

Высокомолекулярные соединения (серия Б), 2020, T. 62, № 1, стр. 68-80
ВЛИЯНИЕ СРЕДЫ СИНТЕЗА ГИДРОКСИАПАТИТА НА СТРУКТУРУ
Т. П. Калмыкова a, b, Ю. В. Костина a, С. О. Ильин a, *, Ю. Г. Богданова b, А. В. Северин b, П. Л. Иванов c, С. В. Антонов a
a Институт нефтехимического синтеза им. А.В. Топчиева Российской академии наук
119991 Москва, Ленинский пр., 29, Россия
b Московский государственный университет имени Ломоносова. Химический факультет
119991 Москва, Ленинские горы, д. 1, стр. 3, Россия
c Институт синтетических полимерных материалов им. Н.С. Ениколопова Российской академии наук
117393 Москва, ул. Профсоюзная, 70, Россия
* E-mail: s.o.ilyin@gmail.com
Поступила в редакцию 06.06.2019
После доработки 09.07.2019
Принята к публикации 02.09.2019
Аннотация
Получены дисперсии на основе гиалуроната натрия и гидроксиапатита, синтезированного как классическим способом в водной среде с получением частиц субмикронного размера, так и в биоактивной среде гиалуронана с формированием наноразмерных частиц. Синтезированные наночастицы гидроксиапатита имеют сложно организованную структуру на основе монодентатного комплекса гиалуронан–Ca2+, которая сохраняется в дисперсии как при добавлении раствора гиалуронана, так и при разбавлении водой. Физико-химическими методами показано существенное различие между энергетическими характеристиками поверхностей биокомпозитов, полученных на основе дисперсий. Рассмотрена реология дисперсий микро- и наночастиц гидроксиапатита, показано влияние на нее концентрации дисперсной фазы, продемонстрировано вязкопластичное поведение дисперсий микрочастиц, которое пропадало при добавлении раствора гиалуронана, приводящего, однако, к потере системой устойчивости. Введение в растворы гиалуронана добавок наночастиц гидроксиапатита оказывало слабое влияние на реологию, флокуляции наночастиц не происходило. Полученные композиции, состоящие из гидроксиапатита и гиалуроната натрия, могут рассматриваться в качестве основы препарата для восстановительного лечения пародонтита.
ВВЕДЕНИЕ
Гидроксиапатит – один из самых хорошо изученных и востребованных в медицине минералов. История его применения в остеопластической хирургии, стоматологии, косметологии и создании препаратов биологического назначения насчитывает более 50 лет [1–3]. Результаты изучения физико-химических свойств фосфатов кальция и развитие технологии производства биосовместимых компонентов на его основе заложили базу для создания широкого спектра новых материалов медицинского назначения: от порошков и гранул до композиционных материалов и сложных по структуре матриксов для клеточных технологий реконструкции тканевых дефектов [4–7]. Самым очевидным является применение фосфатов кальция в стоматологии: костные (пародонтальные) карманы могут быть заполнены порошками или гелями, содержащими гидроксиапатит, после проведения лоскутной операции, что способствует прикреплению соединительной ткани к костной ткани альвеолярных отростков.
В настоящее время известны различные способы синтеза гидроксиапатита [8–10], однако как биологические, так и механические свойства чистых порошков отличаются от свойств природного костного вещества [11]. Уникальные свойства костной ткани связаны с сочетанием в ней органических (белки коллагеновой природы, полисахариды и т.д.) и неорганических (гидроксиапатит и примеси других кальцийфосфатных фаз) составляющих [12, 13], что предопределяет актуальность развития новых методов синтеза композитов, позволяющих получать наноразмерные частицы гидроксиапатита в среде биополимерных матриц.
Наиболее перспективными направлениями исследований и разработок в области биополимерных композитов медицинского назначения являются синтез гибридных органо-неорганических фаз [14, 15] с участием фосфатов кальция и получение наноструктурированных материалов, подобных костной ткани, а также совершенствование технологии и свойств композиций для пластики и регенерации костной ткани in situ [16–18] с целью развития наиболее востребованного в последнее время направления в регенеративной хирургии – инженерии костной ткани с применением клеточных технологий [19–21].
Ранее мы предложили способ синтеза наноразмерных частиц гидроксиапатита кальция в активной среде (водном растворе высокомолекулярного гиалуроната натрия) для создания биополимерных композитов с улучшенными механическими и биологическими свойствами (в том числе биорезистентностью и биорезорбируемостью) [22]. Было показано, что структура кристалла гидроксиапатита изменяется в зависимости от условий синтеза, т.е. гиалуронан выступает как активная среда синтеза наноразмерного гидроксиапатита, служащего основой для создания биополимерного композита.
Цель настоящей работы – получение устойчивых дисперсий наноразмерного гидроксиапатита в среде раствора гиалуронана и биополимерных композитов на их основе в широком диапазоне концентраций компонентов, а также сравнение структурных и физико-химических характеристик этих систем и аналогичных им по составу, но на основе дисперсий субмикронных частиц гидроксиапатита, синтезированных классическим методом в водной среде.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Гиалуронат натрия производства “Shiseido” (Япония) характеризовался молекулярной массой около 1.4 × 106. Для синтеза гидроксиапатита использовали оксид кальция (“Химмед”, Россия) и 30%-ный водный раствор ортофосфорной кислоты (“ИРЕА-2000”, Россия), которые брали в стехиометрическом соотношении для получения соли состава Са10(РО4)6(ОН)2.
С целью определения оптимального соотношения между полимером и частицами минерала в составе дисперсий проводили оценку взаимодействия гиалуронана и катионов кальция. Для этого получали пленки из 0.25%-ных растворов данного полимера, содержащих хлорид кальция. Образцы растворов с мольным соотношением Ca2+ : : гиалуронан = 1 : 10, 1 : 2, 2.5 : 1.0, 5 : 1 и 7.5 : 1.0 наносили на кремниевые подложки и высушивали в течение 1 суток. Для определения энергетических характеристик поверхностей пленок использовали метод смачивания [23]: находили удельную свободную поверхностную энергию γSV, а также ее дисперсионную ($\gamma _{{SV}}^{d}$) и полярную ($\gamma _{{SV}}^{p}$) составляющие двухжидкостным методом в рамках подхода Оуэнса–Вендта–Кабли [24]:
Синтез микроразмерных частиц осуществляли в дистиллированной воде [24], синтез наночастиц проводили в 0.1%-ном водном растворе гиалуронана [22]. Микрочастицы имели сигарообразную форму с диаметром 50 нм и длиной 230–480 нм, наночастицы представляли собой игольчатые нанокристаллы диаметром 6 нм и длиной 60–150 нм. Дисперсии гидроксиапатита, различающиеся типом частиц, а также концентрациями полимера и гидроксиапатита, получали смешением дисперсий синтезированных частиц с дистиллированной водой и водным 2%-ным раствором гиалуронана. При добавлении к дисперсии микрочастиц раствора гиалуронана образцы разжижались, но с течением времени происходило их расслаивание, возможно, из-за адсорбции макромолекул на поверхности частиц и их флокуляции. В случае введения раствора гиалуронана в дисперсию наночастиц образцы сохраняли устойчивость к седиментации и агрегации. Для предотвращения снижения молекулярной массы гиалуроната натрия из-за деятельности бактерий [25] в дисперсии добавляли 0.2 мас. % азида натрия.
Реологические характеристики водных растворов гиалуронана, а также дисперсий микро- и наноразмерных частиц определяли при температуре 25°С на ротационном реометре “DHR-2” (“TA Instruments”, США) с использованием рабочего узла конус–плоскость (диаметр конуса 40 мм, угол между образующей конуса и плоскостью 2°). Находили зависимости вязкости композиций от скорости сдвига, а также зависимости линейных модулей накопления G' и потерь G'' от угловой частоты. Относительная погрешность при измерении реологических характеристик не превышала 5%.
Для определения удельной свободной поверхностной энергии и регистрации ИК-спектров биокомпозитов, формирующихся из дисперсий при удалении воды, были получены пленки методом полива дисперсий на оптические окна из кремния с последующей сушкой при 25°С в течение 1 суток. Использовали дисперсии с концентрацией гиалуронана 0.25% и соотношением гидроксиапатит : гиалуронан = 1 : 4.65. ИК-спектры пленок регистрировали на вакуумном ИК-фурье-спектрометре “IFS 66 v/s” (“Bruker”, США). Спектры обрабатывали с использованием программного обеспечения OPUS (“Bruker”).
Методом фазового анализа рассеянного света измеряли ζ-потенциал дисперсий на анализаторе “Zetasizer Nano ZS” (“Malvern Instruments”, Великобритания). Перед измерением образцы разбавляли в 10, 50 и 100 раз для оценки влияния степени разбавления на величину измеряемого ζ-потенциала.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Определение соотношения реагентов для синтеза наночастиц
Подавить агрегацию наночастиц при их синтезе и хранении можно создав вокруг них оболочку из адсорбированных молекул низко- или высокомолекулярной природы [26–28]. В случае гидроксиапатита, содержащего катионы Ca2+, в качестве стабилизатора должны выступать полианионные макромолекулы, например, гиалуронана. Однако встает вопрос об оптимальном соотношении между полимером и минералом. При этом следует исходить из желательности максимального насыщения полимера катионами кальция, поскольку именно этот элемент входит в состав костной ткани. Таким образом, возникает требование в получении насыщенного комплекса Ca2+–гиалуронан.
В работе [22] показана возможность переионизации заряда двойного электрического слоя частиц гидроксиапатита при их синтезе в активной среде. При получении наночастиц в 0.1%-ном растворе гиалуронана происходит ослабление поляризации связи P–O и локализация электронной плотности на связи Р=О с образованием монодентатного и бидентатного комплексов [29]. Обычно присутствие двухцентровых катионов (например, Ca2+) способствует делокализации, а монодентатных (Na+) – локализации электронной плотности на анионе; однако в нашем случае структура комплекса оказалась зависимой от концентрации гиалуронана.
При синтезе частиц гидроксиапатита в среде раствора гиалуронана карбоксилат-анионы координируются только с катионами Ca2+, что в свою очередь при дальнейшем введении фосфатных групп в среду синтеза вызывает перераспределение электронной плотности на связях O–P=O в молекулах гидроксиапатита. При использовании более концентрированного раствора полиэлектролита происходит координация карбоксилат-анионов не только с катионами Ca2+, но и с фосфатной группой гидроксиапатита, что приводит к делокализации электронной плотности на анионе и наблюдаемому в ИК-спектрах длинноволновому сдвигу максимума полосы поглощения [22, 25].
Таким образом, для создания биоминеральных композиций с предельным замещением катионов натрия в полимере катионами кальция определение соотношения гидроксиапатита и гиалуронана необходимо проводить с учетом возможности переионизации и существования разных комплексов этих соединений. Оптимальное соотношение находили с привлечением двух независимых методов: ИК-спектроскопии и смачивания.
При приготовлении модельных растворов гиалуроната натрия в смеси с CaCl2 исходили из предположения, что оптимальное соотношение компонентов, при котором произойдет насыщение гиалуроната катионами Ca2+, будет стехиометрическим (CaCl2 : гиалуронан = 1 : 2): у гиалуронана как одноосновного полимера один катион Ca2+ замещает максимум два катиона Na+. Наряду со стехиометрическим соотношением Ca2+ : Na+ были исследованы системы с недостаточным и избыточным содержанием Ca2+.
Для серии высушенных образцов растворов гиалуроната натрия в смеси с CaCl2 была найдена зависимость сдвига положения максимума полосы поглощения карбоксилатного аниона от соотношения CaCl2 : гиалуронан (рис. 1а). Зависимость распадается на два линейных участка, что позволяет методом касательных найти значение точки насыщения.
Рис. 1.
Определение насыщения гиалуронана катионами Ca2+ по сдвигу максимума полосы поглощения карбоксилатных анионов (а) и по изменению дисперсионной составляющей удельной свободной поверхностной энергии (б) образцов высушенных растворов гиалуронана и CaCl2. r – соотношение 1/2 Ca2+ : гиалуронан.
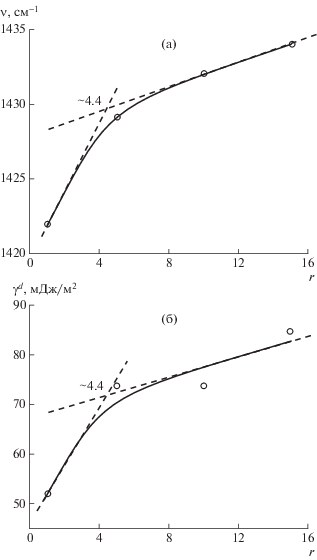
Для проверки правильности измеренного значения точки насыщения применили анализ данных, полученных другим методом – методом смачивания с определением удельной свободной поверхностной энергии и ее полярной и дисперсионной составляющих. При этом удельную свободную поверхностную энергию на границе раздела фаз рассматривали как меру сбалансированности дисперсионных и полярных межмолекулярных взаимодействий: известно, что именно дисперсионная составляющая коррелирует с межмолекулярным взаимодействием, что определяет в том числе и плотность приповерхностных слоев. Экспериментальные данные свидетельствуют о том, что основной вклад в величину поверхностной энергии пленок вносит именно дисперсионная составляющая. Экспериментальная зависимость $\gamma _{{SV}}^{d}$ от соотношения Са2+: гиалуронан и использование метода касательных позволяет определить точку насыщения (рис. 1б).
Оба независимых метода показали одно и то же соотношение (1/2 Ca2+ : гиалуронан ~4.4 : 1), при котором достигается максимально заполнение координационной сферы Ca2+ карбоксилат-анионами. Следовательно, в биополимерных минеральных композициях на основе гидроксиапатита фрагменты ~COO– гиалуронана, образующие координационную сферу Ca2+, будут образовывать насыщенную координационную сферу при этом же соотношении.
Состав предельно насыщенных кальцием композиций
Для получения дисперсий наночастиц с оптимальным соотношением Ca2+ и ~COO– необходимо учесть количество атомов в молекуле гидроксиапатита Ca10(PO4)6(OH)2. Например, для смеси CaCl2 – гиалуронан с мольным соотношением компонентов 1 : 2 одному атому Ca2+ соответствует два атома Na+, следовательно, данному мольному соотношению эквивалентно мольное соотношение гидроксиапатит : гиалуронан = 1 : 20. Аналогично можно рассчитать соотношения минерала и полимера, эквивалентные соотношениям СaCl2 : гиалуронан = 5 : 2 и 10 : 2, и оптимальное соотношение компонентов для получения дисперсии с насыщенной координационной сферой вокруг атомов кальция (табл. 1).
Реологические характеристики
Для синтеза наночастиц важно не только соотношение между реагирующими веществами, но и их концентрация. В нашем случае, поскольку одним из компонентов реакционной смеси является полимер, способный существенно ее загущать, его концентрация будет определяющей. Кроме того, решающее влияние на синтез и устойчивость частиц должна оказывать структура полимерного раствора – наличие или отсутствие в нем макромолекулярных взаимодействий. Определить соответствие той или иной области концентрации растворенного полимера состоянию его раствора (разбавленный, полуразбавленный или концентрированный) можно на основании концентрационной зависимости вязкости.
Кривые течения водных растворов гиалуроната натрия типичны для полимерных систем: вязкость разбавленных растворов от напряжения сдвига не зависит (возможно, из-за достижения недостаточно высоких скоростей сдвига [30, 31]), а вязкость полуразбавленных и концентрированных растворов снижается по мере повышения напряжения (рис. 2). Переход от разбавленных растворов к полуразбавленным происходит при содержании гиалуронана около 0.01 мас. %, что указывает на его весьма высокую молекулярную массу. Для растворов неионогенных полимеров такой переход происходит при концентрации полимера, обратно пропорциональной его характеристической вязкости [32], тогда как для полиэлектролитов, к которым относится гиалуронан, – при содержании полимера в 4 раза больше [33]. Точка перехода позволяет оценить характеристическую вязкость используемого гиалуронана на уровне 400 дл/г, а ММ ≈ 4.5 × 107 [34]. Столь высокая молекулярная масса не характерна для природных линейных полимеров, что свидетельствует об ассоциации макромолекул гиалуронана [35]. Можно полагать, что ассоциация цепей носит случайный характер, так как в противоположном случае (формирования узлов сшивки с неравномерным распределением в среде) разбавленные растворы гиалуроната натрия характеризовались бы наличием предела текучести [36].
Рис. 2.
Кривые течения растворов с концентрацией гиалуронана 0 (1), 0.001 (2), 0.003 (3), 0.01 (4), 0.02 (5), 0.05 (6), 0.1 (7), 0.25 (8), 0.5 (9), 1.0 (10) и 1.5 мас. % (11).

В общем случае для растворов полиэлектролитов можно выделить четыре концентрационные области: разбавленных растворов, полуразбавленных без макромолекулярных переплетений, полуразбавленных с переплетениями и концентрированных. Для каждой из областей зависимость удельной вязкости линейна в логарифмических координатах и имеет свой характерный угол наклона [37, 38]. В нашем случае на концентрационной зависимости можно выделить только три первых участка, соответствующих разбавленным и полуразбавленным растворам (рис. 3). При этом на двух первых удельная вязкость возрастает с повышением концентрации полимера с интенсивностями, типичными для растворов полиэлектролитов: тангенсы углов наклона прямых составляют 1.5 и 0.5 для разбавленных и полуразбавленных растворов без макромолекулярных переплетений. Для растворов с переплетенными цепями типичными являются тангенсы углов наклона прямых, равные 1.5 и 4.7 в случае полуразбавленных и концентрированных растворов соответственно. Вязкость раствора гиалуроната натрия в данной области увеличивается с промежуточным показателем степени 2.6, возможно, из-за размытого перехода между двумя последними концентрационными областями вследствие высокой дисперсности природного полимера или ассоциации макромолекул гиалуронана, приводящей к более интенсивному росту вязкости.
Рис. 3.
Концентрационная зависимость удельной вязкости водного раствора гиалуроната натрия при 25°C. Тангенс угла наклона равен 1.5 (1), 0.5 (2) и 2.6 (3).

С одной стороны, для синтеза наночастиц реакционная среда не должна содержать очень много полимера, чтобы не быть слишком вязкой, замедляющей диффузию реагирующих веществ, а с другой – макромолекулы должны полностью заполнять объем раствора для взаимодействия с формирующимися наночастицами. Таким образом, наиболее оптимальным для синтеза представляется использование граничной концентрации полимера, разделяющей области его полуразбавленных растворов. В нашем случае данная концентрация равна 0.1%. Она и была использована для синтеза наночастиц.
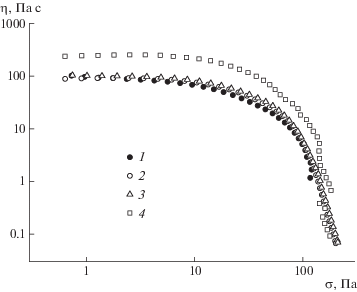
Тем не менее, если рассматривать дисперсии на основе гидроксиапатита в перспективе применения в качестве лекарственного средства, то при их получении рационально использовать наиболее концентрированный раствор гиалуронана, поскольку наличие макромолекулярных переплетений препятствует агрегированию частиц дисперсной фазы [29]. Вязкость при этом не должна превышать ~10 000 Па с, что даст возможность выдавливать препарат, например, через иглу шприца [36]. Вязкость 1.5%-ного раствора гиалуронана низка и составляет 100 Па с, однако в нем присутствуют переплетения цепей. Такое сочетание делает этот раствор подходящей матрицей для введения частиц гидроксиапатита. Согласно закону Эйнштейна, вязкость разбавленных дисперсий возрастает крайне незначительно (в пределах ошибки измерения) при повышении доли дисперсной фазы. И действительно, введение в раствор гиалуронана от 0.1 до 0.5% наночастиц гидроксиапатита не сильно влияет на его вязкость (рис. 4). Ситуация изменяется при добавлении 1.5% наночастиц: вязкость раствора повышается в 2.5 раза. Такой значительный прирост вязкости указывает на структурообразование в системе вследствие взаимодействия наночастиц либо непосредственно между собой, либо с участием макромолекул.
Рис. 4.
Кривые течения дисперсий с концентрацией наночастиц гидроксиапатита 0 (1), 0.1 (2), 0.5 (3) и 1.5 мас. % (4) в среде 1.5%-ного водного раствора гиалуронана.
В случае образования частицами гидроксиапатита перколяционной структуры, она бы проявлялась наличием у образца предела текучести [39]. Благодаря его отсутствию можно заключить, что в образовании структуры участвует гиалуронан, создающий органическую “оболочку” вокруг наночастиц гидроксиапатита при их синтезе [22]. Макромолекулы линейных полимеров, адсорбируясь одновременно на поверхности двух или более твердых частиц, могут связывать их между собой в единую структуру и обусловливать высокую вязкость композиций [40].
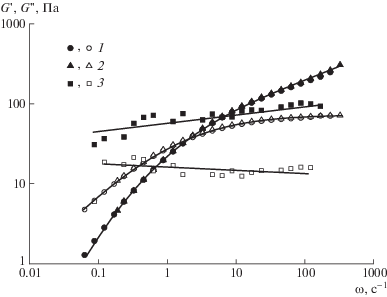
В пользу такого механизма свидетельствует то, что в отсутствие гиалуронана микрочастицы гидроксиапатита образуют в водной среде гель: кривая течения образца дисперсии характеризуется наличием предела текучести с величиной около 1 Па (рис. 5), а его модуль упругости G ' практически не зависит от частоты и превышает модуль потерь G '' (рис. 6). Напротив, раствор гиалуронана и этот же раствор, но содержащий наноразмерные (или микроразмерные, но в течение непродолжительного времени вследствие расслаивания) частицы гидроксиапатита, ведут себя как типичные полимерные вязкоупругие растворы, проявляющие неньютоновский характер течения при высоких напряжениях сдвига (рис. 5). Схожее воздействие, заключающееся в придании агрегативной устойчивости наноразмерным частицам, оказывают и другие высокомолекулярные полимеры, например полиэтиленоксид [41, 42]. При этом частотные зависимости модулей упругости и потерь дисперсий в среде гиалуронана отвечают модели Максвелла и повышаются в области низких частот с показателями 2.0 и 1.0 соответственно (рис. 6).
Рис. 5.
Кривые течения 1.5%-ного раствора гиалуронана (1), этого же раствора с 1.5% наночастиц (2) и 5%-ной дисперсии микрочастиц в воде (3).

Рис. 6.
Частотные зависимости модуля упругости (темные точки) и модуля потерь (светлые) 1.5%-ного раствора гиалуронана (1), этого же раствора с 1.5% наночастиц (2) и 5%-ной дисперсии микрочастиц в воде (3).
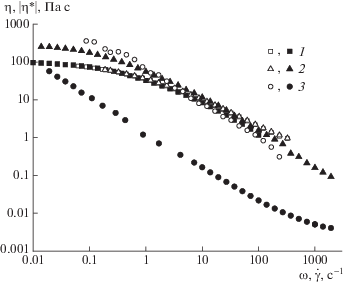
Для оценки природы вязкоупругости образцов можно использовать эмпирическое правило Кокса–Мерца, согласно которому вязкость, измеренная в условиях непрерывного сдвига, совпадает с комплексной вязкостью при численном равенстве скорости сдвига и угловой частоты. Для раствора гиалуронана и дисперсии в нем наночастиц гидроксиапатита данное правило выполняется (рис. 7), указывая на то, что вязкоупругость этих двух систем обусловлена взаимодействием макромолекул, что подтверждает роль полимера в формировании органической оболочки на поверхности частиц гидроксиапатита при синтезе. Напротив, комплексная и стационарная вязкости дисперсии микрочастиц гидроксиапатита в среде воды, не содержащей полимера, сильно различаются, поскольку вязкоупругость этой системы обусловлена коллоидной структурой взаимодействующих микрочастиц.
Рис. 7.
Зависимости вязкости η (темные точки) и комплексной вязкости |η*| (светлые) 1.5%-ного раствора гиалуронана (1), этого же раствора с 1.5% наночастиц (2) и 5%-ной водной дисперсии микрочастиц (3).
Таким образом, реологическое поведение дисперсий наночастиц подобно поведению растворов гиалуронана, тогда как дисперсия микрочастиц в отсутствие полимера является вязкопластичным гелем. Полимер стабилизирует дисперсию наночастиц не столько благодаря загущению дисперсионной среды, сколько вследствие адсорбции на частицах и связыванию их в упругую трехмерную сетку. В пользу образования сетки свидетельствует также найденное выше нестехиометрическое соотношение между гиалуронаном и катионами кальция, при котором достигается максимально заполнение координационной сферы кальция карбоксилат-анионами: несоответствие стехиометрии хорошо соотносится со способностью гиалуронана адсорбироваться одновременно на двух частицах гидроксиапатита.
Макромолекулярная сетка между наночастицами должна сохраняться при удалении дисперсионной среды и формирования из наночастиц массы, заполняющей дефектные места исцеляемых тканей. Ее наличие должно способствовать прочности и эластичности образующегося таким образом биополимерного минерального композита, структуру которого можно оценить по данным ИК-спектроскопии и энергетическим характеристикам поверхности.
ИК-спектроскопия биокомпозитов
На фрагментах ИК-спектров в области поглощения фосфат-аниона гидроксиапатита отчетливо видна разница в количестве выраженных максимумов полос поглощения РО-связей (рис. 8). Для биокомпозитов с наночастицами проявляются максимумы при 1100 и 1080 см–1, тогда как для системы с микрочастицами явно выражен только один пик при 1080 см–1. Учитывая, что ослабление поляризации связи приводит к увеличению частот колебаний, т.е. к сдвигу максимума полосы поглощения в коротковолновую область, а усиление поляризации связи, наоборот, к сдвигу максимума в длинноволновую область [43, 44], в бионанокомпозите содержание монодентатного комплекса гиалуронан–Ca2+ выше, чем в биокомпозите с микроразмерными частицами, который характеризуется в основном существованием бидентатного комплекса. Такое различие в структуре должно отражаться и на физико-химических свойствах биокомпозитов, в том числе на поверхностных свойствах пленок.

Удельная свободная поверхностная энергия биокомпозитов
Для пленок, сформированных из дисперсий как нано-, так и микрочастиц гидроксиапатита в среде гиалуронана, были определены дисперсионная и полярная составляющие удельной свободной поверхностной энергии. Различия в поверхностных характеристиках образцов, полученных при разном соотношении гидроксиапатит : гиалуронан, показаны на рис. 9.
Рис. 9.
Зависимости полярной (а, в) и дисперсионной (б, г) составляющих удельной свободной поверхностной энергии биокомпозитов от соотношения гидроксиапатит : гиалуронан в дисперсиях нано- (а, б) и микрочастиц (в, г) после 1 (темные точки) и 5 дней сушки (светлые).
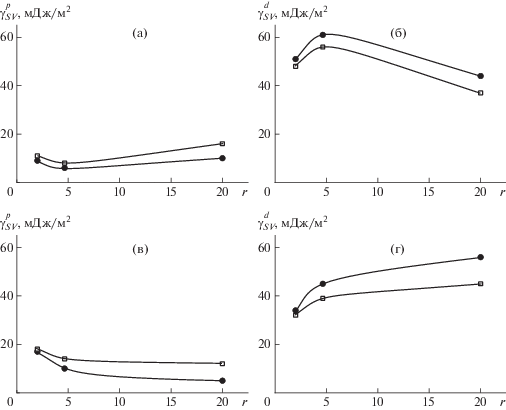
Тенденции зависимости составляющих удельной свободной поверхностной энергии от соотношения компонентов в биокомпозитах различаются для образцов с разным типом частиц и не зависят от продолжительности высушивания. Данный факт подтверждает их структурное различие. Для пленок, полученных из дисперсий наночастиц при соотношении гидроксиапатит : гиалуронан = 4.65, кривые проходят через экстремум, тогда как для пленок на основе дисперсий микрочастиц зависимости монотонны. Можно полагать, что микроразмерные частицы гидроксиапатита окружены органической оболочкой из адсорбированных макромолекул гиалуронана, и при повышении содержания полимера увеличивается лишь размер оболочки, что приводит к ее уплотнению и монотонному росту $\gamma _{{SV}}^{d}$ вследствие усиления дисперсионных взаимодействий. Логичным оказывается и снижение полярной составляющей удельной свободной поверхностной энергии на границе с воздухом: цепи гиалуронана покрывают полярную поверхность минеральных частиц, располагаясь неполярными фрагментами в направлении от их ядра, способствуя таким образом минимизации поверхностной энергии пленки.
Для пленки биополимерного композита на основе наночастиц гидроксиапатита, для которых ранее было показано существование специфических взаимодействий [22], картина другая. Дисперсионная составляющая удельной свободной поверхностной энергии после перехода через соотношение гидроксиапатит : гиалуронан = 4.65, соответствующее насыщению координационной сферы Ca2+ карбоксилат-анионами, уменьшается (происходит разуплотнение поверхностного слоя). Можно полагать, что в данном случае при синтезе гидроксиапатита были сформированы наночастицы (включающие как ядро из гидро-ксиапатита, так и оболочку из гиалуронана) определенной структуры с нестехиометрическим соотношением Ca2+ : COO–, которые сохраняют свою структуру при добавлении гиалуронана. Величина полярной составляющей при этом растет, т.е. в приповерхностном слое пленки из наночастиц гидроксиапатита содержание карбоксилат-анионов выше, чем в случае пленки из микрочастиц. Для проверки этой гипотезы был измерен ζ-потенциал частиц гидроксиапатита в дисперсиях двух типов.
Определение ζ-потенциала дисперсий
С целью нивелирования гидродинамического влияния частиц гидроксиапатита друг на друга образцы для измерения ζ-потенциала готовили разбавлением исходных дисперсий дистиллированной водой или 0.2%-ным раствором азида натрия в 10, 50 и 100 раз. В силу того, что величина и даже знак заряда ζ-потенциала коллоидных частиц зависит от их концентрации, то на примере образца с соотношением Ca2+ : COO– = 4.65 не только сравнили величины ζ-потенциала для дисперсий с разным соотношением гидроксиапатит : гиалуронан, но и проанализировали тенденции изменения ζ-потенциала в зависимости от разбавления. Результаты представлены в табл. 2 и 3.
Таблица 2.
Значения ζ-потенциала дисперсий с разным соотношением гидроксиапатита и гиалуронана
| Тип частиц | Значение ζ-потенциала (мВ) при мольном соотношении гидроксиапатит : гиалуронан | ||
|---|---|---|---|
| 1 : 20 | 1 : 4.65 | 1 : 2 | |
| Нано- | –36 ± 8 | –36 ± 9 | –35 ± 9 |
| Микро- | –23 ± 9 | –21 ± 9 | –31 ± 9 |
Таблица 3.
Значения ζ-потенциала дисперсий с соотношением гидроксиапатит/гиалуронан = 4.65 при разбавлении водой и 0.2%-ным раствором азида натрия
| Тип частиц | Наличие NaN3 | Значение ζ-потенциала (мВ) при разбавлении | ||
|---|---|---|---|---|
| в 10 раз | в 50 раз | в 100 раз | ||
| Нано- | + | –40 ± 5 | –41 ± 5 | –28 ± 4 |
| – | –57 ± 5 | –35 ± 5 | –16 ± 4 | |
| Микро- | + | –42 ± 5 | –23 ± 5 | –32 ± 6 |
| – | –49 ± 5 | –24 ± 6 | –37 ± 4 | |
Поверхностный заряд наночастиц гидроксиапатита выше по абсолютной величине заряда микрочастиц (табл. 2), что указывает на бóльшую коллоидную устойчивость образцов на основе гидроксиапатита, синтезированного в среде раствора гиалуронана. Меньший заряд микрочастиц подтверждает расположение полимерных цепей на поверхности частиц с полярными фрагментами, направленными преимущественно к их ядру. Только в случае избыточного содержания гиалуронана в дисперсии микрочастиц происходит рост абсолютной величины заряда, которая приближается к величине, характерной для наночастиц. Заряд наночастиц не зависит от соотношения полимера и минерала. Это свидетельствует о постоянной, предельной насыщенности карбоксилатными анионами поверхностного слоя этих биоминеральных частиц вне зависимости от концентрации гиалуронана.
Разбавление дисперсий раствором азида натрия не изменяет концентрацию катионов Na+ в дисперсионной среде (поскольку исходные образцы были стабилизированы этой же концентрацией консерванта в дисперсионной среде). Таким образом, концентрацию противоионов в дисперсии можно считать неизменной, как и толщину диффузного слоя частиц. В результате наноразмерные частицы сохраняют высокий заряд при разбавлении как в 10, так и в 50 раз (табл. 3), но теряют заряд при 100-кратном разбавлении, видимо, из-за частичного перехода гиалуронана с поверхности частиц в раствор. Для микрочастиц при десятикратном разбавлении значительно возрастает заряд по абсолютному значению из-за частичной десорбции полимера (ранее покрывавшего неполярными фрагментами цепей поверхность частиц), тогда как разведение системы в 50 и 100 раз снижает абсолютную величину заряда до уровня исходной дисперсии, который сопоставим с уровнем заряда сильно разбавленной дисперсии наночастиц.
Разбавление дисперсий дистиллированной водой способствует снижению концентрации противоионов, что должно приводить к увеличению толщины диффузного слоя. И действительно, десятикратное разбавление дисперсий вне зависимости от размера частиц резко повышает величину их заряда (в том числе по сравнению с зарядом частиц дисперсии, разбавленной раствором азида натрия). Однако дальнейшее разбавление вызывает падение ζ-потенциала, вероятно, в результате десорбции макромолекул гиалуронана.
Разный характер изменения поверхностного заряда нано- и микрочастиц гидроксиапатита дополнительно свидетельствует об их различном строении. Интересным фактом является и то, что величина заряда для дисперсии наночастиц практически неизменна при разбавлении до 50 раз, а у дисперсии микрочастиц она снижается в 1.5 раза, т.е. последняя система менее коагуляционно устойчива. Можно утверждать, что биокомпозитные наночастицы гидроксиапатита являются коллоидными частицами со сложно организованной структурой, которая, и это главное, сохраняется и в дисперсии при ее разбавлении как раствором гиалуронана, так и водой. Структура дисперсии традиционного строения, состоящей из микрочастиц с адсорбированным слоем полимера, зависит от концентрации гиалуронана.
ЗАКЛЮЧЕНИЕ
Создание биополимерных материалов на основе гидроксиапатита контролируемым синтезом одного компонента в активной среде другого приводит к формированию биокомпозита, в котором его составляющие находятся в наиболее биоактивной (“нативной”) форме, при этом активная среда синтеза нивелирует необходимость очистки синтезируемого вещества. Более того, только синтез частиц гидроксиапатита непосредственно в среде гиалуронана обеспечивает им устойчивость к флокуляции, инициируемой добавками макромолекул, и возможность совместного применения этих двух соединений в составе жидких препаратов медицинского назначения. При применении таких препаратов в результате мигрирования из них воды (например, под действием давления окружающих тканей вследствие ее диффузии через клеточные мембраны, непроницаемые для макромолекул и наночастиц) будет происходить формирование полимерных нанокомпозитов с матрицей из гиалуронана, армированного анизотропными наночастицами гидроксиапатита. Кроме того, поскольку устойчивость дисперсиям гидроксиапатита придает отрицательный заряд наночастиц, это может найти отдельное применение при создании препаратов, предназначенных, например, для коагуляции белка крови (фибриногена) благодаря связыванию его с отрицательно заряженной поверхностью биоминеральных наночастиц.
Работа выполнена в рамках Государственного задания ИНХС РАН.
Список литературы
Hydroxyapatite (HAp) for Biomedical Applications / Ed. by M. Mucalo. Sawston: Woodhead Publ., 2015.
Hydroxyapatite Coatings for Biomedical Applications / Ed. by S. Zhang. Boca Raton: CRC Press, 2013.
Habraken W., Habibovic P., Epple M., Bohner M. // Materials Today. 2016. V. 19. № 2. P. 69.
Williams R.J.P. // Philos. Trans. R. Soc. Lond. B. 1984. V. 304. № 1121. P. 411.
Elliott J.C. // Rev. Mineral. Geochem. 2002. V. 48. № 1. P. 427.
Rey C., Combes C., Drouet C., Sfihi H., Barroug A. // Mater. Sci. Eng. C. 2007. V. 27. № 2. P. 198.
Cai Y., Tang R. // J. Mater. Chem. 2008. V. 18. № 32. P. 3775.
Koutsopoulos S. // J. Biomed. Mater. Res. 2002. V. 62. № 4. P. 600.
Ferraz M.P., Monteiro F.J., Manuel C.M. // J. Appl. Biomater. Biomech. 2004. V. 2. № 2. P. 74.
Nayak A.K. // Int. J. Chemtech. Res. 2010. V. 2. № 2. P. 903.
Zakaria S.M., Sharif Zein S.H., Othman M.R., Yang F., Jansen J.A. // Tissue Eng. Part B Rev. 2013. V. 19. № 5. P. 431.
Beniash E. // Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2011. V. 3. № 1. P. 47.
Cazalbou S., Combes C., Eichert D., Rey C. // J. Mater. Chem. 2004. V. 14. № 14. P. 2148.
Novak B.M. // Adv. Mater. 1993. V. 5. № 6. P. 422.
Judeinstein P., Sanchez C. // J. Mater. Chem. 1996. V. 6. № 4. P. 511.
Vallet-Regí M., Arcos D. // Curr. Nanosci. 2006. V. 2. № 3. P. 179.
Costa V.C., Costa H.S., Vasconcelos W.L., Pereira M.D.M., Oréfice R.L., Mansur H.S. // Mater. Res. 2007. V. 10. № 1. P. 21.
Dessì M., Raucci M.G., Zeppetelli S., Ambrosio L. // J. Biomed. Mater. Res. Part A. 2012. V. 100. № 8. P. 2063.
Rezwan K., Chen Q.Z., Blaker J.J., Boccaccini A.R. // Biomaterials. 2006. V. 27. № 18. P. 3413.
Sanchez C., Julián B., Belleville P., Popall M. // J. Mater. Chem. 2005. V. 15. № 35–36. P. 3559.
Barinov S.M. // Russ. Chem. Rev. 2010. V. 79. № 1. P. 13.
Severin A.V., Ivanov P.L., Kostina J.V., Khabarov V.N., Kalmykova T.P., Antonov S.V. // Polymer Science B. 2016. V. 58. № 4. P. 428.
Kloubek J. // Adv. Colloid Interface Sci. 1992. V. 38. P. 99.
Melikhov I.V., Komarov V.F., Severin A.V., Bozhe-vol’nov V.E., Rudin V.N. // Dokl. Phys. Chem. 2000. V. 373. № 1–3. P. 125.
Mondek J., Kalina M., Simulescu V., Pekar M. // Polym. Degrad. Stab. 2015. V. 120. P. 107.
Litmanovich O.E. // Polymer Science C. 2008. V. 50. № 1. P. 63.
Yakimovich N.O., Smirnova L.A., Gracheva T.A., Klychkov K.S., Bityurin N.M., Aleksandrov A.P. // Polymer Science B. 2008. V. 50. № 9–10. P. 238.
Ilyin S., Roumyantseva T., Spiridonova V., Semakov A., Frenkin E., Malkin A., Kulichikhin V. // Soft Matter. 2011. V. 7. № 19. P. 9090.
Nakamoto K. Infrared and Raman Spectra of Inorganic and Coordination Compounds. New York: Wiley, 2009.
Kalashnikov V.N. // J. Rheol. 1994. V. 38. № 5. P. 1385.
Clasen C., Plog J.P., Kulicke W.M., Owens M., Macosko C., Scriven L.E., Verani M., McKinley G.H. // J. Rheol. 2006. V. 50. № 6. P. 849.
Ilyin S.O., Chernikova E.V., Kostina Y.V., Kulichikhin V.G., Malkin, A.Y. // Polymer Science A. 2015. V. 57. № 4. P. 494.
Fouissac E., Milas M., Rinaudo M. // Macromolecules. 1993. V. 26. № 25. P. 6945.
Cleland R.L., Wang J.L. // Biopolymers. 1970. V. 9. № 7. P. 799.
Ghosh S., Li X., Reed C.E., Reed W.F. // Biopolymers. 1990. V. 30. № 11–12. P. 1101.
Ilyin S.O., Kulichikhin V.G., Malkin A.Y. // Rheol. Acta. 2016. V. 55. № 3. P. 223.
Colby R.H. // Rheol. Acta. 2010. V. 49. № 5. P. 425.
Wyatt N.B., Gunther C.M., Liberatore M.W. // Polymer. 2011. V. 52. № 11. P. 2437.
Ilyin S.O., Arinina M.P., Malkin A.Y., Kulichikhin V.G. // Colloid J. 2016. V. 78. № 5. P. 608.
Brantseva T., Antonov S., Kostyuk A., Ignatenko V., Smirnova N., Korolev Y., Tereshin A., Ilyin S. // Eur. Polym. J. 2016. V. 76. P. 228.
Colloid Polym. Sci. 2017. V. 295. № 4. P. 555.
Ilyin S.O., Pupchenkov G.S., Krasheninnikov A.I., Kulichikhin V.G., Malkin A.Y. // Colloid J. 2013. V. 75. № 3. P. 267.
Bellamy L.J. The Infra-Red Spectra of Complex Molecules. London; New York: Methuen & Co. Ltd; Wiley, 1971.
Bellami L.J., Williams R.L. // J. Chem. Soc. 1957. P. 4294.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)