополимера ПММА–блок–поли(метилметакрилат–со-стирол), зафиксированной RI-детектором (рис. 11, кривые 2 и 5).
Реинициирование полимеризации метакриловых эфиров образцами полиметакрилатов позволяет синтезировать широкий спектр блок-сополимеров. Наряду с уже указанными ММА, БМА и ОМА, получены блок-сополимеры с этилметакрилатом (ЭМА) и цетилметакрилатом (ЦМА) (табл. 3; рис. 12). Конверсии добавляемых метакриловых эфиров выше, чем других мономеров вне зависимости от характеристик макроинициатора.
Сохранение унимодальности ММР и низких значений Đ продукта сополимеризации зависит от сочетания природы мономера макроинициатора и прививаемого метакрилового эфира. Образцы ПММА, полученные при участии imQ, позволяют синтезировать блок-сополимеры с унимодальным ММР и значениями Đ ~ 1.3–1.5 (табл. 3) всех использованных метакриловых эфиров. Образцы же ПОМА дают возможность получить унимодальные продукты сополимеризации только с ЦМА. С увеличением размера спиртового остатка происходит явное понижение эффективности полиметакрилатов, используемых в качестве макроинициатора. Следует отметить, что это связано как с различной регулирующей способностью imQ при гомополимеризации метакриловых эфиров, так и с методикой выделения макроинициатора.
ЗАКЛЮЧЕНИЕ
Установлено, что характер влияния 4,6-ди-трет-бутил-N-(2,6-диизопропилфенил)-о-иминобензохинона на кинетические закономерности радикальной полимеризации виниловых мономеров и молекулярно-массовые характеристики полимеров существенно зависит от температурных условий протекания процесса, соотношения инициатор : о-иминобензохинон : мономер и природы мономера. В случае эфиров метакриловой кислоты полимеризация проходит в режиме обратимого ингибирования. Благодаря используемому 4,6-ди-трет-бутил-N-(2,6-диизопропилфенил)-о-иминобензохинону удается синтезировать блок-сополимеры на основе виниловых мономеров в широком диапазоне молекулярных масс, причем в ряде случаев с относительно невысокими коэффициентами полидисперсности и многообразным составом.
Работа выполнена при финансовой поддержке Российского научного фонда (проект № 18-73-10092) и частично (синтез блок-сополимеров) в рамках госзадания Министерства образования и науки (код проекта 4.5706.2017/БЧ).
Авторы выражают признательность И.Д. Гришину за регистрацию и интерпретацию спектров MALDI-TOF, а также обсуждение полученных результатов.
Список литературы
Fukuda T., Goto A. // Polym. Sci., Compr. Ref. 2012. V. 37. № 3. P. 119.
Allan L.E.N., Perry M.R., Shaver M.P. // Prog. Polym. Sci. 2012. V. 37. № 3. P. 127.
Nicolas J., Guillaneuf Y., Lefay C., Bertin D., Gigmes D., Charleux B. // Prog. Polym. Sci. 2013. V. 38. P. 63.
Chernikova E.V., Pokataeva Z.A., Garina E.S., Lachinov M.B., Golubev V.B. // Macromol. Chem. Phys. 2001. V. 202. P. 188.
Kolyakina E.V., Grishin D.F. // Russ. Chem. Rev. 2011. V. 80. № 7. P. 683.
Caille J.R., Debuigne A., Jérôme R. // J. Polym. Sci., Polym. Chem. 2005. V. 43. № 13. P. 2723.
Vaganova L.B., Maleeva A.V., Piskunov A.V., Grishin D.F. // Russ. Chem. Bull., Int. Ed. V. 60. № 8. P. 1620.
Vaganova L.B., Lizyakina O.S., Chegerev M.G., Pisku-nov A.V., Grishin D.F. // Polymer Science B. 2017. V. 59. № 5. P. 506.
Grishin D.F., Vaganova L.B., Lizyakina O.S., Pisku-nov A.V., Chegerev M.G. // Dokl. Chemistry. 2017. V. 475. Part 1. P. 149.
Broere D.L.J., Plessius R., van der Vlugt J.I. // Chem. Soc. Rev. 2015. V. 44. P. 6886.
Энциклопедия полимеров. М.: Советская энциклопедия, 1972. Т. 1.
Синтезы органических препаратов / Под ред. Б.А. Казанского М.: Иностр. лит., 1953.
Гордон А., Форд Р. Спутник химика. М.: Мир, 1976.
Abakumov G.A., Druzhkov N.O., Kurskii Yu.A., Shavyrin A.S. // Russ. Chem. Bull., Int. Ed. 2003. V. 52. № 3. P. 712.
Беленький Б.Г., Виленчик Л.З. Хроматография полимеров. М.: Химия, 1978.
Moad G., Solomon D.H. The Chemistry of Radical Polymerization. Oxford: Elsevier, 2006.
Grishin D.F., Shchepalov A.A., Cherkasov V.K. // J. Polym. Sci., Polym. Phyz. 2005. V. 47. № 9. P. 928.
Abakumov G.A., Druzhkov N.O., Kurskii Yu.A., Abakumova L.G., Shavyrin A.S., Fukin G.K., Poddelskii A.I., Cherkasov V.K., Okhlopkova L.S. // Russ. Chem. Bull., Int. Ed. 2005. V. 47. № 11. P. 2571.
Mishchenko O.G., Spirina I.V., Maslennikov S.V., Piskunov A.V., Meshcheryakova I.N., Panteleev S.V., Kroik R.V. // Russ. J. Gen. Chem. 2014. V. 84. № 4. P. 642.
Chegerev M.G., Piskunov A.V. // Russ. J. Coord. Chem. 2018. V. 44. № 4. P. 258.
Piskunov A.V., Chegerev M.G., Fukin G.K. // J. Organomet. Chem. 2016. V. 803. № 1. P. 51.
Piskunov A.V., Ershova I.V., Fukin G.K., Shavyrin A.S. // Inorg. Chem. Commun. 2013. V. 38. № 12. P. 127.
Piskunov A.V., Meshcheryakova I.N., Fukin G.K., Shavyrin A.S., Cherkasov V.K., Abakumov G.A. // Dalton Trans. 2013. V. 42. № 29. P. 10533.
Zaremski M.Yu., Plutalova A.V., Lachinov M.B., Golubev V.B. // Macromolecules. 2000. V. 33. P. 4365.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)