

Высокомолекулярные соединения (серия Б), 2019, T. 61, № 5, стр. 345-355
ЗЕЛЕНАЯ ХИМИЯ ПОЛИУРЕТАНОВ: СИНТЕЗ, ФУНКЦИОНАЛЬНЫЙ СОСТАВ И РЕАКЦИОННАЯ СПОСОБНОСТЬ ЦИКЛОКАРБОНАТСОДЕРЖАЩИХ ТРИГЛИЦЕРИДОВ ПОДСОЛНЕЧНОГО МАСЛА – ВОЗОБНОВЛЯЕМОГО СЫРЬЯ ДЛЯ НОВЫХ УРЕТАНОВ
М. А. Левина a, *, Д. Г. Милославский b, М. В. Забалов a, М. Л. Придатченко c, А. В. Горшков a, В. Т. Шашкова a, В. Л. Крашенинников a, Р. П. Тигер a
a Институт химической физики им. Н.Н. Семенова Российской академии наук
119991 Москва, ул. Косыгина, 4, Россия
b Казанский национальный исследовательский технологический университет
420015 Казань, ул. К. Маркса, 68, Россия
c Институт энергетических проблем химической физики им. В.Л. Тальрозе Российской академии наук
119991 Москва, Ленинский пр., 38, корп. 2, Россия
* E-mail: mira_lev@mail.ru
Поступила в редакцию 19.04.2019
После доработки 08.05.2019
Принята к публикации 20.05.2019
Аннотация
Описан синтез эпокси- и циклокарбонатсодержащих олигомеров путем окисления подсолнечного масла и последующей каталитической карбонизации за счет реакции с диоксидом углерода. Методом масс-спектрометрии показано, что для олигомеров, представляющих собой триглицериды эпоксид- и циклокарбонатсодержащих производных карбоновых кислот, характерно широкое распределение по составу, отражающее весь набор компонентов, содержащихся в природном сырье. Установлено наличие в олигомерах из подсолнечного масла 16 типов триглицеридов в разнообразных комбинациях в отличие от 25 типов триглицеридов в олигомерах из соевого масла. При этом реакционная способность обоих олигомеров как основных представителей возобновляемого растительного сырья для новых уретанов, установленная путем изучения модельной реакции н-бутиламинолиза, примерно одинакова.
ВВЕДЕНИЕ
Полиуретаны занимают весьма внушительный сегмент мирового рынка полимерных материалов, и их производство уже приближается к 20 млн тонн в год. При этом все компоненты, необходимые для их синтеза – алифатические и ароматические изоцианаты, полиизоцианаты, простые и сложные гидроксилсодержащие полиэфиры (как линейные, так и разветвленные), до сих пор получают из нефтяного сырья.
Между тем, классическая технология производства полиуретанов, разработанная почти 80 лет назад и основанная на реакции групп NCO ди- или полиизоцианатов с группами ОН ди- или полиолов, в экологическом отношении не отвечает современным требованиям. В первую очередь это относится к изоцианатам, которые получают путем реакций первичных аминов с фосгеном – исключительно опасным отравляющим веществом. В известной мере по этой причине производство полиуретанов постепенно стало перемещаться в развивающиеся страны, где требования к промышленной экологии не столь жестки, как в Европе или Северной Америке. Одновременно, как следует из обзорных статей последних лет [1–10], в странах-производителях полиуретанов стали развиваться работы по поиску альтернативных путей синтеза как изоцианатов без использования фосгена, так и самих полиуретанов без изоцианатов, в том числе из ненефтяного сырья. В результате на базе этих исследований возникло новое перспективное направление в области полимерной науки – зеленая химия полиуретанов.
Наиболее удобный и сравнительно простой путь синтеза новых уретанов – реакция первичных аминов с циклокарбонатами:

Изоцианаты как мономеры вообще не участвуют в таком процессе. В отличие от классических полиуретанов продукты данной реакции – гидроксиуретаны содержат первичные (I) и/или вторичные (II) группы ОН в соотношениях, зависящих от строения реагентов и условий проведения процесса. Наличие групп ОН в боковых цепях таких полиуретанов способствует их гидролитической стабильности за счет образования внутри- и межмолекулярных водородных связей и открывает пути для соответствующей модификации полимеров.
Олигомеры с циклокарбонатными группами для новых уретанов чаще всего получают из эпоксидсодержащих предшественников. Для этого используется каталитическая фиксация СО2 эпоксидами [10]. Для зеленой химии полиуретанов важно также, что подобные олигомеры можно получать из возобновляемого сырья – растительных масел путем их окисления с образованием эпоксидсодержащих триглицеридов (ЭТГ) с последующей каталитической карбонизацией последних под действием диоксида углерода. Условия синтеза таких олигомеров из растительного сырья описаны в литературе [11–13]. Содержание ненасыщенных и насыщенных кислот в большинстве растительных масел (соевом, подсолнечном, пальмовом, льняном, оливковом, рапсовом и других) также известно [14, 15], хотя и зависит от региона, в котором произрастает та или иная культура. Следует отметить, что для синтеза полиуретанов из растительных масел принципиальное значение имеет не столько число и содержание в исходных триглицеридах ненасыщенных связей, сколько состав триглицеридов после окисления и карбонизации и их функциональность по эпоксидным и циклокарбонатным группам. Таких данных для триглицеридов конкретных растительных масел в литературе почти нет.
В наших предыдущих статьях [16, 17] описан синтез и функциональный состав циклокарбонатсодержащих триглицеридов (ЦКТГ) соевого масла и изучена их реакционная способность по сравнению с модельными соединениями нефтяного происхождения. Настоящая работа посвящена синтезу ЭТГ подсолнечного масла, их превращению в ЦКТГ, изучению функционального состава соответствующих олигомеров и исследованию их реакционной способности в процессах уретанообразования.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез олигомеров
Использовали рафинированное, вымороженное и дезодорированное подсолнечное масло торговой марки “Каждый день” производства Общества с ограниченной ответственностью “Маслоэкстракционный завод Юг Руси” (Ростов-на-Дону). Кислотное число подсолнечного масла 0.2 мг КОН/г, йодное число 126.3 г I2/100 г. Масло представляло собой смесь триглицеридов ненасыщенных и насыщенных кислот: олеиновой (С 18:1) – 32.3%, линолевой (С 18:2) – 52.3%, линоленовой (С 18:3) – 0.1%, стеариновой (С 18:0) – 3.6%, пальмитиновой (С 16:0) – 6.4%, арахиновой (С 20:0), бегеновой (С 22:0), пальмитолеиновой (С 16:1) в сумме ~ 1%.
Эпоксидирование подсолнечного масла осуществляли пероксидом водорода в присутствии пероксофосфовольфраматного каталитического комплекса, образующегося in situ (способ I), по методике [18], разработанной на примере окисления олеиновой кислоты, или надмуравьиной кислотой, также образующейся in situ (способ II). В обоих случаях окислителем служил водный раствор медицинского пероксида водорода, содержащий 36.8% основного вещества. В способе I в качестве реагентов, участвующих в образовании пероксофосфовольфраматного комплекса, использовали дигидрат вольфрамата натрия и 85%-ный водный раствор о-фосфорной кислоты. Межфазным катализатором служил триоктилметиламмоний хлорид торговой марки Aliquat-336.
При окислении способом I мольное соотношение компонентов [>C=C<] : [Na2WO4] : [H2O2] = = 1.0 : 0.0055 : 2.0 и [Na2WO4] : [H3PO4] : [межфазный катализатор] = 1.0 : 2.5 : 0.75. Реактор представлял собой трехгорлую круглодонную колбу объемом 1000 см3, снабженную обратным холодильником, термометром и механической мешалкой, в которую загружали 300 г подсолнечного масла и межфазный катализатор. В отдельной емкости готовили эпоксидирующую смесь в виде водного раствора пероксида водорода, вольфрамата натрия и ортофосфорной кислоты. Реактор помещали в водяную баню и при перемешивании (300 об/мин) в него вводили эпоксидирующую смесь. Для предотвращения перегрева последнюю добавляли в реактор посредством капельной воронки в течение 0.5 ч. После ввода всей смеси температуру реакционной массы, представляющей собой стойкую эмульсию, поддерживали ~70°С. За реакцией следили по конверсии пероксида водорода во времени (йодометрический анализ). Синтез прекращали при достижении конверсии пероксида водорода в реакционной массе ~90%. По окончании синтеза реакционную массу многократно промывали теплой и холодной водой до нейтральной реакции для освобождения от следов ортофосфорной кислоты, выполняющей роль регулятора pH. После каждой отмывки реакционную массу переносили в делительную воронку для отделения от водной фазы. Для более полного разделения органический слой подвергали центрифугированию (центрифуга ОПН-16, частота вращения ротора 9000 об/мин.).
При окислении способом II мольное соотношение реагентов [>C=C<] : [HCOOH] : [H2O2] = = 1.0 : 0.5 : 2.0. Использовали тот же реактор, что и в способе I, куда загружали 300 г масла и расчетное количество муравьиной кислоты. Затем в течение 1 ч при перемешивании (300 об/мин) в реактор добавляли 36.8%-ный раствор пероксида водорода. Процесс вели при 70°С и прекращали по достижении конверсии активного кислорода (йодометрический анализ) в реакционной смеси ~60%. Продукт выделяли так же, как и в способе I. Продукты эпоксидирования, полученные способами I (за 2.5 ч) и II (за 5.5 ч) характеризовались содержанием эпоксидного кислорода ~6.35%.
Синтез ЦКТГ осуществляли путем реакции ЭТГ с диоксидом углерода по методике [16], используемой нами ранее при получении циклокарбонатсодержащих олигомеров соевого масла и основанной на методике работы [19]. В реактор высокого давления, оснащенный газозахватывающей мешалкой, датчиком температуры, линиями подачи газа, отбора проб и сброса давления (модель “Beluga” серии hpm-b-020, производитель “Premex Reactor AG”) загружали полученное по описанной выше методике эпоксидированное масло (200 г) и катализатор карбонизации – тетрабутиламмоний бромид (ТБАБ), 3 мол. % от содержания эпоксидных групп. Реактор герметизировали при перемешивании и нагревании реакционной массы и по линии подачи газа вводили диоксид углерода. Процесс вели в течение 20 ч при 140°С и давлении СО2 0.8 МПа. Реакция протекала до превращения эпоксидных групп ~95%. Ранее при работе с олигомерами соевого масла [16] продукты карбонизации от катализатора не очищали, и при этом оставался открытым вопрос об их вероятном влиянии на скорость последующего уретанообразования путем реакции циклокарбонатсодержащего олигомера с амином. В настоящей работе в соответствии с рекомендацией статьи [20] после окончания реакции карбонизации с целью разложения остатков катализатора проводили термическую обработку полученного олигомера в автоклаве при 185°С в среде азота при давлении ~0.7 МПа. Высокотемпературное разложение ТБАБ протекает в соответствии с реакцией элиминирования по Гофману с образованием летучих продуктов – трибутиламина, бутилена и HBr, инертных, как полагают [20], в последующей реакции первичных аминов с циклокарбонатными группами олигомера. Влияние концентрации катализатора ТБАБ на скорость уретанообразования изучали в настоящей работе на модельной реакции н-бутиламинолиза этиленкарбоната. Судя по константам скорости аминолиза в отсутствие и в присутствии специально введенных каталитических количеств ТБАБ в диметилсульфоксиде, катализатор карбонизации ЭТГ не оказывает влияния на скорость превращения ЦКТГ в гидроксиуретан в изученных условиях. Так, наблюдаемая константа скорости псевдо-первого порядка при 55°С и [RNH2] = 0.4 моль/л в отсутствие ТБАБ составляет 1.9 × 10–5 с–1, в присутствии 3.5 × 10–3 моль/л ТБАБ (той же концентрации катализатора, которая используется при синтезе ЦКТГ из ЭТГ) константа скорости равна 2.0 × 10–5 с–1. Не было замечено также и разницы в скоростях уретанообразования с участием ЦКТГ, непосредственно полученного в результате синтеза, и олигомера, очищенного от ТБАБ, как описано выше.
Методики анализа олигомеров и кинетики их превращения в уретаны
ММР олигомеров подсолнечного масла, ЭТГ и ЦКТГ исследовали методом ГПХ на приборе “Breeze” фирмы “Waters” с использованием стирогелевых колонок с пористостью 100, 500, 1000 Å и ТГФ хроматографической чистоты в качестве элюента (скорость элюирования 1 мл/мин), калибровка по ПС-стандартам. ИК-спектры исходных и конечных продуктов снимали на ИК-фурье- спектрометре “Tenzor 27” фирмы “Bruker” на cелен-цезиевой подложке.
Состав олигомеров изучали методом масс-спектрометрии в положительной моде на приборе “Bruker amaZon SL” (“Bruker Daltonik GmbH”, Германия) аналогично описанному в нашей предыдущей работе методу анализа триглицеридов соевого масла [16]. Диапазон измерения ММ в масс-анализаторе составлял 200–1900 m/z. Ионизацию электрораспылением выполняли при 2500/500 В, температура осушающего газа азота – 200°С. Ионы накапливались при ICC (Ion Charge Control) 200 000 в течение ~265 мкс. Образцы растворяли в метаноле до концентрации 10–6 мг/мл и вводили в ионизационную камеру со скоростью 300 мкл/ч, использовали метанол хроматографической чистоты 99.9% (“Biosolve”, Нидерланды).
Реакционную способность циклокарбонатсодержащих олигомеров подсолнечного масла изучали на модельной реакции их н-бутиламинолиза в ДМСО при 55°С методом ИК-фурье-спектроскопии на приборе “Tenzor 27” фирмы “Bruker”. Наблюдение за кинетикой велось по убыли оптической плотности на частоте 1796 см–1, расположенной вблизи максимума поглощения циклокарбонатной группы С=О. При концентрации циклокарбонатных групп (1.5–5.0) × 10–2 моль/л в избытке амина в интервале 0.1–0.75 моль/л реакции протекали в режиме псевдопервого порядка. Методика кинетических измерений реакций аминолиза циклокарбонатсодержащих олигомеров описана ранее [17].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
ИК-спектры, ММР и функциональный состав олигомеров
Примеры ИК-спектров образцов исходного подсолнечного масла, ЭТГ и ЦКТГ, представлены на рис. 1. Характер спектров не зависит от способа окисления подсолнечного масла. Один из основных пиков ЦКТГ ~ 1800 см–1 соответствует карбонилу циклокарбонатных фрагментов, образующихся в результате реакции эпоксидных групп с СО2. Изменение оптической плотности на этой частоте во времени использовалось в настоящей работе для наблюдения за протеканием реакции уретанообразования. Как следует из рис. 1, этот пик отсутствует в спектрах подсолнечного масла и ЭТГ, в которых имеется пик 1745 см–1, ответственный за валентные колебания групп С=О сложноэфирной природы. В спектре карбонизованного продукта данный пик расположен на плече пика карбонила циклокарбонатной группы ЦКТГ. Пик при 720 см–1 в спектрах на рис. 1 характеризует колебания групп СН2 углеродных цепей кислотных остатков. В спектре ЭТГ появляются полосы колебаний С–О–С эпоксидных колец (824 и 845 см–1), которые в конечном ЦКТГ отсутствуют. Во всех спектрах имеются пики 2862 и 2930 см–1, соответствующие валентным колебаниям, 1380 и 1465 см–1 – деформационным колебаниям связей С–Н углеводородных цепей триглицеридов. Пики 1654 и 3007 см–1, относящиеся к двойным связям С=С–Н проявляются лишь в спектре исходного подсолнечного масла до его окисления. Пик 1056 см–1 отвечает колебаниям С–О в циклокарбонатной группе и присутствует лишь в спектре ЦКТГ. Пик 1160 см–1 обусловлен колебаниями С–О сложноэфирных групп углеводородных цепей в подсолнечном масле, ЭТГ и ЦКТГ. Во всех спектрах имеется широкая полоса 3400–3600 см–1, ответственная за наличие ассоциированных групп ОН, возникающих в результате окисления и образования побочных продуктов, содержание которых возрастает при переходе от подсолнечного масла к ЭТГ и далее к ЦКТГ, как и при карбонизации соевого масла [16].
Рис. 1.
ИК-спектры исходного подсолнечного масла (1) и продуктов его эпоксидирования (2) и карбонизации (3).

Для гель-хроматограмм (рис. 2) исследуемых олигомеров характерно наличие основного пика, составляющего 99.1% площади хроматограммы подсолнечного масла и 96.5 и 89.7% площади хроматограмм ЭТГ и ЦКТГ соответственно, полученных путем окисления подсолнечного масла способом II (ЭТГ) и последующей карбонизации (ЦКТГ). Полидисперсность ЭТГ, полученного из подсолнечного масла способом I, как и соответствующего ЦКТГ, выше: площадь основного пика 96.1 и 67.7%. В связи с этим эксперименты по составу олигомеров, их функциональности и реакционной способности выполняли главным образом с использованием более монодисперсных образцов ЭТГ и ЦКТГ, полученных путем окисления подсолнечного масла способом II. Значения ММ олигомеров приведены в табл. 1.
Рис. 2.
Кривые ГПХ триглицеридов подсолнечного масла (1) и продуктов его эпоксидирования (2) и карбонизации (3).

Таблица 1.
ММ триглицеридов подсолнечного масла (ТГПМ), ЭТГ и ЦКТГ по данным ГПХ*
| Триглицериды | Мn | Mw |
|---|---|---|
| ТГПМ | 1160 | 1170 |
| ЭТГ | 1180/1170 | 1190/1180 |
| ЦКТГ | 1300/1240 | 1320/1250 |
Следует заметить, что на гель-хроматограммах олигомеров отчетливо проявляется небольшой пик, относящийся, как предполагалось нами [16], к продуктам димеризации триглицеридов. В зависимости от способа окисления подсолнечного масла площадь пика димеров составляет 3–10% от общей площади хроматограмм. Димеры, вероятно, образуются даже и при спонтанном окислении исходного масла и при хранении, о чем свидетельствует наличие соответствующего пика, хотя и меньшей интенсивности, на хроматограммах исходного подсолнечного масла. Гипотетическая возможность раскрытия эпоксидного кольца при окислении подсолнечного масла и образования гидроксильных групп, способствующих димеризации макромолекул ЭТГ, обсуждается в работе [21].
Масс-спектрометрическое исследование олигомеров было предпринято с целью определения состава триглицеридов подсолнечного масла, ЭТГ и ЦКТГ и отличия его от состава тех же производных соевого масла, установленного ранее [16]. На рис. 3 приведены фрагменты масс-спектров исследуемых подсолнечного масла и ЭТГ в интервале М = 850–1000. В спектре наблюдаются пики, относящиеся к молекулярным ионам [M + 23]+ и [M + 18]+ (меньшей интенсивности), которые возникают при взаимодействии триглицеридов с ионами Na+ и NH$_{4}^{ + }$, содержащимися в виде следов в исходном подсолнечном масле.
Рис. 3.
Фрагменты масс-спектров исследуемых подсолнечного масла (а) и ЭТГ (б) в интервале М = 850–1000. См. примечания к табл. 2 и 3.
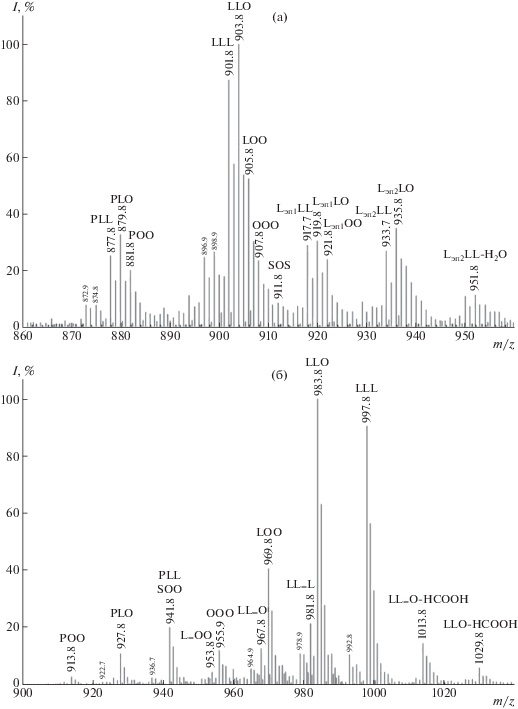
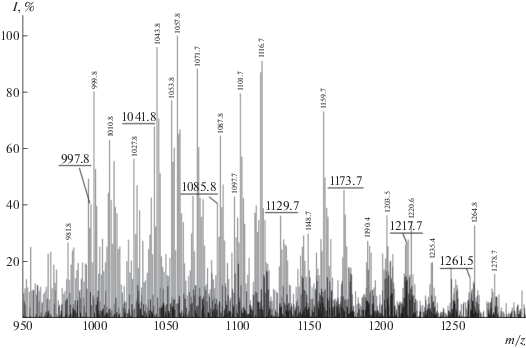
Таблицы 2 и 3 содержат сведения о массе молекулярных ионов, образующихся в результате ионизации подсолнечного масла и ЭТГ. Наряду с исходными данными масс-спектрометрии для установления точного состава молекулярных ионов была проведена фрагментация почти всех пиков по методике, описанной ранее [16]. Это позволило обнаружить наличие в исследуемых объектах примерно 16 различных типов триглицеридов подсолнечного масла, ЭТГ и ЦКТГ. Следует отметить, что состав олигомеров соевого масла по данным предыдущей работы [16] более разнообразен, чем олигомеров подсолнечного масла. Главным отличием является отсутствие в подсолнечном масле и, следовательно, в ЭТГ и ЦКТГ линоленовых фрагментов. В качественном отношении состав триглицеридов исходного подсолнечного масла, установленный в настоящей работе, совпадает с аналогичными масс-спектрометрическими исследованиями подсолнечного масла различного происхождения, имеющимися в литературе [22–24]. В табл. 3 представлены данные по функциональности ЭТГ, которая соответствует максимальной функциональности ЦКТГ в предположении, что все эпоксидные группы при реакции с СО2 превратились в циклокарбонатные. Средняя же функциональность олигомерных ЦКТГ каждого конкретного состава естественно ниже их максимальной функциональности. Это видно из рис. 4 на примере одного из основных пиков ЭТГ (М + 23) = 997.7 состава LLL, который превращается в пик ЦКТГ с одной (1041.7), двумя (1085.6), тремя (1129.6), четырьмя (1173.6), пятью (1217.6) и шестью (1261.6) циклокарбонатными группами. Судя по интенсивности пиков, в продуктах реакции карбонизации больше всего олигомеров с четырьмя циклокарбонатными группами, а меньше всего с шестью.
Таблица 2.
Состав и молекулярная масса М триглицеридов подсолнечного масла (ТГПМ) и их молекулярных ионов [М + 23]+
| Состав ТГПМ* | М | [М + 23]+ | Интенсивность пика, % |
|---|---|---|---|
| PLL | 854.7 | 877.7 | 25 |
| PLO | 856.7 | 879.7 | 33 |
| POO | 858.8 | 881.8 | 20 |
| PLS | 858.8 | 881.8 | |
| LLL | 878.7 | 901.7 | 87 |
| LLO | 880.7 | 903.7 | 100 |
| LOO | 882.8 | 905.8 | 52 |
| LLS | 882.8 | 905.8 | |
| OOO | 884.8 | 907.8 | 24 |
| OLS | 884.8 | 907.8 | |
| SLS | 886.8 | 909.8 | 14 |
| SOO | 886.8 | 909.8 | |
| SOS | 888.8 | 911.8 | 8 |
| Lэп1LL** | 894.7 | 917.7 | 29 |
| Lэп1LO | 896.7 | 919.7 | 30 |
| LLOэп1 | 896.7 | 919.7 | |
| Lэп1OO | 898.8 | 921.8 | 24 |
| LООэп1 | 898.8 | 921.8 | |
| Lэп2LL | 910.7 | 933.7 | 27 |
| Lэп1Lэп1L | 910.7 | 933.7 | |
| ALL | 910.7 | 933.7 | |
| Lэп2LO | 912.7 | 935.7 | 35 |
| Lэп1Lэп1O | 912.7 | 935.7 | |
| Lэп1LOэп1 | 912.7 | 935.7 | |
| ALO | 912.7 | 935.7 | |
| Lэп2LL-H2O*** | 928.7 | 951.7 | 11 |
* Триглицериды кислот: P – пальмитиновой, S – стеариновой , A – арахиновой, O – олеиновой, L – линолевой. Здесь и в табл. 3 в группах триглицеридов c одинаковой ММ подчеркнут состав, содержание которого максимально.
Таблица 3.
Cостав ЭТГ, молекулярная масса М триглицеридов, их молекулярных ионов [М + 23]+ и максимальная функциональность F олигомеров ЭТГ и соответствующих ЦКТГ
| Состав ЭТГ | М | [М + 23]+ | Интенсивность пика, % | F |
|---|---|---|---|---|
| POO | 890.8 | 913.8 | 3 | 2 |
| PLS | 890.8 | 913.8 | 2 | |
| PLO | 904.7 | 927.7 | 11 | 3 |
| PLL | 918.7 | 941.7 | 20 | 4 |
| SLS | 918.7 | 941.7 | 2 | |
| SOO | 918.7 | 941.7 | 2 | |
| L=OO* | 930.8 | 953.8 | 7 | 3 |
| OOO | 932.8 | 955.8 | 11 | 3 |
| OLS | 932.8 | 955.8 | 3 | |
| LL=O* | 944.7 | 967.7 | 13 | 4 |
| LOO | 946.7 | 969.7 | 43 | 4 |
| LLS | 946.7 | 969.7 | 4 | |
| LL=L* | 958.7 | 981.7 | 22 | 5 |
| LLO | 960.7 | 983.7 | 100 | 5 |
| ALO | 960.7 | 983.7 | 3 | |
| LLL | 974.7 | 997.7 | 93 | 6 |
| ALL | 974.7 | 997.7 | 4 | |
| LL=O – HCOOH** | 990.8 | 1013.8 | 12 | 3 |
| LLO – HCOOH** | 1006.7 | 1029.7 | 4 | 4 |
Реакционная способность циклокарбонатсодержащих олигомеров
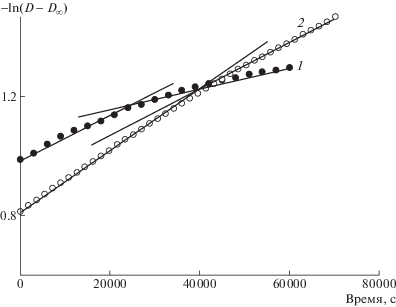
В отличие от аминолиза этиленкарбоната и реакций других модельных соединений (см., например, работы [17, 25, 26]), анаморфозы кинетических кривых псевдопервого порядка превращения ЦКТГ подсолнечного масла не спрямлялись на всем протяжении процесса, как и анаморфозы кинетических кривых реакций с участием ЦКТГ соевого масла (рис. 5). Отклонения, как правило, наблюдались при превращении ~50–70%, причем реакции с участием производных подсолнечного масла хоть и медленно (часто в течение 1 суток и более), но все же доходят до конца в отличие от реакций тех же производных соевого масла, довести которые до конца за обозримые времена не удается [17]. Аномальное поведение кинетических кривых, характерное для процесса аминолиза исследуемых олигомеров, может быть обусловлено разной реакционной способностью циклокарбонатных групп L- и О-фрагментов цепей триглицерида подсолнечного масла (в случае соевого масла также и Ln-фрагментов). Возможно также частичное “блокирование” функциональных групп по ходу реакции за счет наведения межцепных водородных связей с участием образующихся гидроксиуретанов и изменение тем самым подвижности цепей и реакционной способности реагентов. Количественным исследованиям этих вопросов будет посвящено отдельное сообщение.
Рис. 5.
Типичные кинетические анаморфозы в полулогарифмических координатах реакции аминолиза олигомера из соевого масла (1) при [BuNH2] = 0.5 моль/л и олигомера из подсолнечного масла (2) при [BuNH2] = 0.65 моль/л и T = 55°C.
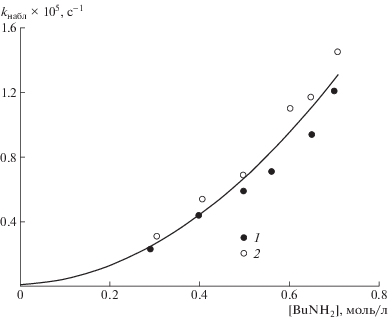
Наблюдаемую константу скорости kнабл рассчитывали как тангенс угла наклона начальных участков анаморфоз кинетических кривых. Как и при аминолизе модельного этиленкарбоната в диоксане, бутаноле и ДМСО [17, 25, 26], связь kнабл с концентрацией амина для изученного в данной работе ЦКТГ и ЦКТГ соевого масла [17] описывается двухпараметрической зависимостью (рис. 6), единой в пределах точности эксперимента для обоих олигомеров:
Рис. 6.
Зависимость наблюдаемой константы скорости псевдопервого порядка реакции ЦКТГ подсолнечного (1) и соевого масла (2) с н-бутиламином в ДМСО от концентрации амина [RNH2] при 55°C. 1 – подсолнечное масло, настоящая работа, 2 – соевое масло, данные работы [17].
ЗАКЛЮЧЕНИЕ
Зеленая химия полиуретанов находится в начальной стадии своего развития, и промышленного производства неизоцианатных уретанов из возобновляемого сырья пока не существует. Рынок же растительного сырья хорошо развивается и его прирост составляет ~3% в год. Суммарный объем производства основных растительных масел в мире в настоящее время приближается к 200 млн тонн в год. При этом по данным Всемирного банка доля подсолнечного масла (8%) несоизмеримо мала по сравнению с производством пальмового (34%), соевого (29%) и даже рапсового (16%) масел. Маловероятно, что триглицериды подсолнечного масла в обозримые сроки будут представлять основной интерес для промышленности новых полиуретанов. В то же время у перечисленных триглицеридов есть одно важное качество, отличающее их от соответствующих подходящих аналогов: в их жирно-кислотных цепях практически нет Ln-фрагментов, в которых содержится сразу три циклокарбонатные группы, отделенные друг от друга лишь одной группой СН2 цепи. С позиций модельных исследований физико-механических и иных свойств будущих гидроксиуретанов из циклокарбонатсодержащего подсолнечного масла по сравнению с триглицеридами других растительных масел они могут представить определенную научную ценность. Кроме того, и это не менее важно, что развитые в настоящей работе методологические принципы исследования функциональности подсолнечного масла с помощью масс-спектрометрии и следующие из кинетических данных сведения об их одинаковой с соевым маслом реакционной способности можно будет применить для изучения аналогичных олигомеров, получаемых из растительного сырья.
Работа выполнена в рамках госзадания (Тема V 45.5, 0082-2014-0015, № АААА-А17-117032750201-9) и при финансовой поддержке Российского фонда фундаментальных исследований (проект 17-03-00146).
Список литературы
Cornille A., Auvergne R., Figovsky O., Boutevin B., Caillol S. // Eur. Polym. J. 2017. V. 87. P. 535.
Rokicki G., Parzuchowski P.G., Mazurek M. // Polym. Adv. Technol. 2015. V. 26. P. 707.
Maisonneuve L., Lamarzelle O., Rix E., Grau E., Cramail H. // Chem. Rev. 2015. V. 115. P. 12407.
Blattmann H., Fleischer M., Bahr M., Mulhaupt R. // Macromol. Rapid Comm. 2014. V. 35. P. 1238.
Figovsky O., Shapovalov L., Leykin A., Birukova R., Potashnikova R. // PU Magazine. 2013. V. 10. P. 1.
Nohra B., Candy L., Blanco J.-F., Guerin C., Raoul Y., Moolaungui Z. // Macromolecules. 2013. V. 46. P. 3771.
Figovsky O., Shapovalov L., Leykin A., Birukova R., Potashnikova R. // Int. Lett. Chem. Phys. Astronomy. 2013. V. 3. P. 52.
Guan J., Song Y., Lin Y., Yin X., Zuo M., Zhao Y., Tao X., Zheng Q. // Ind. Eng. Chem. Res. 2011. V. 50. P. 6517.
Tiger R.P. // Polymer Science B. 2004. V. 46. № 5–6. P. 142.
Garrison T.F., Kessler M.R. // Bio-based Рlant oil Рolymers and Composites / Ed. by S.A. Madbouly, C. Zhang, M.R. Kessler. Amsterdam: Elsevier, 2016. P. 37.
Bahr M., Mulhaupt R. // Green Chem. 2012. V. 14. P. 483.
Javni I., Hong Doo Pyo, Petrovic Z.S. // J. Appl. Polym. Sci. 2013. V. 128. P. 566.
Miloslavsky D., Gotlib E., Figovsky O., Pashin D. // Int. Lett. Chem. Phys. Astronomy. 2014. V. 8. P. 20.
Gunstone F. The Chemistry of Oils and Fats: Sources, Composition, Properties and Uses. Oxford: Blackwell Publ. Ltd., 2004.
Korak N. Vegatable Oil-based Polymers: Properties. Processing and Applications. Philadelphia: Woodhead Publ. Ltd., 2012.
Levina M.A., Miloslavskii D.G., Pridatchenko M.L., Gorshkov A.V., Shashkova V.T., Gotlib E.M., Tiger R.P. // Polymer Science B. 2015. V. 57. № 6. P. 584.
Levina M.A., Zabalov M.V., Krasheninnikov V.G., Tiger R.P. // Polymer Science B. 2018. V. 60. № 5. P. 563.
Miloslavskiy D.G., Cherezova E.N., Ahmedyanova R.A., Liakumovich A.G. // Butlerov Communications. 2012. V. 29. № 3. P. 72.
Li Z., Zhao Y., Yan S., Wang X., Kang M., Wang J., Xiang H. // Catal. Lett. 2008. V. 123. P. 246.
Doll K.M., Erhan S.Z. // Green Chem. 2005. V. 7. P. 849.
Petrovic Z.S., Zlatanic A., Lava C.C., Sinadinovic-Fiser S. // Eur. J. Lipid Sci. Technol. 2002. V. 104. P. 293.
Zeb A., Murkovic M. // Eur. J. Lipid Sci. Technol. 2010. V. 112. P. 844.
Hvattum E. // Rapid Commun. Mass Spectrom. 2001. V. 15. P. 187.
Marzilli L.A., Fay L.B., Dionisi F., Vouros P. // J. Am. Oil Chem. Soc. 2003. V. 80. P. 195.
Levina M.A., Zabalov M.V., Krasheninnikov V.G., Tiger R.P // Polymer Science B. 2017. V. 59. № 5. P. 497.
Zabalov M.V., Levina M.A., Krasheninnikov V.G., Tiger R.P // Russ. Chem. Bull., Int. Ed., 2012. V. 63. P. 1740.
Zabalov M.V., Tiger R.P., Berlin A.A. // Doklady Chemistry. 2011. V. 441. Pt. 2. P. 355.
Zabalov M.V., Tiger R.P., Berlin A.A. // Russ. Chem. Bull., Int. Ed. 2012. V. 61. P. 518.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)