

Высокомолекулярные соединения (серия Б), 2020, T. 62, № 1, стр. 17-25
ИССЛЕДОВАНИЕ КИНЕТИКИ ПОЛИМЕРИЗАЦИИ ЭТИЛЕНА
В. В. Сукулова a, *, А. А. Барабанов a, Т. Б. Микенас a, М. А. Мацько a, В. А. Захаров a, b
a Институт катализа им. Г.К. Борескова Сибирского отделения Российской академии наук
630090 Новосибирск, пр. академика Лаврентьева, 5, Россия
b Новосибирский государственный университет
630090 Новосибирск, Пирогова, 2, Россия
* E-mail: sukulova@catalysis.ru
Поступила в редакцию 14.05.2019
После доработки 13.07.2019
Принята к публикации 20.08.2019
Аннотация
С использованием метода ингибирования полимеризации радиоактивным монооксидом углерода 14СО получены данные о влиянии водорода (агента переноса полимерной цепи) на число активных центров и константу скорости роста при полимеризации этилена на современных высокоактивных титан-магниевых катализаторах различного состава. Найдено, что понижение скорости полимеризации этилена в присутствии водорода связано преимущественно с уменьшением числа активных центров, и эти изменения являются обратимыми при введении и удалении водорода. Предложена схема обратимой реакции временной дезактивации предшественников активных центров, объясняющая полученные результаты. По данным о влиянии концентрации водорода на молекулярную массу полиэтилена и найденным величинам констант скорости роста рассчитана константа скорости переноса полимерной цепи водородом для катализаторов различного состава.
ВВЕДЕНИЕ
Известно, что при полимеризации этилена водород выступает эффективным переносчиком полимерной цепи, поэтому его широко применяют для регулирования молекулярной массы при производстве полиолефинов на катализаторах различного состава, в том числе титан-магниевых (ТМК). Помимо переноса цепи и снижения молекулярной массы полимера водород заметно влияет на активность катализаторов при полимеризации олефинов [1–17]. Характер влияния зависит от состава каталитической системы и типа мономера.
Так, в случае полимеризации пропилена на катализаторах Циглера–Натта присутствие водорода в полимеризационной среде приводит к увеличению активности катализаторов [1–6]. При полимеризации этилена на нанесенных катализаторах, содержащих бис-(имино)пиридиновые комплексы железа и кобальта [7–9] также наблюдается увеличение активности катализаторов в присутствии водорода. Наконец, в случае полимеризации этилена в присутствии водорода на титан-магниевых катализаторах происходит заметное понижение скорости полимеризации [10–16].
При полимеризации пропилена на титан-магниевых катализаторах для объяснения активирующего эффекта водорода в работах [2, 3] предложена схема, включающая реакцию образования временно неактивных (“спящих”) центров после 2,1-присоединения пропилена к растущей полимерной цепи и последующую реактивацию этих центров при взаимодействии с водородом.
Результаты работ [5, 6], свидетельствующие о влиянии водорода на число активных центров CP и константу скорости роста kр, а также данные об этих величинах [7–9] при исследовании полимеризации этилена в отсутствие и присутствии водорода на нанесенных катализаторах, содержащих бис-(имино)пиридиновые комплексы Fe(II) и Co(II), соответствуют этой схеме. В этом случае предполагается, что образование временно неактивных “спящих” центров происходит при 2,1-внедрении в растущую полимерную цепь низкомолекулярных олигомеров, содержащих двойную связь, которые могут образоваться в таких системах при полимеризации этилена. Данные о величинах CP и kр могут дать важную информацию о причинах снижения активности при полимеризации этилена на титан-магниевых катализаторах.
Ранее в работе [17] было исследовано влияние водорода на скорость полимеризации этилена, число активных центров и константу скорости роста при использовании титан-магниевых катализаторов, содержащих очень малое количество титана (≤0.1 мас. %). Эти катализаторы имеют исключительно высокую активность на единицу массы титана за счет высокого числа активных центров [18–20] и являются удобными модельными системами для изучения состава активных центров и механизма полимеризации олефинов. В частности, были получены данные [21] об образовании в этих системах алкилированных моноядерных соединений Ti3+ в качестве предшественников активных центров полимеризации этилена.
В работе [17] для таких модельных катализаторов методом обрыва полимеризации этилена радиоактивным монооксидом углерода 14СО было установлено, что понижение скорости при введении водорода обусловлено, главным образом, уменьшением рассчитываемой величины kр, и предложена схема для объяснения этого необычного результата.
В настоящей работе получены новые данные о влиянии водорода на скорость полимеризации, число активных центров и константы скорости роста при полимеризации этилена на высокоактивных титан-магниевых катализаторах с более высоким содержанием титана (1–2 мас. %). Такие катализаторы имеют высокую активность на единицу массы катализатора, соответствующую известным промышленным образцам. Использованы две модификации таких катализаторов, на которых образуется полиэтилен с различной молекулярной массой при одинаковом содержании водорода. Найдено, что для этих катализаторов, в отличие от модельных катализаторов с низким содержанием титана, понижение скорости полимеризации в присутствии водорода связано преимущественно с уменьшением числа активных центров.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Катализаторы
Носители для катализаторов ТМК-H и ТМК-M получали по методикам [22, 23] при использовании в качестве исходного магнийорганического соединения раствора Mg3Ph4Cl2 в диизоамиловом эфире. Катализаторы ТМК-H и ТМК-M (содержащие 1.0 и 1.7 мас. % титана соответственно) синтезировали путем обработки носителей тетрахлоридом титана. Катализатор ТМК-M содержит в своем составе этокси-группы в количестве 7.5 мас. %. Катализаторы имеют средний размер частиц 4–6 мкм и узкое распределение частиц по размерам.
Полимеризация этилена
Опыты с ингибированием полимеризации этилена радиоактивным монооксидом углерода 14СО проводили в автоклаве из нержавеющей стали объемом 0.5 л. Перед началом полимеризации в реактор загружали катализатор (5.5–6.8 мг) в виде суспензии в гептане в запаянной стеклянной ампуле и выдерживали 90 мин при 80°С и остаточном давлении 2 × 10–2 торр. Полимеризацию проводили в суспензии гептана (150 мл) при постоянном давлении этилена (4 бар) и температуре (80°С). Необходимое количество водорода вводили в автоклав до начала реакции (мольное соотношение [H2]/[C2H4] в газовой фазе реактора варьировали в интервале 0–1). Концентрация активатора AlEt3 в реакторе составляла 2.2 ммоль/л, мольное соотношение [Al]/[Ti] в гептане для катализатора ТМК-М – 170, для катализатора ТМК-Н – 210. Реакцию начинали, разбивая ампулу с катализатором с помощью специального устройства внутри реактора. В нужный момент в реакционную среду вводили 0.5 бар 14CO для ингибирования полимеризации, выдерживали 15 мин и добавляли изопропиловый спирт для разложения компонентов катализатора. В специальных экспериментах с удалением водорода газовую фазу из реактора скачивали и вводили в реактор чистый этилен. Подробное описание эксперимента по полимеризации этилена приведено в работе [17].
Серию опытов с различной концентрацией водорода без обрыва полимеризации осуществляли в стальном реакторе объемом 0.85 л в среде гептана (250 мл) в течение 1 ч. Масса катализатора в этих опытах составляла 5.5–6.8 мг. Остальные условия создавали аналогично радиохимическим опытам.
Концентрацию водорода и этилена в гептане рассчитывали с помощью констант Генри при температуре 80°C: $K_{{\text{H}}}^{{{{{\text{H}}}_{{\text{2}}}}}}$ = 7.64 × 10–3 моль/л бар [24] и $K_{{\text{H}}}^{{{{{\text{C}}}_{{\text{2}}}}{{{\text{H}}}_{{\text{4}}}}}}$ = 0.071 моль/л бар [25, 26] соответственно.
Определение числа активных центров и константы скорости роста
Подробное описание методики определения величин CP и kр с помощью ингибирования полимеризации этилена на ТМК с использованием 14CO представлено в работах [20, 27]. Для удаления побочных радиоактивных продуктов реакции, образующихся при ингибировании полимеризации [5, 6], в настоящей работе проводили двойное переосаждение полимеров в ундекане до достижения постоянной радиоактивности полимеров [28].
Величины CP и kр находили из радиоактивности полимера, измеренной с применением жидкостного сцинтилляционного счетчика “Intertechnique SL-4000”. Расчеты проводили с использованием уравнений, приведенных в работах [17, 28–30].
Определение свойств полимеров
Молекулярно-массовые характеристики полимеров Mn и Mw измерены при помощи метода гель-проникающей хроматографии на приборе “PL 220 С”. Измерения проводили при 160°C; растворителем служил 1,2,4-трихлорбензол; скорость потока составляла 1 см3/мин. Калибровку прибора осуществляли на стандартных образцах полистирола и полиэтилена с узким ММР.
Характеристическую вязкость полимера определяли в декалине при 135°С на вискозиметре Уббелоде. Средневязкостную молекулярную массу рассчитывали по формуле
где η – характеристическая вязкость полимера, K = 67.7 × 10–5 и α = 0.67 – коэффициенты Марка–Хаувинка [31].РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Влияние водорода на величины СP и kр при полимеризации этилена на катализаторах ТМК-М и ТМК-Н
На рис. 1 представлены зависимости скорости полимеризации этилена от времени для катализаторов ТМК-M и ТМК-H в отсутствие и присутствии водорода. Видно, что при введении водорода в полимеризационную среду при мольном соотношении [H2]/[C2H4] = 0.25 в газовой фазе реактора происходит уменьшение скорости полимеризации в 1.7–2.2 раза по сравнению с полимеризацией без водорода. При дальнейшем увеличении мольного соотношения водород : этилен в газовой фазе до единицы, скорость полимеризации продолжает понижаться, но в значительно меньшей степени. При этом для катализатора ТМК-M в присутствии водорода наблюдается небольшое понижение активности в ходе полимеризации, в то время как катализатор ТМК-H сохраняет относительно стабильную кинетическую кривую в присутствии водорода.
Рис. 1.
Влияние водорода на скорость полимеризации этилена w на катализаторах ТМК-М (а) и ТМК-Н (б); мольное соотношение [Н2]/[С2H4] = 0 (1, 4), 0.25 (2, 5) и 1 (3, 6). Момент введения 14CO показан стрелками. Здесь и на рис. 2, 3 номера кривых соответствуют номерам опытов в таблицах.
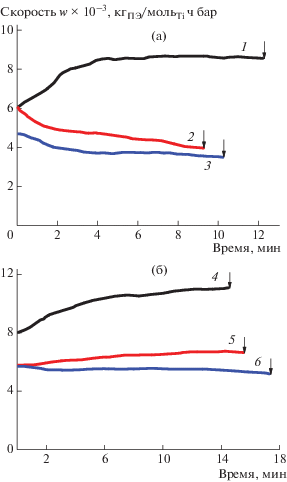
В табл. 1 приведены величины СР и kр, полученные при полимеризации этилена в отсутствие и присутствии водорода на исследованных катализаторах. Для обеих модификаций катализаторов число активных центров в отсутствие водорода (опыты 1, 4) имеет близкое значение (0.116 и 0.130 моль/мольTi для катализаторов ТМК-M и ТМК-H соответственно). Понижение скорости полимеризации со временем в присутствии водорода для катализатора ТМК-М (рис. 1а) может быть обусловлено уменьшением числа активных центров. Следует отметить, что активные центры катализатора ТМК-М очень чувствительны к введению водорода, т.е. даже при небольшом его количестве ([H2]/[C2H4] = 0.25) их число понижается с 0.116 до 0.062 моль/мольTi (опыты 1, 2), в отличие от катализатора ТМК-Н, в котором введение аналогичного количества водорода не приводит к существенному уменьшению числа активных центров (опыты 4, 5) и к нестационарности кинетической кривой (рис. 1б).
Таблица 1.
Влияние водорода на число активных центров и константу скорости роста при полимеризации этилена на катализаторах TMК-M и TMК-H
| Опыт, № | Мольное соотношение Н2/С2H4 в газовой фазе | w* × 10–3, кгПЭ/мольTi ч бар | СP, моль/мольTi | kр × 10–3, л/моль с |
|---|---|---|---|---|
| TMК-M | ||||
| 1 | 0 | 8.5 | 0.116 | 10.2 |
| 2 | 0.25 | 4.0 | 0.062 | 9.0 |
| 3 | 1 | 3.5 | 0.050 | 9.6 |
| TMК-H | ||||
| 4 | 0 | 11.2 | 0.130 | 11.9 |
| 5 | 0.25 | 6.6 | 0.110 | 9.0 |
| 6 | 1 | 5.2 | 0.069 | 10.8 |
| 7** | 0 → 0.25 → 0 | 12.6 | 0.160 | 11.2 |
Введение водорода в полимеризационную среду при мольном соотношении [H2]/[C2H4] = 1 (табл. 1, ср. опыты 1, 3 и 4, 6 для катализаторов ТМК-M и ТМК-H соответственно) приводит в обоих случаях к заметному понижению числа активных центров (в 2.3 и 1.9 раза для катализаторов ТМК-M и ТМК-H соответственно). Величины kр при введении водорода меняются незначительно и находятся в пределах (10.2–9.0) × 103 л/моль с для катализатора ТМК-M и (11.9–9.0) × 103 л/моль с для катализатора ТМК-H. При этом видна небольшая тенденция к снижению величины kр в 1.1.–1.2 раза при полимеризации в присутствии водорода.
Для проверки обратимости изменения величин СP и kр в присутствии водорода, был проведен специальный опыт с введением и удалением водорода из реакционной среды, при полимеризации этилена на катализаторе ТМК-H, обладающем высокой активностью и стационарной скоростью полимеризации. В этом опыте полимеризацию этилена начинали без водорода, затем вводили его при мольном соотношении [H2]/[C2H4] = 0.25 в газовой фазе. Наблюдали уменьшение скорости полимеризации в 3 раза. При удалении водорода из полимеризационной среды, скорость полимеризации возвращалась на прежний уровень (рис. 2). Так, полученные в табл. 1 в опыте 7 величины СР и kр после удаления водорода были близки к значениям в опыте 4, проведенном без водорода. Таким образом, изменения скорости полимеризации и величин СР и kр при введении водорода обусловлены обратимыми процессами трансформации активных центров в ходе полимеризации.
Рис. 2.
Изменение скорости от времени при полимеризации этилена на катализаторе ТМК-Н при введении и удалении водорода в процессе полимеризации.

Ранее было найдено [17], что для модельных титан-магниевых катализаторов с низким содержанием титана (≤0.1 мас. %) уменьшение скорости полимеризации в присутствии водорода происходит преимущественно за счет величины kр. Вместе с тем, при высоком отношении [H2]/[C2H4] = 1 видно небольшое понижение числа активных центров. Также в работе [17] была предложена схема трансформации активных центров при полимеризации этилена в присутствии водорода, объясняющая полученные результаты:
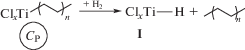


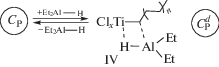
Схема включает реакции образования поверхностного гидридного соединения титана ClxTi–H в результате взаимодействия водорода с активным центром СР, содержащим растущую полимерную цепь (I); взаимодействия триэтилалюминия с гидридным соединением титана с образованием диэтил-алюминийгидрида AlEt2H и предшественника активных центров ClxTi–Et (II); обратимой адсорбции диэтилалюминийгидрида на предшественниках активных центров, присутствующих в реакционной среде; эта реакция приводит к понижению числа активных центров при полимеризации в присутствии водорода (III); обратимой адсорбции AlEt2H на активных центрах СР с образованием временно неактивных (“спящих”) центров $С_{{\text{P}}}^{d}$, содержащих связь Ti-полимер (IV). При ингибировании полимеризации введением 14СО, радиоактивная метка входит как в растущую полимерную цепь в активных центрах СР, так и в полимерную цепь в “спящих” центрах. В этом случае число радиоактивных меток в полимере является суммой величин (СР + $С_{{\text{P}}}^{d}$) и расчет величины kр по числу радиоактивных меток приводит к более низким значениям величины kр по сравнению с опытами, проведенными в отсутствие водорода. Такой результат был получен в работе [17] при исследовании влияния водорода на скорость полимеризации этилена для катализаторов с низким содержанием титана. Так, скорость полимеризации этилена в присутствии водорода понижается преимущественно за счет уменьшения рассчитываемой величины kр, вследствие образования временно неактивных (“спящих”) центров по реакции (IV). При высоком содержании водорода дополнительно наблюдается снижение величины СР за счет реакции (III).
В случае катализаторов с высоким содержанием титана, использованных в настоящей работе, понижение скорости полимеризации в присутствии водорода происходит за счет уменьшения числа активных центров в широкой области содержания водорода в соответствии с реакцией (III).
Как отмечено в работе [22], катализатор ТМК-М обладает повышенной чувствительностью к регулированию молекулярной массы водородом по сравнению с катализатором ТМК-Н. Поэтому можно предположить, что при введении водорода в случае катализатора ТМК-М образуется больше гидридов AlEt2H, и в соответствии с реакцией (II) значительная часть активных центров может находиться в виде предшественников СPr при невысокой концентрации водорода. Это приводит к тому, что для катализатора ТМК-M понижение скорости полимеризации этилена происходит в основном за счет уменьшения величины СP (ср. опыты 1 и 2 в табл. 1) и блокирования предшественников активных центров по реакции (III). Для катализатора ТМК-H уменьшение скорости полимеризации при невысоком содержании водорода [H2]/[C2H4] = 0.25 (опыт 5) происходит как за счет уменьшения величины СР, так и за счет уменьшения рассчитываемой величины константы скорости роста (ср. опыты 4 и 5). Таким образом, в этом случае оба процесса – блокирование предшественников активных центров по реакции (III) и блокирование самих активных центров по реакции (IV) – проходят в близком соотношении. При увеличении содержания водорода преобладает процесс блокирования предшественников активных центров по реакции (III), поскольку в этих условиях их доля растет вместе с увеличением концентрации диэтилалюминийгидрида. В результате, в этих условиях (опыты 5 и 6) наблюдается значительное понижение числа активных центров.
В целом, полученные в настоящем исследовании сведения о влиянии водорода на величины СP и kр для катализаторов ТМК-H и ТМК-M, а также результаты работы [17] для модельных катализаторов с низким содержанием титана соответствуют схеме превращений активных центров при полимеризации этилена в присутствии водорода. Однако эти катализаторы существенно отличаются относительным вкладом реакций временной дезактивации предшественников активных центров (III) и дезактивации активных центров (IV) при полимеризации в присутствии водорода. Такое различие приводит к разным выводам относительно роли изменения числа активных центров и рассчитываемой величины константы скорости роста в наблюдаемом эффекте уменьшения скорости полимеризации этилена в присутствии водорода.
Влияние соотношения [H2]/[C2H4] на молекулярно-массовые характеристики полиэтилена, полученного на катализаторах ТМК-М и ТМК-Н, и расчет константы скорости переноса цепи с водородом
В табл. 2 показано влияние концентрации водорода (мольное соотношение [H2]/[C2H4] в газовой фазе в реакторе при постоянной концентрации этилена) на молекулярную массу и молекулярно-массовое распределение полиэтилена, полученного на титан-магниевых катализаторах ТМК-M и ТМК-H.
Таблица 2.
Данные о молекулярно-массовых характеристиках полимеров, полученных на катализаторах ТМК-M и ТМК-H при разном мольном соотношении [H2]/[C2H4] в газовой фазе
| Опыт, № | Мольное соотношение Н2/С2H4 в газовой фазе | w* × 10–3, кгПЭ/мольTi ч бар | Mn | Mw | Đ |
|---|---|---|---|---|---|
| TМК-M | |||||
| 1 | 0 | 7.0 | 220** | 1100*** | – |
| 2 | 0.125 | 4.7 | 44 | 220 | 5.0 |
| 3 | 0.25 | 4.2 | 26 | 130 | 5.0 |
| 4 | 0.5 | 3.9 | 14 | 85 | 6.1 |
| TМК-H | |||||
| 5 | 0 | 9.6 | 347** | 1700*** | – |
| 6 | 0.125 | 5.6 | 63 | 310 | 4.9 |
| 7 | 0.25 | 6.5 | 43 | 210 | 4.9 |
| 8 | 0.5 | 5.7 | 30 | 150 | 5.0 |
В отсутствие водорода образуются полимеры с высокой молекулярной массой, которую трудно определить методом гель-проникающей хроматографии (табл. 2, опыты 1 и 5). В этом случае измеряли характеристическую вязкость получаемых полимеров и рассчитывали средневязкостную молекулярную массу Mη, близкую к величине Mw. Величины Mn в данных опытах рассчитывали по формуле ${{M}_{n}} = {{M}_{{\eta }}}{\text{/}}D$, где D – полидисперсность в опытах с минимальным количеством водорода (опыты 2 и 6).
При полимеризации в отсутствие водорода величины Mη для полимера, полученного на катализаторе ТМК-М, в 1.5 раза ниже по сравнению с величиной Мη для полимера, полученного на катализаторе ТМК-H. Введение водорода в обоих случаях влечет резкое снижение молекулярной массы полимеров.
Ранее было показано, что при полимеризации этилена на ТМК в присутствии водорода основной реакцией ограничения цепи является перенос цепи с водородом [32]. С учетом этих данных, известное выражение для степени полимеризации [33] можно преобразовать к следующему более простому виду:
(2)
$\frac{1}{{\overline {{{P}_{n}}} }} - \frac{1}{{\overline {P_{n}^{0}} }} = \frac{{k_{{tr}}^{{\text{H}}}{{{[{{{\text{H}}}_{2}}]}}^{m}}}}{{{{k}_{{\text{р}}}}[{{{\text{C}}}_{2}}{{{\text{H}}}_{4}}]}},$Таблица 3.
Константы скорости роста и переноса цепи водородом при полимеризации этилена на катализаторах ТМК-М и ТМК-Н
| Опыт, № | Катализатор | $k_{{tr}}^{{\text{H}}}$/kр | kр × 10–3, л/моль с | $k_{{tr}}^{{\text{H}}}$, л/моль с |
|---|---|---|---|---|
| 1 | TМК-M | 0.036 | 10.2 | 370 |
| 2 | TМК-H | 0.017 | 11.9 | 200 |
Примечание. Зависимость для определения величины $k_{{tr}}^{{\text{H}}}$/kр представлена на рис. 3.
На рис. 3 приведена зависимость (1/Pn – 1/$P_{n}^{0}$) от соотношения [H2]/[C2H4], построенная по данным о влиянии концентрации водорода на молекулярную массу полимеров, полученных на катализаторах TМК-M и TМК-H (табл. 2). Из этой зависимости найдены $k_{{tr}}^{{\text{H}}}$/kр (табл. 3). Используя величины константы скорости роста, полученные в опытах без водорода (10.2 × 103 и 11.9 × 103 л/моль с для катализаторов ТМК-M и ТМК-H соответственно), были рассчитаны величины $k_{{tr}}^{{\text{H}}}$ (табл. 3). Видно, что константа скорости переноса цепи с водородом для модифицированного катализатора ТМК-M (370 л/моль с) в 1.8 раза выше, чем величина $k_{{tr}}^{{\text{H}}}$ для катализатора ТМК-H (200 л/моль с), которая близка к величине $k_{{tr}}^{{\text{H}}}$, полученной в работе [17] для модельного катализатора ТМК-А с низким содержанием титана и не модифицированного этокси-группами (160 л /моль с).

ЗАКЛЮЧЕНИЕ
Получены данные о влиянии водорода на число активных центров и константы скорости роста цепи при полимеризации этилена на высокоактивных титан-магниевых катализаторах ТМК-М и ТМК-Н, содержащих 1.0–1.7 мас. % Ti. Найдено, что снижение скорости полимеризации при вводе водорода в реакционную среду преимущественно связано с уменьшением числа активных центров. С этой точки зрения, катализаторы ТМК-М и ТМК-Н отличаются от ранее исследованных модельных катализаторов с низким содержанием титана (<0.1 мас. %), в которых понижение скорости полимеризации происходит в основном за счет рассчитываемой величины константы скорости роста.
Показано, что уменьшение величины CP для катализатора ТМК-М происходит при вводе даже небольшого количества водорода (мольное соотношение в газовой фазе [H2]/[C2H4] = 0.25), в то время как в случае катализатора ТМК-Н введение аналогичного количества водорода приводит к небольшому понижению величин CP и kр в равной степени, и только при мольном соотношении [H2]/[C2H4] = 1 заметно понижение числа активных центров. Установлено, что уменьшение величины СР при введении водорода в полимеризационную среду является обратимым процессом. Полученные данные могут быть объяснены с использованием схемы, включающей реакции образования гидридов титана в результате взаимодействия активных центров с водородом; взаимодействия гидридов титана с AlEt3 с образованием молекул AlEt2H их обратимой адсорбцией как на активных центрах, так и на их предшественниках.
Авторы выражают благодарность М.П. Ваниной за анализ молекулярно-массовых характеристик образцов полимеров.
Работа выполнена в рамках Госзадания Института катализа Сибирского отделения РАН и частично поддержана Министерством науки и высшего образования Российской Федерации.
Список литературы
Guastalla G., Giannini U. // Makromol. Chem. Rapid Commun. 1983. V. 4. P. 519.
Busico V., Cipullo R., Corradini P. // Makromol. Chem. Rapid Commun. 1992. V. 13. P. 15.
Chadwick J.C., Miedema A., Sudmeijer O. // Makromol. Chem. 1994. V. 195. P. 167.
Mori H., Tashino K., Terano M. // Macromol. Chem. Phys. 1995. V. 196. P. 651.
Bukatov G.D., Goncharov V.S., Zakharov V.A. // Macromol. Chem. 1986. V. 187. P. 1041.
Bukatov G.D., Zakharov V.A. // Macromol. Chem. Phys. 2001. V. 202. P. 2003.
Barabanov A.A., Bukatov G.D., Zakharov V.A., Semikolenova N.V., Mikenas T.B., Echevskaja L.G., Matsko M.A. // Macromol. Chem. Phys. 2006. V. 207. P. 1368.
Barabanov A.A., Bukatov G.D., Zakharov V.A. // J. Polym. Sci., Polym. Chem. 2008. V. 46. P. 6621.
Barabanov A.A., Bukatov G.D., Zakharov V.A., Semikolenova N.V., Echevskaja L.G., Matsko M.A. // Macromol. Chem. Phys. 2008. V. 209. P. 2510.
Albizzati E., Giannini U., Morini G., Galimberti M., Barino I., Scardamaglia R. // Macromol. Symp. 1995. V. 89. P. 73.
Soares J.B.P., Hamielec A.E. // Polymer. 1996. V. 37. P. 4607.
Echevskaya L.G., Matsko M.A., Mikenas T.B., Nikitin V.E., Zakharov V.A. // J. Appl. Polym. Sci. 2006. V. 102. P. 5436.
Grieveson B.M. // Macromol. Chem. 1965. V. 84. P. 93.
Kissin Yu.V. // J. Polym. Sci., Polym. Chem. 2001. V. 39. P. 1681.
Kissin Yu.V., Rishina L.A. // Polymer Science A. 2008. V. 50. № 11. P. 1101.
Kissin Yu.V., Mink R.I., Nowlin T.E., Brandolini A.J. // Top. Catal. 1999. V. 7. P. 69.
Sukulova V.V., Barabanov A.A., Mikenas T.B., Matsko M.A., Zakharov V.A. // J. Mol. Catal. 2018. V. 445. P. 299.
Nikolaeva M.I., Mikenas T.B., Matsko M.A., Echevskaya L.G., Zakharov V.A. // J. Appl. Polym. Sci. 2010. V. 115. P. 2432.
Zakharov V.A., Bukatov G.D., Barabanov A.A. // Macromol. Symp. 2004. V. 213. P. 19.
Barabanov A.A., Sukulova V.V., Matsko M.A., Zakharov V.A. // J. Mol. Catal. Chem. 2015. V. 396. P. 328.
Koshevoy E.I., Mikenas T.B., Zakharov V.A., Shubin A.A., Barabanov A.A. // J. Phys. Chem. C. 2016. V. 120. P. 1121.
Mikenas T.B., Koshevoy E.I., Cherepanova S.V., Zakharov V.A. // J. Polym. Sci, Part A, Polym. Chem. 2016. V. 54. P. 2545.
Mikenas T.B., Koshevoy E.I., Zakharov V.A. // J. Polym. Sci, Part A, Polym. Chem. 2017, V. 55. P. 2298.
Веселовская Е.И., Северова Н.Н., Дунтов Ф.И. Сополимеры этилена. Москва: Химия, 1983. 1–224 p.
Kissin Yu.V. // J. Polym. Sci, Part A, Polym. Chem. 2003. V. 41. P. 1745.
Meshkova I.N., Ushakova T.M., Gul′tceva N.M. // Polymer Science A. 2004. V. 46. № 12. P. 1213.
Barabanov A.A., Zakharov V.A. // Catal. Commun. 2014. V. 45. P. 79.
Barabanov A.A., Bukatov G.D., Zakharov V.A., Semikolenova N.V., Echevskaja L.G., Matsko M.A. // Macromol. Chem. Phys. 2005. V. 206. P. 2292.
Barabanov A.A., Semikolenova N.V., Matsko M.A., Echevskaja L.G., Zakharov V.A. // Polymer. 2010. V. 51. P. 3354.
Barabanov A.A., Semikolenova N.V., Bukatov G.D., Matsko M.A., Zakharov V.A. // J. Polym. Res. 2012. V. 19. № 11. P. 9998.
Wilson T.P., Hurley C.P. // J. Polym. Sci., Polym. Symp. 1963. V. 1. P. 281.
Nikolaeva M.I., Mikenas T.B., Matsko M.A., Echevskaya L.G., Zakharov V.A. // J. Appl. Polym. Sci. 2011. V. 122. № 5. P. 3092.
Natta G., Pasquon I. // Adv. Catal. 1959. V. 11. P. 1.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)