

Высокомолекулярные соединения (серия Б), 2020, T. 62, № 1, стр. 53-67
СИНТЕЗ И СВОЙСТВА СУЛЬФИРОВАННЫХ СОПОЛИМЕРОВ ОКСАДИАЗОЛА, ДИОКСИФЕНОКСАТИИНА И ДИФЕНИЛОКСИДА
А. Е. Ядыкова a, В. С. Ященко b, В. В. Макарова a, Ю. В. Матвеенко b, А. В. Костюк a, С. О. Ильин a, *
a Институт нефтехимического синтеза им. А.В. Топчиева Российской академии наук
119991 Москва, Ленинский пр., 29, Россия
b Институт химии новых материалов Национальной академии наук Беларуси
220141 Минск, ул. Франциска Скорины, 36, Белоруссия
* E-mail: s.o.ilyin@gmail.com
Поступила в редакцию 20.06.2019
После доработки 21.08.2019
Принята к публикации 18.09.2019
Аннотация
Получены сульфированные сополимеры 1,3,4-оксадиазола, дифенилоксида и 10,10-диоксифеноксатиина для использования в качестве полимерных суперабсорбентов. Однореакторный синтез сополимеров осуществляли в среде олеума, исходными реагентами служили 4,4'-оксидибензойная кислота и гидразин сульфат. В результате были получены сополимеры с одинаковой долей оксадиазолового фрагмента в составе цепи и различающиеся соотношением фрагментов, содержащих сульфокислотные группы: фрагментов дифенилоксида c двумя сульфокислотными группами и фрагментов диоксифеноксатиина с одной сульфокислотной группой и одной сульфонильной группой. Сополимеры оказались способными к стократному набуханию в водной среде и ограниченно в ней растворялись при добавлении полярных апротонных сорастворителей или повышении pH. Соотношение сульфокислотных и сульфонильных групп в составе сополимера влияло на его набухание в воде и вязкоупругость гидрогелей, находящихся в мезофазном состоянии при высоком содержании сополимера.
ВВЕДЕНИЕ
Сульфированные полимеры представляют интерес для создания протонпроводящих мембран [1, 2] и применения в качестве суперабсорбентов [3, 4]. Последнее обусловлено особенностью сульфированных полимеров – их высокой гидрофильностью. Для протонпроводящих мембран, эксплуатируемых при температуре ниже 100°C, высокая гигроскопичность может являться недостатком [5]. Однако она же может стать достоинством, если рассматривать такие полимеры для применения в качестве суперабсорбентов. Кроме того, благодаря наличию анионных групп имеется и еще одна область использования таких материалов – как суперадсорбентов.
Полимерные суперадсорбенты применяют для извлечения из водной среды красителей [6–9], катионов тяжелых металлов [10–14] и минерального масла [15–17]. Полимерные суперабсорбенты обычно получают на основе полиакриламида или солей полиакриловой кислоты [18, 19], которые формируют в воде гидрогели. С одной стороны, обычные области приложения суперабсорбентов (средства гигиены, медицина [20, 21], добыча нефти [22], сельское хозяйство [23]) не предусматривают необходимость в высокой термостойкости макромолекул. Однако, с другой стороны, хорошая термостабильность открывает возможность для их многократного применения. Например, для проведения очистки загрязненных вод с использованием гидрогеля, который после применения может быть извлечен, регенерирован, высушен и использован вновь.
Для многократного применения сульфированных полимеров с целью удаления красителей и ионов металлов из водных сред важна их термостабильность. Хорошей долговечности полимерных абсорбентов в условиях высоких температур добиваются синтезом перфторированных углеводородов с концевыми сульфокислотными группами в основной цепи и ее боковых ответвлениях [24] и сульфированием жесткоцепных полимеров [25]. Росту термостойкости также способствует введение в состав макромолекулярной цепи ароматических фрагментов, в том числе гетероциклических группировок ароматического характера, в частности фрагментов дифенилоксида и 1,3,4-оксадиазола [26].
Синтез сополимеров 1,3,4-оксадиазола можно осуществлять однореакторным методом с использованием реакции поликонденсации ароматической дикарбоновой кислоты с гидразин сульфатом в дымящей серной кислоте [27]. Кроме того, ранее была показана возможность однореакторного синтеза сульфированных сополимеров, содержащих фрагменты п-фенилен-1,3,4-оксадиазола, 10,10-диоксифеноксатиина и дифенилоксида с использованием в качестве реагентов 4,4'-оксидибензойной и терефталевой кислот, гидразин сульфата и олеума [28].
Цель настоящей работы – однореакторный синтез сульфированных сополимеров, содержащих фрагменты 1,3,4-оксадиазола, 10,10-диоксифеноксатиина и дифенилоксида, пригодных к использованию в качестве суперабсорбентов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы
Для синтеза сополимеров использовали 4,4'-оксидибензойную кислоту (ОДБК, “ChemicalPoint”, Германия), 98.5%-ный гидразин сульфат (Китай) и 20%-ный олеум (Россия). Растворителями служили дистиллированная вода, диметилсульфоксид (“Panreac”, Испания) и тетрагидрофуран (“Компонент-реактив”, Россия). Все реактивы были химически чистыми и применяли без проведения дополнительной очистки. Синтезировали сополимеры, содержащие только фрагменты 1,3,4-оксадиазола и дифенилоксида (образец S0), фрагменты 1,3,4-оксадиазола и 10,10-диоксифеноксатиина (S100) и содержащие все три фрагмента одновременно (S25, S50 и S75). В последнем случае соотношение между фрагментами дифенилоксида и диоксифеноксатиина варьировали от 25 до 75 мас. %. Во всех случаях содержание оксадиазольных фрагментов в сополимерах составляло 50 мол. %. Таким образом, число в условном обозначении образцов равно массовой концентрации звеньев, включающих фрагмент диоксифеноксатиина и фрагмент оксадиазола:
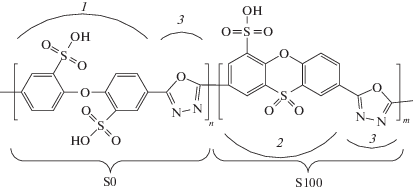
Растворы и гели сополимеров изучали в воде, водном растворе NaOH с концентрацией 0.003 моль/л (pH 14.5), 33%-ном водном растворе ТГФ, а также в 20–40%-ных водных растворах ДМСО. Растворы приготавливали посредством перемешивания компонентов на магнитной мешалке при 75°C не менее 24 ч, гели готовили при набухании полимера при 75°C в течение недели.
Методы
Cпектры ЯМР 1Н записывали на спектрометре “Avance-500” (“Bruker”, США) в средах дейтерированных диметилсульфоксида (DMSO-d6) и серной кислоты (D2SO4) при частоте 500 МГц, внутренний стандарт тетраметилсилан.
Поведение сополимеров при контакте с водой изучали методом лазерной интерферометрии, который позволяет определить концентрационный профиль диффундирующего вещества в области диффузионной зоны, оценить скорость его изменения и тем самым охарактеризовать взаимодиффузию полимера и растворителя и рассчитать их равновесную концентрацию на границе раздела фаз в случае их ограниченной совместимости [29–32]. Для этого пленку полимера размером 5 × 5 × 0.1 мм помещали между двумя стеклами 20 × 15 × 4 мм, на поверхность которых была нанесена методом вакуумного напыления полупроницаемая пленка из золота толщиной 20 нм. С помощью фторопластовой пленки толщиной около 150 мкм, помещаемой между стеклами с одной из сторон, между стеклами создавали клиновидный зазор. В результате при пропускании света через стекла на их покрытой золотом поверхности возникала интерференционная картина, которая зависела от показателя преломления среды между стеклами. Подробное описание методики было изложено ранее [33, 34]. Микрофотографии диффузионных зон получали с использованием оптического микроскопа с объективом, обеспечивающим увеличение в 3.5 раза, и цифровой камеры с матрицей “Sony IMX226”, имеющей размер 1/1.7'' и разрешение 12 мегапикселей. Источником света служил диодный лазер с длиной волны 532 нм.
Для оценки набухания сополимеров их предварительно высушенные пленки помещали в воду на 24 ч при 25°C, затем извлекали, аккуратно снимали влагу с поверхности фильтровальной бумагой и взвешивали, после чего отжимали между листами фильтровальной бумаги, и снова взвешивали. За изменением линейной длины пленки полимера размером 5 × 5 × 0.1 мм, помещенной между двумя препаратными стеклами и приведенной в контакт с водой, следили при помощи оптического микроскопа.
Реологические исследования выполняли на ротационном реометре DHR-2 (“TA Instruments”, США), для гелей использовали измерительный узел плоскость–плоскость (диаметр плоскостей 8 мм, расстояние между плоскостями 500 мкм), а для растворов конус–плоскость (диаметр плоскости 40 мм, угол между образующей конуса и плоскостью 2°). Кривые течения образцов получали посредством ступенчатого повышения скорости сдвига в диапазоне 10–4–103 с–1, время измерения вязкости при каждой скорости сдвига составляло не менее 1 мин. Линейную вязкоупругость образцов исследовали при амплитуде относительной деформации 0.1% и варьировании угловой частоты в интервале 0.0628–628 с–1. Температурные зависимости модулей упругости и потерь получали со скоростью нагревания образца 5 град/мин. Относительная погрешность определения реологических характеристик не превышала 5%.
Термограммы сополимеров регистрировали на дифференциальном сканирующем калориметре MDSC 2920 (“TA Instruments”, США) в среде аргона со скоростью повышения температуры 10 град/мин.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Синтез мономера
Для синтеза 2,8-дикарбокси-10,10-диоксо-10λ6-феноксатиин-4-сульфоната натрия в одногорлую колбу объемом 50 мл, снабженную магнитной мешалкой и обратным холодильником, поместили 2 г (7.75 ммоля) ОДБК и 20 мл олеума. Реакционную смесь перемешивали при 120°С в течение 3.5 ч, затем охладили и добавили избыток насыщенного раствора NaCl, после чего выпал осадок белого цвета. Схема реакции приведена ниже.

Невысокий выход продукта обусловлен хорошей растворимостью натриевой соли сульфокислоты в воде, что затрудняет ее выделение. Проведение реакции при более низких температурах приводило к смеси продуктов различной степени замещения, выделение и идентификация которых была затруднена вследствие хорошей растворимости в воде.
Полученное вещество отфильтровали, дважды промыли ледяной водой (2 × 10 мл) и высушили в эксикаторе в присутствии хлорида кальция. Выход соединения, представляющего собой белые кристаллы с Tm > 310°С (разл.) составил 1.15 г (35%). Спектр ЯМР 1Н (500 МГц, DMSO-d6), δ, м.д. (J, Гц) (рис. 1): 7.78 (1Н, д, J = 9, Н-6), 8.37 (1Н, дд, J1 = 9, J2 = 2, Н-7), 8.44 (1Н, д, J = 2, Ar-H), 8.48 (1Н, д, J = 2, Ar–H), 8.64 (1Н, д, J = 2, Ar–H). Наличие трех однопротонных дублетов дальнего порядка с малой (J = 2 Гц) константой спин-спинового взаимодействия, а также дублета дублетов (J1 = 9 и J2 = 2 Гц) и дублета (J = 9 Гц), позволяет однозначно определить положение сульфогруппы в феноксатиине.

Синтез сополимеров
Бинарный сополимер, содержащий фрагменты 10,10-диоксифеноксатиина и 1,3,4-оксадиазола (S100), получали однореакторным методом. ОДБК прогревали в 50 мл олеума при 120°C в течение 2 ч для получения олеумного раствора 10,10-диоксо-4-(сульфо)-10λ6-феноксатиин-2,8-дикарбоной кислоты согласно приведенной выше схеме. В полученный раствор после охлаждения до 50°C добавляли гидразин сульфат (использовали 20%-ный избыток) и проводили реакцию поликонденсации при 120°С.
Для получения бинарного сополимера с фрагментами дифенилоксида и 1,3,4-оксадиазола (S0) гидразин сульфат (20%-ный избыток) и ОДБК растворяли в 50 мл олеума при 50°C, затем температуру плавно поднимали до 120°C для проведения поликонденсации.
Синтез тройных сополимеров, содержащих одновременно фрагменты дифенилоксида, диоксифеноксатиина и оксадиазола, осуществляли следующим способом. Первоначально ОДБК прогревали в олеуме при температуре 120°С в течение 2 ч. В этих условиях образовывался раствор 10,10-диоксо-4-(сульфо)-10λ6-феноксатиин-2,8-дикарбоной кислоты в олеуме, который затем охлаждали до 50°C. Далее в него добавляли новую порцию ОДБК и гидразин сульфат, а затем проводили поликонденсацию при 120°C.
Соотношение мономеров во всех случаях рассчитывали таким образом, чтобы концентрация полимера в полученном растворе составляла 5–6%. Завершенность процесса определяли по изменению нагрузки на лопасти перемешивающего устройства. Реакцию останавливали при одном и том же значении нагрузки для всех получаемых полимеров одновременным охлаждением и разбавлением 96%-ной серной кислотой, количество которой рассчитывали таким образом, чтобы присутствующая в серной кислоте вода связала избыток SO3. Полученные сополимеры осаждали в раствор гидрокарбоната натрия и затем промывали водой для удаления остатков солей. Сушку образцов выполняли в эскикаторе над щелочью в течение недели.
Образцы представляли собой нерегулярные сополимеры с разной массовой долей дифенилоксидных и диоксифеноксатииновых звеньев. Характеристическая вязкость полимеров была примерно одинаковой и составляла около 1.01–1.19 дл/г в серной кислоте при 25°С. Различие в строении сополимеров проявлялось при анализе спектров ЯМР 1Н их растворов в дейтерированной серной кислоте. В спектре образца S0 имеется три группы сигналов протонов дифенилоксидного фрагмента сополимера. Протоны, находящиеся в орто-положении к сульфогруппам, проявляются в виде двухпротоного дублета при 10.0 м.д. Уширенный двухпротонный синглет в области 8.68 м.д. отвечает орто-протонам при эфирной группе, а двухпротонный уширенный синглет в области 9.67 м.д. относится к двум протонам звена ОБСФ, находящихся в пара-положении к сульфогруппе в ароматическом кольце (рис. 2а).

В случае сополимера S100 спектр ЯМР 1Н имеет более сложный характер, однако дает возможность идентифицировать сигналы всех пяти протонов диоксифеноксатииновых фрагментов (рис. 2б). Так, сигналы трех протонов, находящихся в орто-положении к электроноакцепторным группам, проявляются в более слабом поле в виде двух мультиплетов при 10.42–10.35 и 10.29–10.16 м.д. Уширенные синглеты при 9.78 и 9.13 м.д. относятся к оставшимся двум протонам феноксатиинового гетероцикла.
Набухание сополимеров
Согласно данным лазерной интерферометрии при комнатной температуре, все сополимеры ограниченно совместимы с водой: на всех интерферограммах присутствует фазовая граница (рис. 3a, 4а). Сильные загибы полос со стороны полимера свидетельствуют об интенсивном проникновении в него воды, т.е. о процессе набухания. По консистенции набухшая масса полимера представляла собой гель. При этом в области воды искривления полос очень слабые, что указывает на крайне незначительную растворимость полимеров.
Рис. 3.
Микрофотографии зон взаимной диффузии сополимера S0 и воды при 26 (a) и 47°C (б, в). Фотографии получены в условиях освещения зеленым лазером (а, б) и в скрещенных поляроидах и белом свете (в).

Рис. 4.
Микрофотографии зон взаимной диффузии сополимера S100 и воды при 26 (a) и 80°C (б, в). Фотографии получены в условиях освещения зеленым лазером (а, б) и в скрещенных поляроидах и белом свете (в).
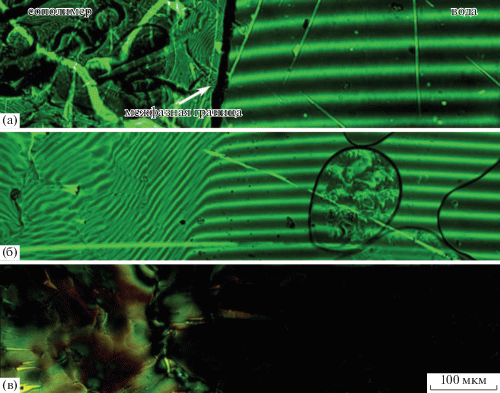
Фазовая граница исчезает при температуре выше 47°С (рис. 3б, 4б), что предполагает полную растворимость сополимеров при высоких температурах. Тем не менее, построить фазовые диаграммы не представляется возможным, поскольку количество интерференционных полос интенсивно меняется как при повышении температуры, так и во времени из-за набухания и упругости полимерных пленок, приводящих к непостоянству толщины зазора между стеклами в диффузионной ячейке.
Анализ диффузионной зоны в скрещенных поляроидах как в свете лазера, так и в белом свете показывает наличие жидкокристаллической фазы в водных растворах (рис. 3в, 4в). При этом из-за радужной окраски системы S0–вода можно предположить формирование холестерической мезофазы, тогда как микрофотография системы S100–вода соответствует наличию в ней обычной нематической фазы. Формирование мезофазы в воде различными полиэлектролитами, в том числе сульфированными, является обычным событием и объясняется снижением молекулярной подвижности и изменением конформации цепей из-за взаимодействия молекул растворителя с ионными группами полимера [35]. Как правило, образование мезофазы происходит при содержании полиэлектролита 25–77%. В данном же случае жидкокристаллическое состояние проявляется при содержании сополимера в воде выше 15%; вероятно, меньшая концентрация обусловлена жесткоцепным строением сополимеров.
Сополимеры проявляют свойства суперабсорбентов. При контакте с водой они начинают быстро набухать, что сопровождается увеличением их линейных размеров Δl. Это позволяет оценить коэффициент диффузии воды в полимер согласно соотношению [33, 36]
где t – время, а коэффициент α принимает значение, равное шести в случае полимера, растворяющегося в процессе диффузии, и равное π для ограниченно набухающих систем, к которым относятся данные полимеры.Действительно, зависимости изменения длины пленок линеаризуются в координатах Δl – t0.5 (рис. 5), что позволяет по тангенсу угла наклона прямых рассчитать коэффициент диффузии воды (табл. 1). Быстрее всего водой насыщаются полимеры с бόльшим количеством сульфокислотных групп. Бинарный сополимер с фрагментами дифенилоксида набухает в 10 раз быстрее, чем бинарный сополимер, содержащий фрагменты 10,10-диоксифеноксатиина. При этом зависимость коэффициента диффузии от содержания фрагментов дифенилоксида в сополимере нелинейная: линейной является зависимость логарифма коэффициента диффузии.
Рис. 5.
Изменение длины пленки сополимера S0 (1), S25 (2), S50 (3), S75 (4) и S100 (5), помещенной между двумя препаратными стеклами и приведенной в контакт с полубесконечным количеством воды.

Таблица 1.
Некоторые характеристики сополимеров
| Образец | Коэффициент диффузии воды D × 1011, м2/с | Предельное набухание сополимеров m/m0, г/г |
|---|---|---|
| S100 | 1.1 | 100.9 |
| S75 | 2.4 | 120.3 |
| S50 | 3.2 | 119.1 |
| S25 | 4.7 | 98.7 |
| S0 | 10 | 82.8 |
Нелинейной оказалась зависимость предельной степени набухания сополимеров от соотношения звеньев, которую оценивали как отношение массы полимера до и после набухания m/m0 (табл. 1). Несмотря на высокую скорость набухания, сополимер S0 набирает меньше воды по сравнению с другими образцами; тем не менее, его масса почти в 83 раза возрастает, что соответствует содержанию полимера в набухшем геле около 1.2%. По консистенции гель данного полимера мягкий, не способный к поддержанию формы. Напротив, масса сополимера S100 увеличивается при набухании в 101 раз, а гель является крепким, поддерживающим свою форму. Тройные сополимеры набухают в 100–120 раз с образованием гелей промежуточной консистенции. При сдавливании набухших гелей между листами фильтровальной бумаги они отдают воду, но не всю: остаточное содержание воды в отжатых гелях вне зависимости от строения полимера составляет около 45–55%.
Предельное набухание сополимеров меньше характерного для несшитых гидрогелей полиакриловой (m/m0 = 300–820) и полиметакриловой кислот (210) [37, 38], а также сополимеров акриловых кислот с акриламидом (10–800) [39, 40]. Оно сопоставимо с набуханием сшитой полиакриловой кислоты (36–300 в зависимости от сшивателя и плотности сшивки) [37, 41] и существенно превосходит значения, присущие гелям сшитого полиакриламида (8–56) [42, 43], сшитой полистиролсульфоновой кислоты (37–66) [44], ее сополимеров (4–45) [45] и ионообменных смол на основе сульфированного полистирола (0.9–2.1) [46, 47].
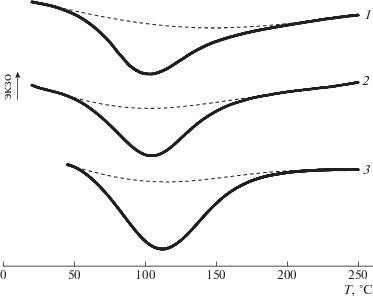
Сополимеры являются крайне гигроскопичными материалами и легко набирают влагу даже из сухого воздуха. На термограммах сополимеров при их нагревании в области 50–200°С обнаруживается эндотермический пик (рис. 6), соответствующий потере связанной воды. Согласно тепловому эффекту, бинарный сополимер с фрагментами диоксифеноксатиина набирает в 2 раза больше воды (ΔHS100 = 360 Дж/г, T = 112°C, cH2O = = 13.6 мас. %), чем второй бинарный сополимер (ΔHS0 = 131 Дж/г, T = 99°C, cH2O = 5.8 мас. %), и отдает ее при более высокой температуре. Связь между содержанием мономерных звеньев в цепи и гигроскопичностью нелинейна: сополимер с равным содержанием диоксифеноксатииновых и дифенилоксидных фрагментов набирает меньше воды, чем в случае аддитивного влияния звеньев (ΔHS50 = 149 Дж/г, T = 106°C, cH2O = 6.6 мас. %). Таким образом, фрагменты дифенилоксида снижают гигроскопичность, возможно, из-за формирования внутри- и межмолекулярных водородных связей между сульфонильными и сульфокислотными группами макромолекул. Образование таких связей оказывается более энергетически выгодным по сравнению с образованием связей с водой. По уровню гигроскопичности сополимеры схожи с глицерином (cH2O = 8% при 25°C и относительной влажности 20%) [48] и ДМСО (16%) [49].
Реология растворов и гелей
Все сополимеры в водной среде образуют гели (набухшие массы полимера): их модуль упругости практически не зависит от частоты деформирования и превышает модуль потерь (рис. 7а). Бинарный сополимер с фрагментами дифенилоксида образует слабый гель, в тысячу раз менее упругий по сравнению с прочими. Частотные зависимости модуля потерь для остальных гелей на основе полимеров, содержащих фрагменты диоксифеноксатиина, одинаковы, а их модуль упругости возрастает с увеличением содержания этого фрагмента. Зависимость модуля упругости от соотношения между мономерными звеньями в составе цепи линеаризуется в полулогарифмических координатах (рис. 7б), причем экстраполяция логарифма модуля упругости к нулевому содержанию диоксифеноксатииновых фрагментов дает величину модуля намного большую по сравнению с измеряемой экспериментально. Таким образом, наличие фрагментов диоксифеноксатиина обусловливает качественно иной принцип гелеобразования, заключающийся в их взаимодействии между собой. Чем больше этих фрагментов, тем больше число узлов сетки геля в единице объема и тем выше его упругость (по абсолютной величине модуля гели сополимеров более чем на десятичный порядок превосходят гели полиакриловой кислоты [50]). Дифенилоксидные фрагменты более гидрофильны благодаря вхождению в их состав одновременно двух сульфокислотных групп, наличие которых тоже приводит к специфическому межцепному взаимодействию (хотя и более слабому) и гелеобразованию: об этом свидетельствуют зависимости G'(ω) и G''(ω) системы вода–S0 и нелинейная зависимость G' от содержания фрагментов диоксифеноксатиина.
Рис. 7.
Частотные зависимости модуля упругости G' (темные точки) и модуля потерь G'' (светлые) (а), а также зависимость модуля упругости от содержания фрагментов диоксифеноксатиина (б) для гидрогелей сополимеров S0 (1), S25 (2), S50 (3), S75 (4) и S100 (5) с концентрацией 5 г/дл при 25°C.

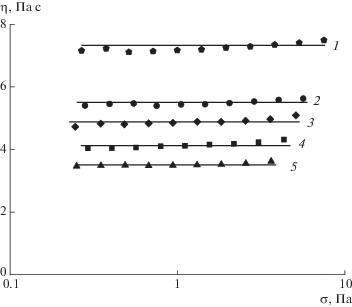
Растворимость сополимеров при нормальных условиях в воде крайне низка. Добавление к гелям щелочи (до pH 11.5) способствует замещению протонов сульфокислотных групп полимеров катионами натрия. В результате полимеры образуют растворы, а не гели, но их растворимость остается ограниченной и составляет около 7–8 г/дл. Иными словами, перевод макромолекул в анионную форму позволяет получить только разбавленные растворы, не проявляющие вязкоупругость. Вязкость таких растворов не зависит от напряжения сдвига (рис. 8): они являются ньютоновскими жидкостями. При этом бóльшая вязкость характерна для раствора бинарного сополимера с дифенилоксидными фрагментами, вероятно, из-за более высокого содержания в нем анионных групп, которые способствуют разворачиванию цепей полимера, приводя к большему объему, занимаемому макромолекулами. С понижением содержания сульфонатных групп в составе цепи вязкость растворов уменьшается.
Рис. 8.
Кривые течения растворов сополимера S0 (1), S25 (2), S50 (3), S75 (4) и S100 (5) с концентрацией 5 г/дл в среде 0.003 М водного раствора гидроксида натрия с pH 11.5 при 25°C.
Возможность осуществления золь-гель-перехода изменением уровня pH является преимуществом данных образцов перед другими сульфированными полимерами, которые применяют, как правило, в сшитом виде и перевод которых в солевую форму не придает им растворимости, а только несколько снижает их предельное набухание из-за худшей гидратации катионов металлов [44, 46, 47, 51]. Для придания растворимости сополимерам достаточно перевести небольшую часть их сульфокислотных групп в сульфонатную форму: в нашем случае, исходя из количества использованной щелочи, не более 2.4% сульфокислотных групп сополимера S0 и вдвое меньшее количества групп сополимера S100. Вероятно, это связано с хорошей диссоциацией сульфокислотных групп даже в изначально протонированном состоянии (действительно, органические сульфокислоты являются сильными, например, pKa бензолсульфокислоты равен –2.8 [52]).
Другой способ улучшить растворимость сополимеров – добавление к воде полярных апротонных сорастворителей, например ТГФ или более полярного ДМСО, но в количестве не более 40–45 об. % (ни ДМСО, ни ТГФ, ни другие индивидуальные жидкости за исключением серной кислоты не являются растворителями сополимеров).
Добавление тетрагидрофурана к гидрогелям не приводит к формированию из них растворов (за исключением слабого геля сополимера S0), но способствует снижению компонент комплексного модуля тем сильнее, чем больше дифенилоксидных фрагментов в цепи сополимера (рис. 9a). Так, модуль упругости сополимера S100 остается практически неизменным, тогда как для сополимера S25 он снижается в 100 раз.
Рис. 9.
Частотные зависимости модуля упругости G ' (темные точки) и модуля потерь G '' (светлые) (а) и кривые течения (б) систем, содержащих 5 г/дл сополимера S0 (1), S25 (2), S50 (3), S75 (4) и S100 (5) в среде 33%-ного водного раствора ТГФ при 25°C.
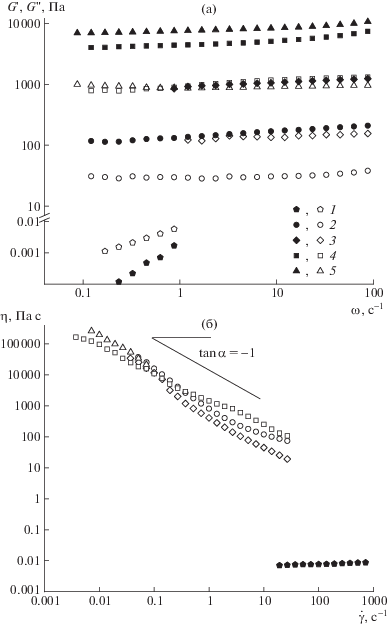
Кривые течения гелей представляют собой близкие к линейным зависимости, тангенс угла наклона которых равен примерно –1 (рис. 9б). Это означает, что вне зависимости от скорости сдвига образцы текут при одном и том же напряжении – пределе текучести, который характеризует прочность структуры геля. В то же время строение макромолекул и вязкоупругость геля не оказывают заметного влияния на его прочность. Бинарный сополимер с фрагментами дифенилоксида в водном растворе ТГФ формирует раствор, который является вязкоупругой ньютоновской жидкостью, подчиняющейся модели Максвелла – G' ~ ω2 и G'' ~ ω. Таким образом, введение ТГФ в воду улучшает сольватирование дифенилоксидных фрагментов макромолекул, но не препятствует ассоциации фрагментов диоксифеноксатиина.
Добавление ДМСО к гидрогелям действует схожим образом: по мере повышения содержания ДМСО вязкоупругость геля немного снижается (рис. 10a); однако содержание ДМСО более 45–50 об. % инициирует фазовый распад (осаждение полимера). Рост температуры приводит к снижению значений компонент комплексного модуля (рис. 11a), причем в случае использования 30–40%-ного водного раствора ДМСО падение модулей более существенно. В случае повышения температуры до 95°C образцы с низким содержанием ДМСО остаются гелями (для них G ' > G '', рис. 10б), а с более высоким – переходят в состояние структурированных растворов. Модуль упругости таких растворов при низких угловых частотах принимает постоянное значение и превышает модуль потерь, тогда как при увеличении частоты оба модуля возрастают и принимают примерно равное значение. По-видимому, структурированность растворов обусловлена ассоциацией фрагментов диоксифеноксатиина, которая также приводит к неньютоновскому течению образцов (рис. 11б). Для структурированных растворов характерно наличие области течения с постоянной относительно высокой вязкостью при низких напряжениях. Затем по мере повышения напряжения происходит разрушение макромолекулярных ассоциатов, сопровождающееся уменьшением вязкости до постоянного уровня. Напряжение, вызывающее резкое снижение вязкости, характеризует прочность ассоциатов и может быть трактовано как предел текучести. Похожее неньютоновское поведение, а также характер зависимостей G '(ω) и G ''(ω), проявляют и другие структурирующиеся растворы, например, поли-акрилонитрила [53–55] и сополимера винилацетата и винилового спирта [56].
Рис. 10.
Частотные зависимости модулей упругости (темные точки) и потерь (светлые) образцов, содержащих 5 г/дл сополимера S25 в среде водного раствора ДМСО с концентрацией 0 (1), 20 (2), 30 (3) и 40 об. % (4) при 25 (а) и 95°C (б).

Рис. 11.
Температурные зависимости модуля упругости (темные точки) и модуля потерь (светлые) (а), а также кривые течения (б) систем, содержащих 5 г/дл сополимера S25 в среде водного раствора ДМСО с концентрацией 0 (1), 20 (2), 30 (3) и 40 об. % (4).
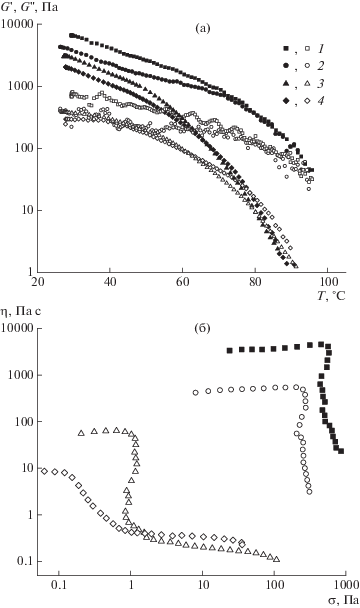
Для гелей сополимера также характерно наличие области низких напряжений, при которых экспериментально наблюдается деформирование гелей с постоянной высокой эффективной вязкостью (рис. 11б), а также предела текучести, при достижении которого структура геля разрушается [57]. Различие структурированных растворов и гелей состоит в том, что первые способны к течению при высоких напряжениях сдвига, тогда как вторые при таких условиях необратимо разрушаются.
ЗАКЛЮЧЕНИЕ
Высокая гигроскопичность и наличие многочисленных анионных групп определяют возможность приложения сульфированных сополимеров 1,3,4-оксадиазола, дифенилоксида и 10,10-диоксифеноксатиина для связывания воды или многовалентных катионов. Данные сополимеры имеют ряд преимуществ перед другими полимерами схожего назначения. Прежде всего их можно синтезировать из доступных реагентов однореакторным методом без проведения стадии сульфирования. По уровню влагопоглощения сополимеры уступают только несшитой полиакриловой кислоте и ее производным, но благодаря жесткости цепей формируют более упругие гели и характеризуются высокой термостойкостью (начало разложения при 460–475°C). Линейное строение сополимеров позволяет их перерабатывать через растворы, возможности чего лишены наиболее распространенные абсорбенты и ионообменные смолы на основе сшитых полимеров. Круг растворителей сильно ограничен, что означает стойкость сополимеров к органическим средам. Все это в совокупности делает данные полимеры пригодными для применения в качестве суперсорбентов, способных к длительной эксплуатации в условиях многократно повторяющегося цикла набора–отдачи воды.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (код проекта 17-53-04002 Бел_мол_а) и Белорусского республиканского фонда фундаментальных исследований (грант X17PM-013).
Список литературы
Bose S., Kuila T., Nguyen T.X.H., Kim N.H., La K.T., Lee J.H. // Prog. Polym. Sci. 2011. V. 36. № 6. P. 813.
Gao H., Lian K. // RSC Adv. 2014. V. 4. № 62. P. 33091.
Kamal M.S., Sultan A.S., Al-Mubaiyedh U.A., Hussein I.A. // Polym. Rev. 2015. V. 55. № 3. P.491.
Staple T.L., Chatterjee P.K. // Elsevier. 2002. V. 13. P. 283.
Li H., Tang Y., Wang Z., Shi Z., Wu S., Song D., Liu Z. // J. Power Sources. 2008. V. 178. № 1. P. 103.
Dhodapkar R., Rao N.N., Pande S.P., Nandy T., Devotta S. // React. Funct. Polym. 2007. V. 67. № 6. P. 540.
Sivashankar R., Sathya A.B., Vasantharaj K., Sivasubramanian V. // Environ. Nanotechnol. Monit. Manage. 2014. V. 1. P. 36.
Hu X. S., Liang R., Sun G. // J. Mater. Chem. A. 2018. V. 6. № 36. P. 17612.
Chen D., Wang L., Ma Y., Yang W. // NPG Asia Mater. 2016. V. 8. № 8. P. 301.
Wang X., Lu S., Chen L., Li J., Dai S., Wang X. // J. Radioanal. Nucl. Chem. 2015. V. 306. № 2. P. 497.
Zhao G., Zhang H., Fan Q., Ren X., Li J., Chen Y., Wang X. // J. Hazard. Mater. 2010. V. 173. № 1–3. P. 661.
Shao D., Hou G., Li J., Wen T., Ren X., Wang X. // Chem. Eng. J. 2014. V. 255. P. 604.
Hayati B., Maleki A., Najafi F., Daraei H., Gharibi F., McKay G. // J. Mol. Liq. 2016. V. 224. P. 1032.
Manatunga D.C., de Silva R.M., de Silva K.N., Ratnaweera R. // RSC Adv. 2016. V. 6. № 107. P. 105618.
Rao K.V., Mohapatra S., Maji T.K., George S.J. // Chem. Eur. J. 2012. V. 18. № 15. V. 4505.
Ono T., Sugimoto T., Shinkai S., Sada K. // Nat. Mater. 2007. V. 6. № 6. P. 429.
Jin H.X., Dong B., Wu B., Zhou M.H. // Polym. Plast. Technol. Eng. 2012. V. 51. № 2. P. 154.
Zohuriaan-Mehr M.J., Kabiri K. // Iran. Polym. J. 2008. V. 17. № 6. P. 451.
Kabiri K., Omidian H., Zohuriaan-Mehr M.J., Doroudiani S. // Polym. Compos. 2011. V. 32. № 2. P. 277.
Sannino A., Esposito A., Rosa A.D., Cozzolino A., Ambrosio L., Nicolais L. // J. Biomed. Mater. Res. A. 2003. V. 67. № 3. P. 1016.
Ilyin S.O., Kulichikhin V.G., Malkin A.Y. // Rheol. Acta. 2016. V. 55. № 3. P. 223.
Abdulbaki M., Huh C., Sepehrnoori K., Delshad M., Varavei A. // J. Petrol. Sci. Eng. 2014. V. 122. P. 741.
Zohuriaan-Mehr M.J., Omidian H., Doroudiani S., Kabiri K. // J. Mater. Sci. 2010. V. 45. № 21. P. 5711.
Rikukawa M., Sanui K. // Prog. Polym. Sci. 2000. V. 25. № 10. P. 1463.
Roziere J., Jones D.J. // Annu. Rev. Mater. Res. 2003. V. 33. № 1. P. 503.
Korshak V.V., Vinogradov S.V. // Russ. Chem. Rev. 1968. V. 37. № 11. P. 885.
Janietz S., Schulz B. // Eur. Polym. J. 1996. V. 32. № 4. P. 465–474.
Yashchenko V.S., Pap A.A., Matveenko Y.V., Ol’khovik V.K. // Polymer Science B. 2016. V. 58. № 5. P. 529.
Bairamov D.F., Chalykh A.E., Feldstein M.M., Siegel R.A., Platé N.A. // J. Appl. Polym. Sci. 2002. V. 85. № 5. P. 1128.
Mráček A. // Int. J. Mol. Sci. 2010. V. 11. P. 532.
Bayramov D.F., Singh P., Cleary G.W., Siegel R.A., Chalykh A.E., Feldstein M.M. // Polym. Int. 2008. V. 57. № 5. P. 785.
Ilyin S.O., Makarova V.V., Anokhina T.S., Ignatenko V.Y., Brantseva T.V., Volkov A.V., Antonov S.V. // Cellulose. 2018. V. 25. № 4. P. 2515.
Malkin A., Askadsky A., Chalykh A., Kovriga V. Experimental Methods of Polymer Physics. Moscow: Mir, 1983.
Makarova V., Kulichikhin V. In Interferometry. Research and Applications in Science and Technology; Ed. by I. Padron. InTech: Rijeka, 2012. P. 395.
Hatakeyama H., Hatakeyama T. // Thermochim. Acta. 1998. V. 308. № 1–2. P. 3.
Mráček A., Benešová K., Minařík A., Urban P., Lapčík L. // J. Biomed. Mater. Res. A. 2007. V. 83. № 1. P. 184.
Kabiri K., Omidian H., Hashemi S.A. Zohuriaan-Mehr M.J. // Eur. Polym. J. 2003. V. 39. № 7. P. 1341
Philippova O.E. // Polymer Science C. 2000. V. 42. № 2. P. 208.
Omidian H., Hashemi S.A., Sammes P.G., Meldrum I. // Polymer. 1998. V. 39. № 26. P. 6697.
Shabadrov P.A., Safronov A.P. // Polymer Science A. 2018. V. 60. № 5. P. 628.
Zhang S., Chen H., Liu S., Guo J. // Energy Fuels. 2017. V. 31. № 2. P. 1825.
Karadaǧ E., Üzüm Ö.B., Saraydin D. // Eur. Polym. J. 2002. V. 38. № 11. P. 2133.
Philippova O.E., Zaroslov Y.D., Khokhlov A.R., Wegner G. // Macromol. Symp. 2003. V. 200. № 1. P. 45.
Xu L., Li X., Zhai M., Huang L., Peng J., Li J., Wei G. // J. Phys. Chem. B. 2007. V. 111. № 13. P. 3391.
Valencia J., Piérola I.F. // Eur. Polym. J. 2001. V. 37. № 12. P. 2345.
Toteja R.S.D., Jangida B.L., Sundaresan M., Venkataramani B. // Langmuir. 1997. V. 13. № 11. P. 2980.
Van Wart H.E., Janauer G.E. // J. Phys. Chem. 1974. V. 78. № 4. P. 411.
Miner C.S., Dalton N.N. Glycerol. New York: Reinhold Publ. Corp., 1953.
LeBel R.G., Goring D.A.I. // J. Chem. Eng. Data. 1962. V. 7. № 1. P. 100.
Kim J.Y., Song J.Y., Lee E.J., Park S.K. // Colloid Polym. Sci. 2003. V. 281. № 7. P. 614.
Kelly C.P., Cramer C.J., Truhlar D.G. // J. Phys. Chem. B. 2006. V. 110. № 32. P. 16066.
Guthrie J.P. // Can. J. Chem. 1978. V. 56. № 17. P. 2342.
Malkin A., Ilyin S., Roumyantseva T., Kulichikhin V. // Macromolecules. 2013. V. 46. № 1. P. 257.
Ilyin S.O., Kulichikhin V.G., Malkin A.Y. // Polymer Science A. 2013. V. 55. № 8. P. 503.
Kulichikhin V.G., Ilyin S.O., Mironova M.V., Berkovich A.K., Nifantyev I.E., Malkin A.Ya. // Adv. Polym. Technol. 2018. V. 37. № 4. P. 1076.
Ilyin S.O., Malkin A.Y., Kulichikhin V.G., Denisova Y.I., Krentsel L.B., Shandryuk G.A., Litmanovich A.D., Litmanovich E.A., Bondarenko G.N., Kudryavtsev Y.V. // Macromolecules. 2014. V. 47. №.14. P. 4790.
Malkin A., Kulichikhin V., Ilyin S. // Rheol. Acta. 2017. V. 56. № 3. P. 177.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)