

Высокомолекулярные соединения (серия Б), 2020, T. 62, № 1, стр. 3-16
КИНЕТИЧЕСКИЕ ОСОБЕННОСТИ ЭЛЕМЕНТАРНЫХ АКТОВ РАДИКАЛЬНОЙ ПОЛИМЕРИЗАЦИИ МЕТИЛМЕТАКРИЛАТА В УСЛОВИЯХ ОБРАТИМОГО ИНГИБИРОВАНИЯ НИТРОКСИЛАМИ
М. Ю. Заремский a, В. В. Одинцова a, *
a Московский государственный университет имени М.В. Ломоносова. Химический факультет
119991 Москва, Ленинские горы, Россия
* E-mail: viktoriaodintsova@outlook.com
Поступила в редакцию 20.06.2019
После доработки 02.09.2019
Принята к публикации 16.09.2019
Аннотация
Исследованы кинетические особенности элементарных актов инициирования, реинициирования и обратимого обрыва в радикальной полимеризации метилметакрилата с участием нитроксилов пиперидиновой и имидазолиновой природы. Определены значения констант скорости указанных реакций, а также константы равновесия между “спящими” и растущими цепями. Во всех изученных системах наблюдается понижение значений константы скорости реинициирования и константы равновесия в ходе полимеризации. Активность имидазолиновых нитроксилов в реакции обрыва оказалась на порядок ниже пиперидиновых, а константа взаимодействия обоих типов нитроксилов с радикалами роста метилметакрилата – на два с половиной порядка ниже, чем с их низкомолекулярными аналогами.
ВВЕДЕНИЕ
Радикальная полимеризация с участием нитроксилов в условиях обратимого ингибирования применяется для контролируемого синтеза большого числа виниловых полимеров. Однако в случае ММА этот процесс, как правило, протекает в затухающем режиме и приводит к образованию “мертвого” полимера. Хотя в печати периодически появляются сведения об осуществлении полимеризации ММА по механизму обратимого ингибирования с участием нитроксильных радикалов, на практике область “живого” процесса обычно не выходит за рамки 30–50% конверсии [1–9] или ограничивается получением олигомеров [10, 11]. Понять причину такого явления можно, если бы были известны основные кинетические параметры этого процесса: константы скорости реакций реинициирования kd и обрыва на нитроксилах kx и их отношение – константа равновесия К = kd/kx, а также константа скорости реакции диспропорционирования kdisp. К сожалению, такие параметры определены только для модельных низкомолекулярных веществ. Количественная информация о реакциях, протекающих непосредственно в полимеризационных процессах, крайне мала и ограничивается нитроксилами 2,2,6,6-тетраметилпиперидин-1-оксил и SG-1.
Хорошо известно, что аддукты нитроксилов с низкомолекулярными радикалами, моделирующими радикал роста ПММА, нестойки [12]. Эта же особенность характерна и для высокомолекулярных аддуктов ПММА–нитроксил. Среди всех известных полимерных нитроксильных аддуктов они выделяются самыми высокими значениями констант реинициирования. Период полураспада аддукта ПММА–ТЕМПО при 120°С равен 20 с [13], что в 50 раз меньше, чем у ПС–ТЕМПО. Для ПММА–SG-1 и ПС–SG-1 такое различие превышает 250 раз. (К сожалению, все приводимые в литературе [13–15] значения получены из кинетического анализа полимеризации ММА с участием SG-1. Нет ни одного экспериментально измеренного значения kd для аддукта ПММА– SG-1.) Следовательно, с точки зрения реакции реинициирования никаких препятствий для реализации механизма обратимого ингибирования нет.
Напротив, сведения о кинетике обратной реакции, т.е. взаимодействия радикала ПММА или его низкомолекулярных аналогов с нитроксилами, весьма противоречивы. Согласно литературным данным, низкомолекулярные аналоги радикалов ПММА и ПС либо не отличаются по активности в реакции с нитроксилами (это характерно для SG-1 [16, 17]), либо первый оказывается в несколько раз активнее второго, как в случае ТЕМПО или имидазолиновых нитроксилов [16, 17].
Однако в реакциях полимерных радикалов с нитроксилами наблюдается обратная зависимость: активность радикала ПММА на порядок ниже, чем ПС. По нашим данным, константа скорости взаимодействия радикала ПММА с ТЕМПО составляет 2 × 106 л/моль с, радикала ПС – 1.5 × 107 л/моль с [18]. Для реакции с нитроксилом SG-1 эти величины имеют порядок 104 и 105 л/моль с для ПММА и ПС соответственно [1]. Иными словами, реакционная способность радикала ПММА существенно ниже его низкомолекулярных аналогов. В ряду радикалов (СН3)(СООСН3)(СН3)С•–(C6H5)(CH3)2C–MMA•–ПMMA• константа скорости их взаимодействия с ТЕМПО понижается на два с половиной порядка [15].
Вторая особенность реакции взаимодействия радикалов ПММА с нитроксилами – понижение константы скорости обрыва с ростом температуры (в 5 раз при переходе от 20 до 45°С [1]).
Эти два обстоятельства свидетельствуют о том, что нитроксильные радикалы как ловушки радикалов роста ММА “работают плохо”. Попытки описания полимеризации ММА с участием нитроксилов без учета реальных величин константы скорости обрыва [1, 13, 17, 19] существенно снижают их предсказательную силу.
Ситуация осложняется еще и тем, что взаимодействие радикалов роста ММА с нитроксилами сопровождается диспропорционированием, приводящим к образованию “мeртвых” цепей и гидроксиламина. Например, для ТЕМПО вероятность такого пути равна 1/5 по сравнению с обратимой рекомбинацией [15]. В свою очередь, гидроксиламин вступает в реакцию вторичного ингибирования, которая также приводит к формированию “мeртвых” цепей. Заметим, что существуют нитроксильные радикалы, не склонные к диспропорционированию, например SG-1, но и для них обратимое ингибирование при полимеризации ММА также не реализуется [1, 20].
Ранее экспериментально было найдено, что при использовании алкоксиаминов, т.е. аддуктов радикала (СН3)(СН3)(СОOEt)С• c нитроксилами имидазолинового ряда, реакция диспропорционирования на стадии инициирования не происходит. Поэтому реализация механизма обратимого ингибирования при полимеризации ММА с такими алкоксиаминами наиболее вероятна. Первые экспериментальные подтверждения этому были получены недавно [21].
Цель данной работы – определить количественные характеристики полимеризации ММА в условиях обратимого ингибирования имидазолиновыми и некоторыми другими нитроксилами. При этом основное внимание здесь уделено реакции радикала ПММА с нитроксилами как наименее изученному процессу.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В настоящей работе метилметакрилат, стирол, бензол, хлорбензол, ацетонитрил, трифторуксусный ангидрид (ТФА) очищали по стандартной методике, ДАК – перекристаллизацией из этанола. ТЕМПО, ди-трет-бутилнитроксил (ДТБН) и С-фенил-N-трет-бутилнитрон (ФБН) фирмы “Sigma”, а также оксид серебра Ag2O квалификации х.ч. дополнительной очистке не подвергали. Алкоксиамины 2-(4-гидрокси-дициклогексанспиро-2,6-пиперидин-1-оксил)этилизобутират (A-1), 2-(1-гидроксиэтилциклопентанспиро-2-3-фенил-5,5-диэтил-имидазолин-1-оксил)этил-изобутират (А-2) и 2-(2,5,5-триэтил-3-индол-5-пара-фенилкарбинилимидазолин-1-оксил)этил-изобутират (A-3) синтезировали и охарактеризовали по методике [22] в лаборатории азотистых соединений Новосибирского института органической химии им. Н.Н. Ворожцова Сибирского отделения РАН с использованием соответствующих нитроксильных радикалов: 4-гидрокси-дициклогексанспиро-2,6-пиперидин-1-оксил (N-1), 1-гидрокси-этилциклопентанспиро-2-3-фенил-5,5-диэтилимидазолин-1-оксил (N-2) и 2,5,5-триэтил-3-индол-5-пара-фенилкарбинил-имидазолин-1-оксил:
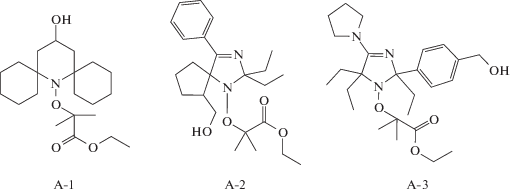
Для синтеза макроинициаторов получали растворы алкоксиаминов в ММА. При необходимости к раствору алкоксиамина в ММА добавляли стирол, ДАК и ТФА. Реакционную смесь заливали в ампулу, дегазировали на вакуумной установке путем трехкратного цикла заморозка–вакуумирование–разморозка до остаточного давления 5 × 10–3 мм рт.ст., после чего ампулу отпаивали. Образец помещали в термостат, разогретый до заданной температуры (45–80°С) и выдерживали в течение требуемого времени. Полимер выделяли лиофильным высушиванием в вакууме из бензола.
Для определения константы скорости инициирования готовили растворы алкоксиаминов в хлорбензоле (10–4 моль/л). Смеси разливали в ампулы, помещали в датчик ЭПР-спектрометра “Bruker EMX”, нагревали в ячейке резонатора до 60, 70 или 90°С и регистрировали спектры. В качестве “ловушки” алкильных радикалов использовали растворенный кислород.
Для определения константы реинициирования готовили раствор макроинициатора (10–4 моль/л) в бензоле, после чего добавляли ФБН (0.4 моль/л). Реакционную смесь разливали в ампулы, дегазировали, ампулы запаивали и помещали в термостат при температуре 60–90°С, количество выделившегося нитроксила регистрировали с помощью радиоспектрометра RE-1307.
Молекулярно-массовые характеристики образцов определяли методом ГПХ по полистирольным стандартам с пересчетом на ПММА по формуле MПММА = 0.644 × $М_{{{\text{ПС}}}}^{{1.019}}$ на жидкостном хроматографе “Waters” с рефрактометрическим детектором, тремя колонками, наполненными ультрастирогелем с размером пор 104, 105 Å и линейной колонкой. Анализ ГПХ проводили в ТГФ при 35°С и скорости потока 0.9 мл/мин. Хроматограммы обрабатывали на интеграторе “Data Module-730”.
Количество гидроксиламина, образующегося в реакционной смеси по ходу полимеризации, определяли по известной методике [18]. После проведения полимеризации ([алкоксиамин] = = 20 ммоль/л, 80°С) ампулу вскрывали, добавляли оксид серебра, смесь вакуумировали и отпаивали. Ампулу выдерживали 1.5–2 ч при комнатной температуре, затем измеряли количество выделившегося нитроксила методом ЭПР. Количество гидроксиламина, образующегося при диспропорционировании алкоксиаминов в инертной среде, узнавали аналогичным образом, используя раствор алкоксиамина в бензоле.
Константу скорости обратимого обрыва находили методом конкурентного ингибирования при 40°С, используя предварительно дегазированный раствор ТЕМПО и нитроксила N-2 в метилметакрилате.
Скорость полимеризации определяли методом калориметрии при 40°С на дифференциальном автоматическом микрокалориметре ДАК-1-1а в режиме прямой регистрации скорости тепловыделения в изотермических условиях. В рабочую ячейку помещали ампулу с исследуемой смесью, в компенсационную ячейку сравнения – ампулу с приблизительно равным количеством уже заполимеризовавшегося мономера. Для расчета скорости полимеризации использовали известное значение энтальпии полимеризации ММА.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Определение констант скорости инициирования и реинициирования
Реакцию инициирования изучали на примере распада трех алкоксиаминов А-1, А-2 и А-3 в хлорбензоле при 60, 70 и 90°С. Эти алкоксиамины представляют собой аддукты имидазолиновых нитроксилов и замещенного ТЕМПО с низкомолекулярным радикалом, моделирующим радикал роста ММА.
Для определения константы скорости инициирования kd регистрировали накопление нитроксила при распаде алкоксиаминов в термостатируемой ячейке спектрометра ЭПР. Конкурентным ингибитором служил растворенный в образце кислород:
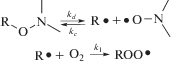
Константу скорости рассчитывали из анализа кинетической кривой накопления нитроксила в координатах уравнения первого порядка:
а также из наклона начального линейного участка кривой накопления нитроксила при низких значениях температуры: d[N]/dt = –d[A]/dt = kd [A]0, где [N] и [A]0 – текущая и начальная концентрация нитроксила N и аддукта A, t – время.Оба способа дают значения, различающиеся не более чем на 30%.
На рис. 1 приведены кинетические кривые накопления нитроксильных радикалов при распаде аддуктов А-1, А-2 и А-3 в координатах уравнения (1). Полученные из анализа кривых значения констант инициирования и энергии активации представлены в табл. 1.
Рис. 1.
Зависимости концентрации нитроксильного радикала от времени в координатах уравнения (1) при распаде алкоксиамина А-1 (а), А-2 (б) и А-3 (в) в растворе хлорбензола при температуре 60 (1), 70 (2) и 90°С (3).
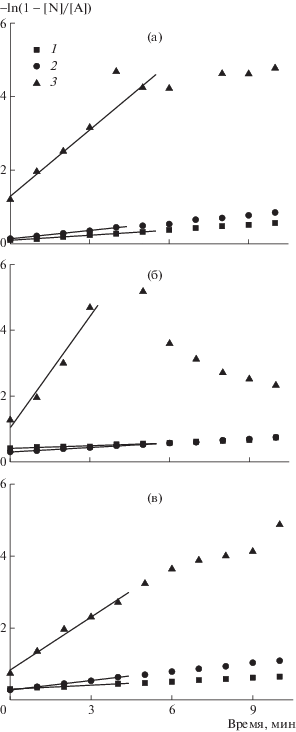
Таблица 1.
Константы скорости инициирования для алкоксиаминов и энергия активации инициирования
| Алкоксиамин | kd × 104, с–1 | Ea, кДж/моль | ||
|---|---|---|---|---|
| 60°С | 70°С | 90°С | ||
| А-1 | 7.5 | 13.0 | 135 | 100 ± 15 |
| А-2 | 4.4 | 7.6 | 190 | 130 ± 30 |
| А-3 | 6.1 | 15.0 | 82 | 85 ± 10 |
Как видно, инициаторы А-1 пиперидиновой и А-2 имидазолиновой природы достаточно активны и близки по своим кинетическим параметрам. Последний из инициаторов – А-3 отличается нетипично низкой для алкоксиаминовых инициаторов энергией активации. Поэтому все дальнейшие исследования проводили с первыми двумя соединениями.
Для определения константы скорости реинициирования, т.е. распада полимерных аддуктов, получили ряд образцов ПММА–N-1 и ПММА–N-2 полимеризацией метилметакрилата под действием А-1 и А-2 в разных условиях (табл. 2). Затем проводили аналогичные эксперименты с использованием в качестве конкурентного ингибитора ФБН. Поскольку ПММА–N-2 активен даже при комнатной температуре, то его выделяли методом лиофильной сушки и не переосаждали, так как происходит существенная потеря его активности. В этом случае полимерный аддукт содержит большое количество остаточного свободного нитроксила, который сам по себе является хорошим конкурентным ингибитором, поэтому в качестве конкурента брали спиновую ловушку ФБН. Концентрацию ловушки (0.4 моль/л) подбирали опытным путем так, чтобы начальная скорость накопления нитроксила не зависела от концентрации ФБН. При анализе кинетики реакции рост концентрации нитроксила делили пополам, поскольку на один акт распада макроинициатора ПММА выделяется два нитроксильных радикала. При выполнении этих условий, скорость накопления нитроксильного радикала и скорость распада инициатора совпадают.
Таблица 2.
Условия получения и молекулярно-массовые характеристики образцов ПММА для определения константы реинициирования
| Макро-инициатор | Время, мин | [Ai], ммоль/л | Mn × 10–3 | Ð |
|---|---|---|---|---|
| ПММА–N-2 | ||||
| (1) | 10 | 3 | 9.3 | 2.3 |
| (2) | 10 | 10 | 10.0 | 1.5 |
| (3) | 13 | 10 | 11.1 | 1.4 |
| (4) | 20 | 3 | 21.6 | 1.5 |
| (5) | 120 | 30 | 7.6 | 1.9 |
| (2а)* | 10 | 10 | 11.7 | 1.5 |
| ПММА–N-1 | ||||
| (1) | 180 | 10 | 5.9 | 1.3 |
| (2)** | 40 | – | 8.9 | 3.4 |
Кинетические кривые распада ПММА–N-2 (2) приведены на рис. 2, а значения константы распада – в табл. 3. Рассчитанная энергия активации распада составляет 128 ± 19 кДж/моль, что типично для нитроксильных макроинициаторов [23].
Рис. 2.
Кинетические кривые накопления нитроксила при распаде ПММА–N-2 (2) в присутствии ФБН при температуре Т = 60 (1), 70 (2), 72 (3), 80 (4) и 90°С (5). [ПММА–N-2 (2)] = 10–4 моль/л.
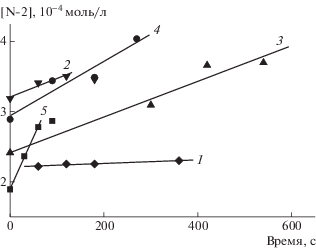
Таблица 3.
Величины константы распада макроинициаторов на основе ПММА
| Макро-инициатор | Т, °С | kd × 103, c–1 |
|---|---|---|
| ПММА–N-2 | ||
| (2) | 90 | 12.0 |
| 80 | 2.5 | |
| 72 | 1.8 | |
| 70 | 0.51 | |
| 60 | 0.25 | |
| (1) | 80 | 3.6 |
| (3) | 80 | 1.3–2.0 |
| (4) | 80 | 2.2 |
| (5) | 80 | 0.9 |
| (2а) | 80 | 1.8 |
| ПММА–N-1 | ||
| (1) | 80 | <0.3 |
| (2) | 80 | <0.2 |
| 60 | <0.03 | |
Сопоставление значений kd и Ea для низкомолекулярного алкоксиамина А-2 и полимера на его основе показывает, что как инициаторы они обладают примерно одинаковой активностью. Однако величина константы распада макроинициатора падает в ходе полимеризации в 3–4 раза, что, вероятно, связано с постепенным накоплением “мертвых” цепей (показано ниже).
Напротив, полимер ПММА–N-1 имеет существенно меньшую (близкую к нулевой) инициирующую способность, т.е. практически все цепи этого полимеры “мeртвые”.
Реакции диспропорционирования при полимеризации метилметакрилата
Предполагаемое накопление “мeртвых” цепей в ПММА–N-1 и ПММА–N-2 связано с реакцией диспропорционирования радикалов роста с нитроксилами, которое приводит к образованию гидроксиламина и макромолекул с концевой двойной связью:
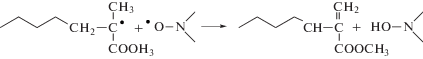
Вначале исследовали данную возможность для самих алкоксиаминов в инертной среде. За образованием гидроксиламина следили с помощью реакции серебряного зеркала [18]. Для чего нагревали раствор А-2 в бензоле в вакууме в течение 5 мин. Затем образец вскрывали, добавляли к нему избыток окиси серебра, и выдерживали в течение 1.5–2 ч. При наличии в среде гидроксиламина должна протекать реакция выделения нитроксила и образования серебряного зеркала.
Однако, как показал анализ ЭПР продуктов реакции, концентрация нитроксила до и после добавления окиси серебра с точностью до ошибки измерения совпадала (табл. 4). Это свидетельствует об отсутствии реакции диспропорционирования между низкомолекулярными радикалами – продуктами распада А-2.
Таблица 4.
Результаты реакции серебряного зеркала
| Система | Время, мин | Концентрация N (ммоль/л) до/после добавления Ag2O | Содержание гидроксиламина, % |
|---|---|---|---|
| A-2–бензол | 5 | 0.43/0.36 | ~0 |
| А-2–ММА | 5 | 1.0/1.8 | 2.8 |
| 20 | 2.4/1.1 | 16 | |
| 40 | 1.6/5.3 | 13 | |
| 60 | 5.0/4.6 | 17 | |
| А-1–бензол | 35 | 0.2/1.1 | 7 |
| 90 | 1.6/2.3 | 6 | |
| А-1–ММА | 9 | 1.8/1.6 | 38 |
| 15 | 8.8/20 | ~100 |
При замене инертной среды реакции на мономер ММА, картина меняется: уже через 5 мин после начала реакции система содержит 3% гидроксиламина, концентрация которого повышается до ~15% через 20 мин и далее не меняется. Этот результат позволяет заключить, что даже в том случае, когда продукты распада алкоксиаминового инициатора сами не могут диспропорционировать, такая возможность появляется, как только в системе образуются радикалы роста ММА.
Такую же закономерность наблюдали и при исследовании алкоксиамина А-1. При его термолизе в инертной среде вероятность диспропорционирования невелика, хотя в среде мономера уже через 15 мин почти весь нитроксил превращается в гидроксиламин.
Таким образом, можно сделать следующие выводы. Во-первых, вероятность диспропорционирования нитроксила пиперидиновой природы существенно выше, чем имидазолиновой. Во-вторых, реакция диспропорционирования в мономере происходит намного быстрее, чем в инертном растворителе, т.е. вероятность диспропорционирования для высокомолекулярного радикала роста существенно выше, чем для низкомолекулярного. В-третьих, в случае полимеризации, инициированной А-1, высокая вероятность диспропорционирования приводит практически к полной гибели растущих цепей уже через 15 мин после начала полимеризации. Именно с этим связана “нулевая” активность полученного ПММА– N-1 в качестве макроинициатора. Наконец, накопление “мертвых” цепей за счет диспропорционирования в случае полимеризации ММА с участием А-2 влечет снижение инициирующей способности ПММА–N-2. Однако наблюдаемое понижение константы скорости инициирования намного больше ожидаемого за счет гибели цепей по реакции диспропорционирования.
Константа скорости обратимого обрыва kx
Скорость диспропорционирования между радикалом роста и нитроксилом тем выше, чем больше общая скорость их взаимодействия. Поэтому знание величины константы скорости обрыва kx радикалов ПММА необходимо для понимания механизма полимеризации. Для ее нахождения применяли два общепринятых подхода – линеаризацию распределения макромолекул по длине цепи и конкурентное ингибирование [24, 25].
Первый способ позволяет найти kx непосредственно в условиях полимеризации. Для этого используют линеаризацию распределения макромолекул по длине цепи P(M), которую получают преобразованием кривых ГПХ G(RT) полимера [26]:
Величину kx находят путем дифференцирования функции lnP(M) по длине цепи i, используя соотношение
(2)
$\mathop {\lim }\limits_{i \to \infty } \frac{{d\ln P}}{{di}} = - \frac{{{{k}_{x}}[X]}}{{k_{p}^{{}}[M]}}$Если полимер получен в стационарных условиях при постоянной концентрации свободного нитроксила Х, то участок зависимости ln(P), отвечающий высокомолекулярной ветви кривой ГПХ, представляет собой прямую линию с тангенсом угла наклона –(kx[X]/kp[M])m (m – молекулярная масса звена). Ранее, данным методом была определена величина kx = (1.5–2.0) × 106 л/моль с для реакции радикала ПММА с нитроксилом ТЕМПО [24].
Оказалось, что при замене ТЕМПО на его аналог с двумя циклогексильными заместителями (N-1) скорость реакции нитроксила с радикалом ПММА практически не изменяется. Найденное из тангенса угла наклона линейных участков кривых зависимости lnP(M) = f(M) (рис. 3) значение kx составляет 1.0 × 106 л/моль с (табл. 5). Измерение данной константы в реакции полимеризации, инициированной алкоксиамином А-1, и в полимеризации под действием системы ДАК–N-1 приводит практически к одинаковым результатам.
Рис. 3.
Зависимость lnP(M) от М для ПММА, полученного в системе ММА–А-1 при 45°С (1) и ММА–А-1–ДАК при 80°C (2); время полимеризации 20 (1) и 45 мин (2). [A-1] = 10 ммоль/л; [ДАК] = 10 ммоль/л.
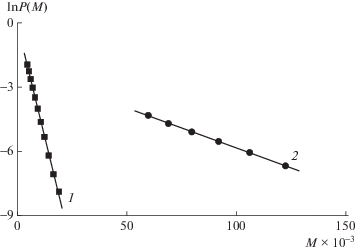
Таблица 5.
Определение константы kx взаимодействия нитроксилов с радикалом ПММА
| Система | Концентрация, ммоль/л | Т, оС | kx × 10–6, л/моль с | 〈kx〉 × 10–6, л/моль с |
|---|---|---|---|---|
| A-1 | 10 | 45 | 1.2 | 1.0 ± 0.1 |
| A-1–ДАК | 10 | 80 | 0.95 | |
| N-1–ДАК | 10 | 90 | 0.88 | |
| 0.89 | ||||
| A-1–ТФА | 10 | 80 | 0.28 | 0.27 ± 0.03 |
| A-1 | 10 | 80 | 0.21 | |
| ТФА | 25 | 80 | 0.21 | |
| A-1 | 10 | 80 | 0.29 | |
| ТФА | 50 | 80 | 0.29 | |
| A-1 | 10 | 80 | 3.9 | |
| 10% cтирола | 10 | 80 | 4.1 | |
| ДТБН–ДАК | 10 | 90 | 1.2 | 3.2 ± 1.7 |
| ДТБН | 10 | 90 | 4.4 | |
| ДАК | 12 | 90 | 4.0 | |
| A-2 | 3 | 80 | 0.19 | 0.16 ± 0.02 |
| 10 | 80 | 0.15 | ||
| 10 | 80 | 0.13 | ||
| 30 | 80 | 0.14 | ||
| A-2–ДАК | 10 | 80 | 0.17 | |
| A-2–ТФА | 10 | 80 | 0.11 | 0.11 |
| 0.11 |
Несколько большую активность в реакции с радикалами ПММА имеет ациклический нитроксил ДТБН, что очевидно связано с его меньшими размерами.
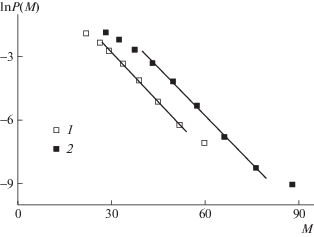
Напротив, при переходе к пятичленному стерически нагруженному имидазолиновому нитроксилу N-2 наблюдается падение константы обрыва. Об этом свидетельствует существенно больший наклон аналогичных зависимостей lnP(M) от М для ПММА, полученного в разных условиях в присутствии N-2 (рис. 4). Усредненное по пяти опытам значение константы kx на порядок ниже, чем для нитроксила ТЕМПО.
Рис. 4.
Зависимость lnP(M) от М для ПММА, полученного в системе ММА–A-2 при 80°C; время полимеризации 20 (1, 3, 4) и 13 мин (2). [A-2] = 10 (1, 2), 30 (3) и 3 ммоль/л (4).

Интересно, что добавление всего 10% стирола к ММА резко увеличивает скорость обрыва радикалов роста на нитроксилах. Как видно из табл. 5, значение kx при этом повышается до 4 × 106 л/моль с. Такое поведение следует из общей концепции радикальной сополимеризации по механизму обратимого ингибирования [27], т.е. при добавлении 5–10% стирола к ММА основные кинетические характеристики процесса становятся близкими к таковым для полимеризации стирола.
Второй подход, который мы использовали для нахождения kx – метод конкурентного ингибирования – заключается в том, что проводят полимеризацию в присутствии двух нитроксилов А и В, для одного из которых (“эталона”) известна константа kx1 скорости взаимодействия с радикалом роста мономера. Процесс ведут при низкой температуре, чтобы исключить вероятность обратной реакции распада аддукта полимер–нитроксил и при низких значениях концентрации ингибиторов, чтобы до момента обрыва успели вырасти полимерные радикалы. Измеряя с помощью метода ЭПР относительную скорость расхода обоих ингибиторов, находят искомую константу kx2 из соотношения
(3)
$~d[{\text{A}}]{\text{/}}d[{\text{B}}] = {{k}_{x}}_{1}\left[ {\text{А}} \right]{\text{/}}{{k}_{x}}_{2}\left[ {\text{В}} \right]~$Применимость данного метода к полимеризации ММА с участием нитроксилов ограничена выбором “эталонов” с известной константой обрыва. Таким веществом может служить только ТЕМПО, поскольку для него надежно установлено значение kx1. Нитроксил SG-1, для которого также известна величина kx1, здесь не подходит, так как его активность настолько низкая (kx ~ 104 л/моль c), что он не является конкурентом даже для реакции необратимого квадратичного обрыва.
На рис. 5 представлено изменение спектров ЭПР в системе ММА–ТЕМПО–N-2–ДАК при полимеризации при 40°С. Видно, что оба нитроксила имеют одинаковую структуру спектров – триплет синглетов, однако заметное различие в константах расщепления (aN, TEMПО = 15.4 Э и aN, N-2 = 14.0 Э) позволяет по низкополевой компоненте спектра определить расход каждого нитроксила в ходе реакции. Полученные кинетические кривые представлены на рис. 6, а рассчитанные отношения констант обрыва приведены в табл. 6. Используя известное значение kx1 = 1.6 × × 106 л/моль с [24], получаем среднюю величину для N-2 kx2 = 5.0 × 105 л/моль с. Это означает, что активность N-2 как ингибитора на полпорядка ниже, чем ТЕМПО.
Рис. 5.
Изменение спектров ЭПР в системе ММА–ТЕМПО–N-2–ДАК по ходу полимеризации ММА при 40°С; время регистрации 2 (1), 5 (2), 8 (3), 11 (4) и 17 мин (5).
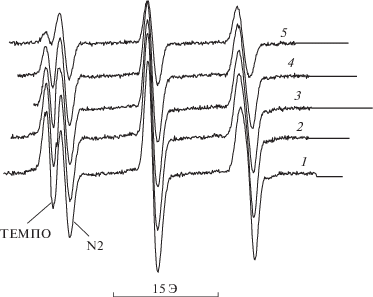
Рис. 6.
Зависимость амплитуды сигнала А нитроксилов ТЕМПО (1, 3, 5, 7) и N-2 (2, 4, 6, 8) при полимеризации ММА при 40°С в системах ТЕМПО–N-2–ДАК. [ТЕМПО] = 0.055 (1, 2), 0.16 (3, 4, 7, 8) и 0.22 ммоль/л (5, 6); [ДАК] = 100 (1, 2) и 500 ммоль/л (3–8); [N-2] = 0.0025 (1, 2), 0.0075 (3, 4) и 0.22 ммоль/л (5–8).
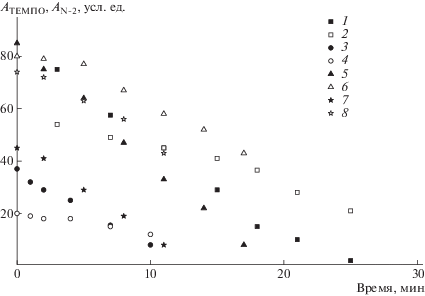
Таблица 6.
Отношение констант скорости взаимодействия радикалов роста метилметакрилата с ТЕМПО (kx1) и N-2 (kx2) в системе мономер–ТЕМПО–N-2–ДАК, Т = 40°С
| [TЕМПО], ммоль/л | [N-2], ммоль/л | [ДАК], ммоль/л | kx1/ kx2 |
|---|---|---|---|
| 0.055 | 0.025 | 100 | 3.5 ± 0.2 |
| 0.16 | 0.075 | 500 | 3.5 ± 0.6 |
| 0.16 | 0.22 | 500 | 3.3 ± 0.6 |
| 0.22 | 0.22 | 500 | 3.0 ± 0.3 |
Определить kx для N-1 и ДТБН данным методом нельзя, поскольку их спектры ЭПР и спектр ТЕМПО практически совпадают.
Из полученных результатов следуют два важных заключения: первое, активность имидазолиновых нитроксилов на порядок ниже пиперидиновых и, второе, сопоставление значений констант kx для реакции N-2 с радикалом ПММА и с его низкомолекулярным аналогом kx =1 × 108 л/моль c [3] показывает, что они различаются на два с половиной порядка. Для нитроксила ТЕМПО, как представлено выше, эта разница также составляет два с половиной порядка. Таким образом, резкое падение активности полиметакрилатных радикалов в реакции с нитроксилами, является общим правилом.
Константа равновесия между спящими и растущими цепями
Из найденных значений констант скорости реинициирования и обрыва следует, что величина константы равновесия K = kd/kx между растущими и “спящими” цепями ПММА должна уменьшаться в ходе полимеризации ММА с участием N-2 от 2 × 10-8 до 5 × 10-9 л/моль с. Определение константы равновесия из прямого эксперимента, т.е. путем нахождения равновесных значений концентрации нитроксила N, макрорадикала P и аддуктов A из данных ЭПР и калориметрии: K = = [N][P]/[A], дает близкие значения (табл. 7).
Таблица 7.
Равновесная концентрация радикалов роста и нитроксилов, а также константа равновесия
| Алкоксиамин | Концентрация алкоксиамина, ммоль/л | Время, мин | [N], ммоль/л | [Р] × 108, моль/л | K × 108, л/моль |
|---|---|---|---|---|---|
| A-2 | 30 | 17 | 3.1 | 13 | 1.3 |
| 30 | 3.3 | 8.3 | 0.91 | ||
| 45 | 3.4 | 5.0 | 0.57 | ||
| 60 | 3.4 | 3.2 | 0.37 | ||
| 70 | 3.3 | 2.3 | 0.26 | ||
| 20 | 20 | 2.5 | 8.8 | 1.1 | |
| 40 | 3.6 | 4.1 | 0.73 | ||
| 60 | 3.2 | 2.0 | 0.32 | ||
| 10 | 20 | 1.5 | 7.7 | 1.1 | |
| 30 | 1.6 | 6.2 | 1.0 | ||
| 40 | 1.7 | 4.6 | 0.77 | ||
| 60 | 1.9 | 2.5 | 0.42 | ||
| 70 | 1.7 | 1.8 | 0.32 | ||
| 9 | 20 | 1.4 | 6.6 | 1.1 | |
| 30 | 1.6 | 4.8 | 0.82 | ||
| 40 | 1.7 | 3.4 | 0.62 | ||
| 50 | 1.7 | 2.2 | 0.42 | ||
| 60 | 1.7 | 1.5 | 0.29 | ||
| 5 | 25 | 1.1 | 5.0 | 1.1 | |
| 45 | 1.1 | 2.6 | 0.56 | ||
| 65 | 1.3 | 1.4 | 0.35 | ||
| 3 | 20 | 0.57 | 5.5 | 1.04 | |
| 25 | 0.64 | 4.3 | 0.92 | ||
| 45 | 0.66 | 2.0 | 0.44 | ||
| 65 | 0.76 | 1.0 | 0.24 | ||
| A-2–ДАК | 0.01–0.01 | 20 | 0.56 | 11.8 | 0.53 |
| 25 | 0.42 | 11.5 | 0.38 | ||
| 35 | 0.27 | 10.7 | 0.23 | ||
| 45 | 0.22 | 10.5 | 0.18 | ||
| A-1 | 30 | 25 | 2.5 | 1.0 | 0.084 |
| 50 | 2.6 | 0.20 | 0.018 | ||
| 70 | 2.3 | 0.071 | 0.0057 | ||
| 100 | 2.2 | 0.020 | 0.0015 | ||
| 10 | 25 | 3.4 | 0.58 | 0.18 | |
| 40 | 4.1 | 0.11 | 0.045 | ||
| 75 | 2.6 | 0.012 | 0.0036 | ||
| 3 | 25 | 0.35 | 0.29 | 0.033 | |
| 55 | 0.28 | 0.033 | 0.0031 | ||
| A-1–ДАК | 10–5 | 30 | 0.17 | 9.7 | 0.16 |
| 40 | 0.061 | 7.1 | 0.044 | ||
| 60 | 0.029 | 3.7 | 0.011 | ||
| 80 | 0.019 | 1.8 | 0.0035 |
Видно, что при полимеризации под действием нитроксила N-1 константа равновесия оказывается на один–два порядка ниже, чем при использовании N-2, что сочетается с найденными выше константами элементарных актов реинициирования и обрыва цепи.
Хорошее согласие между значениями констант реинициирования, обрыва на нитроксилах и равновесия, определенных разными методами, подтверждает достоверность этих значений.
Влияние ускоряющих добавок на полимеризацию ММА под действием нитроксилов
Чтобы предотвратить затухание полимеризации ММА с участием нитроксилов, т.е. сдвинуть равновесия в сторону образования растущих цепей используют ряд ускоряющих добавок [4]. Наиболее эффективными среди них являются высокополярные соединения (малононитрил, трифторуксусный ангидрид, соли металлов) и радикальные инициаторы, которые распадаются существенно медленнее, чем основной алкоксиамин. Природа действия первых окончательно не ясна. Радикальные же инициаторы, поставляя дополнительные активные радикалы в систему, связывают свободный нитроксил и тем самым сдвигают равновесие в сторону диссоциации.
В настоящей работе исследовали как добавки ТФА и ДАК влияют на количественные параметры полимеризации ММА под действием нитроксилов N-1 и N-2.
Известно, что полярные соединения понижают значение kx, поскольку способствуют перемещению спиновой плотности на атом азота в нитроксиле, уменьшая его активность [28]. Ранее этот эффект был описан в реакциях нитроксилов с полимерными радикалами в полярных растворителях [29]. Тот же эффект мы наблюдали и при небольших добавках ТФА в полимеризат, содержащий нитроксилы N-1 и N-2.
На рис. 7 показаны зависимости lnP(M) от М для ПММА, полученного в присутствии нитроксила N-1 и ТФА в количестве от эквивалента до пятикратного избытка по отношению к нитроксилу. Оказалось, что добавление ТФА понижает активность N-1 в 3–4 раза (табл. 8). При этом константа обрыва практически не зависит от количества введeнной добавки.
Рис. 7.
Зависимость lnP(M) от М для ПММА, полученного в системе ММА–A1–ТФА при 80°C; время полимеризации 17 (1), 20 (2) и 10 мин (3). [A-1] = = 10 ммоль/л; [ТФА] = 10 (1) и 50 ммоль/л (2, 3).

Таблица 8.
Равновесная концентрация радикалов роста и нитроксилов, а также константа равновесия; [A] = = 10 ммоль/л, Т = 80°С
| Алкоксиамин | Добавка | Концентрация, ммоль/л | Время, мин | [N], ммоль/л | [Р] × 108, моль/л | К × 108, л/моль |
|---|---|---|---|---|---|---|
| A-1 | – | – | 25 | 3.4 | 0.58 | 0.18 |
| ТФА | 10 | 0.64 | 10.8 | 0.69 | ||
| 50 | 0.57 | 12.9 | 0.73 | |||
| ДАК | 5 | 0.06 | 10.9 | 0.06 | ||
| – | – | 30 | 3.6 | 0.33 | 0.12 | |
| ТФА | 10 | 0.57 | 6.6 | 0.37 | ||
| 50 | 0.57 | 7.8 | 0.44 | |||
| ДАК | 5 | 0.06 | 9.38 | 0.06 | ||
| – | – | 35 | 3.7 | 0.19 | 0.05 | |
| ТФА | 10 | 0.51 | 3.9 | 0.20 | ||
| 50 | 0.57 | 5.3 | 0.30 | |||
| ДАК | 5 | 0.06 | 8.6 | 0.05 | ||
| A-2 | – | – | 20 | 1.50 | 7.7 | 1.1 |
| ТФА | 10 | 1.57 | 7.2 | 1.1 | ||
| ДАК | 10 | 0.56 | 11.8 | 0.51 | ||
| – | – | 30 | 1.6 | 6.2 | 1.0 | |
| ТФА | 10 | 1.6 | 5.9 | 0.93 | ||
| ДАК | 10 | 0.35 | 8.5 | 0.30 | ||
| – | – | 40 | 1.7 | 4.6 | 0.77 | |
| ТФА | 10 | 1.6 | 4.6 | 0.72 | ||
| ДАК | 10 | 0.25 | 8.2 | 0.21 |
Падение константы обратимого обрыва в присутствии ТФА вызывает повышение константы равновесия. Это приводит к тому, что скорость полимеризации ММА увеличивается, и выход полимера возрастает с 3–5 до 25%.
Влияние ТФА на активность нитроксила N-2 выражено слабее. Найденное по наклону аналогичных зависимостей (рис. 8) значение kx лишь в 1.5 раза ниже, чем в отсутствие добавки (табл. 8). На равновесные концентрации радикалов роста и нитроксила, а следовательно, и на константу равновесия введение ТФА не влияет.
Рис. 8.
Зависимости lnP(M) от М для ПММА, полученного в системе ММА–A-2–ТФА при 80°C; время полимеризации 20 (1) и 30 мин (2). [A-2] = 10 ммоль/л; [ТФА] = 10 ммоль/л.
Напротив, добавление ДАК, хотя и приводит к заметному повышению равновесной концентрации радикалов роста и понижению концентрации свободного нитроксила, но величина константы равновесия при этом уменьшается. Это характерно для полимеризации с участием обоих нитроксилов N-1 и N-2. Очевидно, это связано с уменьшением количества “живых” цепей вследствие увеличения вероятности квадратичного обрыва. При добавлении 0.5–1.0 эквивалента ДАК величина К падает в 2–3 раза в обеих системах.
На скорость обрыва цепей такое количество добавленного ДАК не влияет. Об этом свидетельствует одно и то же значение констант обрыва как в присутствии ДАК, так и в его отсутствие. Это может быть результатом двух причин: либо основной обрыв макрорадикалов происходит по реакции с нитроксилами, а не друг с другом; либо значения скорости обратимого и квадратичного обрыва близки.
Однако добавление ДАК влияет на константу реинициирования. Из сопоставления скорости распада образцов ПММА–N-2 (1) и ПММА–N-2 (2а) (см. табл. 2) видно, что она понижается в 2 раза. Это хорошо согласуется с общим уменьшением константы равновесия вследствие накопления “мертвых” макромолекул в последнем.
Таким образом, добавление ДАК и ТФА приводит к повышению равновесной концентрации радикалов роста и понижению концентрации нитроксила. Это обстоятельство уменьшает “затухающий” характер полимеризации, однако не предотвращает его до конца.
ЗАКЛЮЧЕНИЕ
Проведенное исследование было направлено на выявление особенностей элементарных актов полимеризации ММА под действием различных нитроксилов. Обнаруженные закономерности помогают понять “упорное сопротивление” ММА полимеризоваться по механизму обратимого ингибирования с участием нитроксилов и преодолеть это “сопротивление”.
Аддукты ПММА–нитроксил достаточно активны как инициаторы (на порядок активнее соответствующих аддуктов ПС–нитроксил и на два порядка – аддуктов полиакрилат–нитроксил). Природа нитроксила в этом процессе не играет принципиальной роли. Реакция реиницирования успешно происходит и в случае ТЕМПО, и его производных, и имидазолиновых нитроксилов. С этой точки зрения, никаких препятствий для реализации механизма обратимого ингибирования нет. Напротив, реакцию стоит проводить при пониженных значениях температуры с целью уменьшения скорости реинициирования, чтобы снизить вероятность квадратичного обрыва образующихся макрорадикалов.
Константа равновесия между растущими и спящими цепями находится в интервале 10–8–10–10 моль/л, что также удовлетворяет требованиям к реализации “живого” механизма полимеризации.
Однако как в случае производных ТЕМПО, так и имидазолиновых радикалов, полимеризация протекает в затухающем режиме, и эффективная константа равновесия уменьшается в несколько раз. Причем в первом случае затухание приводит к превращению растущих цепей в “мертвый” полимер практически сразу.
Основное препятствие к реализации механизма обратимого ингибирования полимеризации ММА состоит в диспропорционировании радикалов роста и нитроксилов, в каждом акте которого образуется “мертвая цепь” и гидроксиламин. Именно поэтому ведется постоянный поиск новых алкоксиаминовых инициаторов, которые бы не давали продуктов диспропорционирования. К ним относятся, например, имидазолиновые алкоксиамины. Полученные данные показывают, что и для них характерно затухание полимеризации ММА. Следовательно, отсутствие диспропорционирования радикалов инициатора – необходимое, но не достаточное условие. После того как первичные радикалы превращаются в радикалы роста ММА, они начинают вступать в реакцию диспропорционирования даже с имидазолиновыми нитроксилами. Только, эта побочная реакция для них существенно менее вероятна, чем для производных ТЕМПО. Поэтому имидазолиновые нитроксилы позволяют существенно повысить выход ПММА и улучшить контроль его молекулярно-массовых характеристик.
Низкая вероятность диспропорционирования имидазолиновых радикалов с радикалами роста ММА, по-видимому, объясняется их общей низкой активностью в качестве ингибиторов полимеризации ММА, если сравнивать с производными ТЕМПО. Константа скорости взаимодействия радикалов ПММА с первыми на порядок ниже, чем со вторыми и на два порядка ниже константы их квадратичного обрыва. Это означает, что при полимеризации ММА под действием имидазолиновых нитроксилов весьма велика вероятность квадратичной гибели растущих цепей, по крайней мере, в начале полимеризации, пока в системе еще не накопилось достаточное количество свободных нитроксилов11.
Таким образом, реакция диспропорционирования позволяет понять, почему невозможна реализация режима обратимого ингибирования полимеризации ММА в случае производных ТЕМПО. Только она не может объяснить затухание полимеризации ММА с участием имидазолиновых нитроксилов. Согласно нашим данным только 15% растущих цепей становятся “мертвыми” вследствие диспропорцинирования. Это должно было бы понизить эффективную константу равновесия по ходу процесса всего на 15%; однако она падает в 3–4 раза. Такое падение не связано с каким-либо изменением в скорости реинициирования с ростом степени полимеризации аддукта ПММА–нитроксил, как это следует из табл. 1 и 2. Также оно не связано и с накоплением в ПММА “мертвых” молекул за счет квадратичного обрыва, поскольку доля таких молекул не превышает 10–15% (оценено как отношение концентрации свободного нитроксила в конце полимеризации к концентрации исходного алкоксиамина). Причина этого пока не ясна и требует дальнейших исследований.
Проведенные эксперименты показывают, что если нельзя избежать “затухания” нитроксильной полимеризации ММА, то его можно существенно ограничить. Введение добавок ТФА или ДАК сдвигает равновесие между растущими и спящими цепями в сторону первых. Окончательный ответ на возможность проведения контролируемой нитроксильной полимеризации ММА может дать математическое моделирование кинетики процесса и ММР продуктов с использованием значений констант элементарных реакций, в том числе представленных в настоящей работе.
Авторы выражают благодарность Е.С. Гариной за анализ полимеров методом ГПХ и Д.А. Пархоменко за помощь в определении константы инициирования.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (код проекта 18-03-00707).
Список литературы
Guillaneuf Y., Gigmes D., Marque S.R.A., Tordo P., Bertin D. // Macromol. Chem. Phys. 2006. V. 207. P. 1278.
Edeleva M., Marque S.R.A., Kabytaev K., Guillaneuf Y., Gigmes D., Bagryanskaya E. // J. Polym. Sci., Polym. Chem. 2013. V. 51. P. 1323.
Еделева М.В. Дис. … канд. хим. наук. Новосибирск: НИОХ СО РАН, 2011.
Guegain E., Guillaneuf Y., Nicolas Y. // Macromol. Rapid Commun. 2015. V. 36. P. 1227.
Greene A.C., Grubbs R.B. // Macromolecules. 2010. V. 43. № 24. P. 10320.
Grignard B., Phan T., Bertin D., Gigmes D., Jerome C., Detrembleur C. // Polym. Chem. 2010. № 1. P. 837.
Yoshida E. // Coll. Polym. Sci. 2011. V. 289. P. 1625.
Ansong O.E., Jansen S., Wei Y., Pomrink G., Li S., Patel A. // Polym. Int. 2008. V. 57. P. 863.
Zhu Z., Shan G., Pan P. // RSC Adv. 2016. V. 6. P. 73842.
Ballard N., Aguirre M., Simula A., Agirre A., Leiza J.R., Asua J.M., van Es S. // ACS Macro Lett. 2016. V. 5. P. 1019.
Simula A., Ballard N., Aguirre M., Leiza J.R., van Es S., Asua J.M. // Eur. Polym. J. 2019. V. 110. P. 319.
Bertin D., Gigmes D., Marque S.R.A., Tordo P. // Macromolecules. 2005. V. 38. № 7. P. 2638.
Fischer H. // ACS Symp. 2003. V. 854. P. 10.
Beaudoin E., Bertin D., Gigmes D., Marque S.R.A., Siri D., Tordo P. // Eur. J. Org. Chem. 2006. № 7. P. 1755.
Ananchenko G.S., Souaille M., Fischer H., Le Mercier C., Tordo P. // J. Polym. Sci., Polym. Chem. 2002. V. 40. P. 3264.
Ananchenko G.S., Fischer H. // J. Polym. Sci., Polym. Chem. 2001. V. 39. P. 3604.
Bagryanskaya E.G., Marque S.R.A. // Chem. Rev. 2014. V. 114. P. 5011.
Zaremski M.Yu., Zhaksylykov A.B., Orlova A.P., Garina E.S., Badun G.A., Lachinov M.B., Golubev V.B. // Polymer Science A. 2005. V. 47. № 6. P. 526.
Dire C., Belleney J., Nicolas J., Bertin D., Magnet S., Charleux B. // J. Polym. Sci., Polym. Chem. 2008. V. 46. P. 6333.
Charleux B., Nicolas J., Guerret O. // Macromolecules. 2005. V. 38. P. 5485.
Еделева М.В., Marque S.R.A., Багрянская Е.Г. // Успехи химии. 2018. Т. 87. № 4. С. 328.
Matyjaszewski K., Woodworth B.E., Zhang X., Gaynor S.G., Metzner Z. // Macromolecules. 1998. V. 31. P. 5955.
Zaremski M.Yu. // Polymer Science C. 2015. V. 57. № 1. P. 65.
Zaremski M.Yu. // Polymer Science A. 2006. V. 48. № 3. P. 234.
Borisova O.V., Zaremskii M.Yu., Golubev V.B., Plutalova A.V., Borisov O.V., Billon L. // Polymer Science B. 2013. V. 55. № 9–10. P. 508.
Moad G., Moad C.L. // Macromolecules. 1996. V. 29. № 24. P. 7727.
Nicolas J., Dire C., Mueller L., Belleney J., Charleux B., Marque S.R.A., Bertin D., Magnet S., Couvreur L. // Macromolecules. 2006. V. 39. № 24. P. 8274.
Beckwith A.L.J., Bowry V.W., Ingold K.U. // J. Am. Chem. Soc. 1992. V. 114. № 13. P. 4983.
Zaremski M., Borisova O., Chen Xin, Golubev V.B., Billon L. // J. Polym. Sci., Polym. Chem. 2012. V. 50. P. 3437.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)