

Высокомолекулярные соединения (серия Б), 2020, T. 62, № 4, стр. 315-320
Метанолиз отходов поликарбонатов как метод регенерации мономеров для их получения
А. Я. Самуилов a, М. В. Коршунов a, Я. Д. Самуилов a, *
a Казанский национальный исследовательский технологический университет
420015 Казань, К. Маркса, 68, Россия
* E-mail: ysamuilov@yandex.ru
Поступила в редакцию 23.10.2019
После доработки 10.01.2020
Принята к публикации 20.01.2020
Аннотация
Изучены факторы, влияющие на метанолиз поликарбонатов при катализе метилатом натрия. Скорость превращений возрастает с увеличением основных свойств растворителей, скорость метанолиза в зависимости от количества растворителя (тетрагидрофурана) описывается экстремальной зависимостью. Изменение концентрации метилата натрия и температуры мало влияет на метанолиз поликарбонатов.
ВВЕДЕНИЕ
Масса полимерных отходов в мире в настоящее время достигает 300 млн т в год [1]. Среди них немаловажное значение имеют отходы поликарбонатов. Мировое производство ПК в 2016 г. достигло почти 5 млн т [2] и объемы его производства возрастают из года в год. Получаемые на основе 2,2-бис-(4-гидроксифенил)пропана (бисфенол А) ПК представляют особую опасность для окружающей среды [3], что обусловлено высокой токсичностью бисфенола А [4, 5], образующегося при гидролизе ПК. В связи с этим утилизация отходов ПК методом захоронения, который пока широко используют в разных странах для утилизации полимерных отходов [6], является совершенно неприемлемым. Предложен ряд других методов утилизации отходов ПК: механический [7–10], некаталитический [11–14] и каталитический пиролиз [15, 16]. Механические методы переработки отходов ПК сопровождаются существенным понижением их физико-механических показателей. Пиролитические методы приводят к образованию сложной смеси продуктов деструкции ПК и мало применимы для переработки отходов ПК.
Гидролиз ПК на основе бисфенола А принципиально должен приводить к образованию бисфенола А и углекислого газа. Данный подход усложняется тем, что ПК совершенно не растворимы в воде, поэтому реакции необходимо проводить при повышенных температурах, когда ПК находятся в жидком состоянии. Многие реакции в воде протекают более эффективно, когда она находится в сверхкритическом состоянии [17, 18]. Сверхкритическая вода была использована для осуществления гидролиза ПК. В работе [19] исследован гидролиз ПК при 430°С и показано, что через 1 ч с выходом 88.9% образуется смесь, состоящая из п-изопропилфенола, п-изопропенилфенола, фенола и бисфенола А. При изучении гидролиза ПК в субкритической воде [20] было обнаружено, что при 300°С образуется смесь продуктов, состоящая из бисфенола А, фенола, п-трет-бутилфенола, п-изопропенилфенола, 1,2,4,5-тетраметилбензола, 1,2,4-триметил-5-изопропилбензола. Максимальный выход бисфенола А (40%) достигается через 30 мин. Увеличение продолжительности и температуры реакции приводит к резкому снижению выхода бисфенола А [20]. Представленные данные указывают на то, что гидролиз ПК протекает неселективно, и он осложняется побочными процессами, обусловленными термической нестабильностью бисфенола А.
Использование ионных жидкостей как растворителей в ряде случаев позволяет проводить химические превращения в мягких условиях [21, 22]. Ионные жидкости как среды были использованы и при гидролизе ПК [23, 24]. В работе [23] изучен гидролиз ПК в присутствии ацетата 1-бутил-3-метилимидазолия. При 140°С и массовом соотношении Н2О : ПК = 0.70 : 1.00 и конверсии ПК 95.4% за 3 ч выход бисфенола А составил 91.8%. Гидролиз описывается кинетическим уравнением первого порядка по ПК. Авторы работы [23] полагают, что стадией, определяющей скорость превращения, является не акт взаимодействия молекул воды с карбонатными фрагментами, а переход ПК в раствор. Ионные жидкости имеют высокую стоимость, поэтому их использование для утилизации отходов ПК проблематично.
Другим методом утилизации отходов ПК является их алкоголиз, приводящий к образованию бисфенола А и диалкилкарбонатов. Эти реакции проводят в присутствии кислот и оснований Льюиса как катализаторов [25–28]. Ионные жидкости способствуют алкоголизу ПК [25, 26]. В работе [29] изучен катализируемый едким натром метанолиз ПК. В чистом метаноле при 60°С через 330 мин выход бисфенола А составил 7%. При использовании смеси метанол : толуол (1:1) через 70 мин при этой же температуре выход бисфенола А достигал 96% [29]. Некаталитический метанолиз ПК протекает медленнее. В работе [30] показано, что при мольном соотношении метанол : элементарное звено ПК, равном 71, при 160°С через 180 мин образуется бисфенол А с выходом 60%. В реакции наблюдается индукционный период. Проведенное рассмотрение показывает, что разрабатываются различные подходы к утилизации отходов ПК. Наибольший интерес с практической точки зрения представляет метанолиз ПК, поскольку он позволяет получать бисфенол А и диметилкарбонат, которые являются мономерами для получения ПК. Из анализа литературных данных неясно, какие условия, какой катализатор являются лучшими для проведения указанного процесса.
В связи с этим представляют интерес данные работы [31], в которой методом функционала плотности B3LYP/6-311++G(df, p) рассмотрены механизмы модельной реакции переэтерификации диэтилкарбоната метанолом. Полученные данные указывают на то, что некаталитические превращения с участием ассоциатов метанола, катализируемые ацетатом цинка, характеризуются высокими барьерами свободной энергии и могут протекать только при повышенных температурах. Стадии метанолиза, катализируемых метилатом натрия, включают образование пред- и послереакционных комплексов [31]:
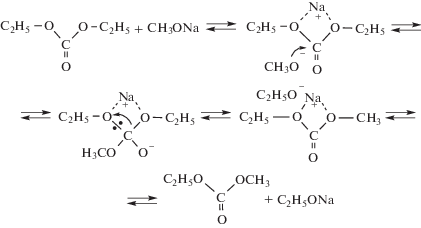
Элементарные стадии имеют малые барьеры свободной энергии, что является причиной быстрого протекания превращения [31]. Цель настоящей работы – выявление факторов, влияющих на скорость метанолиза ПК в присутствии метилата натрия.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовали ПК на основе бисфенола А производства Публичного акционерного общества “Казаньоргсинтез” марки РС-003 (TУ 2226-173-00203335-2007). Гранулы ПК предварительно механически измельчали.
Метанол кипятили над гидридом кальция и перегоняли при атмосферном давлении в атмосфере аргона. Едкий натр (“Sigma-Aldrich”) с содержанием основного вещества 97% применяли без дополнительной очистки. Метилат натрия генерировали в реакционной среде взаимодействием едкого натра с метанолом [32, 33]. Растворители очищали известными методами, они имели константы, совпадающие с литературными.
Метанолиз поликарбонатов в ТГФ
В двугорлую колбу объемом 50 мл, снабженную магнитной мешалкой, термометром, обратным холодильником добавляли 5 г (0.02 моля в расчете на элементарное звено) ПК, 0.1 г (0.0025 моля) гидроксида натрия, 4.8 г (6 мл, 0.15 моля) метанола и 10 г (11 мл, 0.14 моля) ТГФ. Реакционную массу перемешивали при 40°С в течение 10 мин. Колбу охлаждали до комнатной температуры и в нее вводили 25 мл дистиллированной воды. Выпавший твердый осадок фильтровали, промывали водой и сушили. Высушенный продукт экстрагировали метанолом (3 раза по 10 мл). Нерастворимую часть отделяли. Метанол удаляли упариванием. Полученное твердое вещество высушивали до постоянного веса.
Установление структуры полученных веществ
Строение полученного вещества устанавливали методом элементного анализа (элементный анализатор “Vario El cube”), спектроскопии ЯМР 1Н (спектрометр “Bruker Avance 400 MHz”) и хромато-масс-спектроскопии (газовый хроматограф с масс-селективным детектором “Маэстро ГХ 7820”). По данным этих методов, полученное вещество является бисфенолом А. Выход 3.6 г (80%). Тпл = = 158–159°С (по лит. данным [34] Тпл = 160°С).
Спектр ЯМР 1Н, дейтерохлороформ, δН, м.д.: 1.80 с (6Н, СН3), 4.95 с (2Н, ОН), 6.80–7.60 м (8 Н, С6Н4). Масс-спектр, m/z (Iотн, %): 228 (18) [M]+, 213 (100), 197 (4), 119 (4), 91 (21), 65 (7). Эти же методы использовали для установления структуры всех полученных в настоящей работе соединений.
Для доказательства образования в ходе метанолиза ПК диметилкарбоната летучую часть реакционной смеси после проведения процесса отгоняли; методом газовой хроматографии (хроматограф “КристалЛюкс-4000М”) обнаруживали метанол, диметилкарбонат и ТГФ.
Метанолиз ПК в других растворителях проводили по описанному выше методу. Кинетические эксперименты при разной температуре выполняли в закрытых колбах, помещенных в термостат. Периодически колбы извлекали, и реакционную смесь обрабатывали, как указано выше.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Влияние природы растворителей на метанолиз ПК
Необходимость изучения влияния природы растворителей на метанолиз ПК вызвана тем, что стадией, лимитирующей скорость реакции, в процессах гидролиза [23], метанолиза [35, 36] ПК является переход макромолекул в раствор. Переход макромолекул ПК в раствор затруднен тем, что между ними существуют межмолекулярные взаимодействия различной природы. Концевые гидроксильные группы в ароматических ядрах ПК образуют водородные связи [37]. Рассмотрение кристаллической структуры модельного соединения (дифенилкарбоната) указывает на существование между ароматическими ядрами π–π-взаимодействий [38]. Кристаллическая структура другого модельного соединения 1,3-фенилен-бис-фенилкарбоната имеет слоистую структуру [39]. Отдельные слои связаны друг с другом за счет π–π-взаимодействий ароматических ядер. Полагают, что эти взаимодействия в основном являются дисперсионными [40, 41]. По данным работы [42], элементарные ячейки ПК на основе бисфенола А содержат цепи макромолекул, связанные друг с другом π–π-взаимодействиями ароматических ядер и водородными связями за счет взаимодействия связей С–Н в метильных фрагментах с неподеленными парами электронов атомов кислорода карбонатной группы.
Систему водородных связей в ПК на основе бисфенола А можно было бы разрушить, используя π- и n-донорные растворители. Можно было ожидать, что с повышением их основности реакции метанолиза будут протекать быстрее. В табл. 1 приведены полученные нами данные по выходу бисфенола А в метанолизе ПК в различных растворителях.
Таблица 1.
Выход бисфенола А при метанолизе поликарбоната в различных растворителях (время реакции 10 мин, температура 40°С, масса поликарбоната 5 г (0.02 моля в расчете на элементарное звено), NaOH 0.15 г (3.7 × 10–3 моля), СН3ОН 5 г (0.16 моля), масса растворителя 10 г)
| Растворитель | Выход бисфенола А, % | Сродство к протону растворителя, кДж/моль [43] |
|---|---|---|
| Метанол | 0 | 754.3 |
| Толуол | 30 | 784.0 |
| 1,4-Диоксан | 75 | 797.4 |
| ТГФ | 80 | 822.1 |
В качестве меры основности растворителей использованы величины их сродства к протону [43]. Из табл. 1 следует, что основность растворителей оказывает сильное влияние на метанолиз ПК. В метаноле, растворителе с пониженной основностью, алкоголиз практически не протекает. В ТГФ, растворителе с наибольшей основностью среди использованных, реакция протекает наиболее полно. Имеются указания на то [44], что в толуольных растворах ПК (0.5–4.0 мас. %) сохраняется существенная агрегация. Полученные данные свидетельствуют о возможности успешного проведения алкоголиза ПК при значительном разрушения ассоциированной структуры ПК. Если она мало разрушена, то реакционные центры ПК остаются мало доступными для атаки алкоголят-ионами. Следствием является медленное протекание реакции.
Зависимость скорости метанолиза ПК от их концентрации в ТГФ
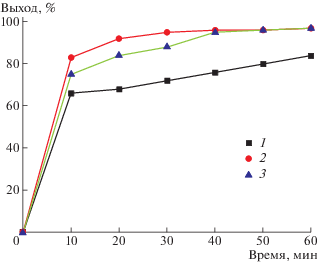
В соответствии с законом действующих масс скорость химических реакций прямо пропорциональна произведению текущих концентраций реагирующих веществ. Можно было предполагать, что в изучаемом метанолизе ПК возможны отклонения от этого закона. При высоких концентрациях ПК в растворе может происходить неполное разрушение ассоциированной структуры ПК. Следствием этого должно являться понижение скорости алкоголиза с повышением концентрации ПК в растворе. При малых концентрациях ПК в растворе скорости реакций должны понижаться из-за уменьшения концентрации карбонатных центров. На рис. 1 приведены кинетические кривые метанолиза ПК в ТГФ при разной концентрации полимера в растворе. Как видно, наибольшая степень превращения при метанолизе наблюдается при массовом соотношении ТГФ : : ПК, равным 2 : 1. И увеличение, и уменьшение концентрации ПК приводит к уменьшению выхода бисфенола А.
Рис. 1.
Зависимость выхода бисфенола А от продолжительности метанолиза ПК при 40°С и количестве NaOH 10 г. Количество ТГФ 5 (1), 10 (2) и 15 г (3). Здесь и на рис. 2 и 3 масса ПК и метанола 5 г.
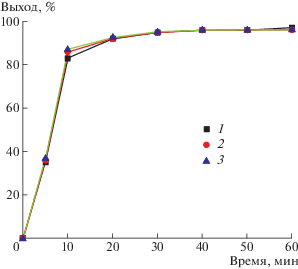
Зависимость скорости метанолиза ПК от концентрации NaOH
Скорость каталитических реакций прямо пропорциональна концентрации катализаторов [45]. Вопреки ожиданиям, в катализируемом метилатом натрия метанолизе ПК выход бисфенола А практически не зависит от концентрации NaOH (рис. 2).
Рис. 2.
Зависимость выхода бисфенола А от времени метанолиза ПК. Количество NaOH 0.10 (1), 0.20 (2) и 0.25 г (3). Количество ТГФ 10 г, Т = 40°С.
Полученный результат указывает на то, что лимитирующей в метанолизе ПК является не стадия взаимодействия метилата натрия с карбонатными фрагментами, а какая-то другая, в которой метилат натрия участия не принимает. На наш взгляд, такой стадией может быть разрушение ассоциированной структуры ПК. Данные работы [31] свидетельствуют о том, что катализируемый метилатом натрия алкоголиз органических карбонатов должен протекать быстро. Результаты, приведенные на рис. 2, указывают на то, что в растворе ПК в ТГФ значительная часть карбонатных фрагментов, видимо, существует в виде ассоциатов, которые находятся в равновесии с неассоциированными формами.
Влияние температуры на метанолиз ПК в ТГФ
В работе [31] установлено, что элементарные стадии метанолиза диэтилкарбоната в присутствии метилата натрия характеризуются малыми величинами энтальпий активации. Скорость подобных реакций слабо меняется при изменении температуры. Мы изучили влияние температуры на метанолиз ПК в присутствии метилата натрия в среде тетрагидрофурана (рис. 3).
Рис. 3.
Зависимость выхода бисфенола А от времени метанолиза ПК в ТГФ. Количество NaOH и ТГФ 10 г; Т = 40 (1), 50 (2) и 60°С (3).
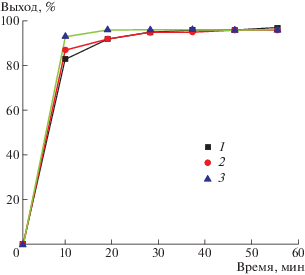
Полученные результаты указывают на действительно незначительное влияние температуры на метанолиз ПК в присутствии оснований. Наблюдается лишь небольшое возрастание выхода бисфенола А с повышением температуры. Это согласуется с тем, что энтальпии активации элементарных стадий метанолиза ПК малы. Если в ПК ассоциированная структура разрушена, то его метанолиз в присутствии алкоголятов как катализаторов мало чувствителен к температуре реакции.
ЗАКЛЮЧЕНИЕ
Изучение метанолиза ПК в условиях катализа алкоголятом натрия приводит к выводам, имеющим важное прикладное значение.
Успешное проведение процесса возможно тогда, когда принимаются меры по предварительному разрушению ассоциированной структуры ПК. Для их разрушения необходимо использовать растворители с повышенной основностью.
Существуют оптимальные концентрации ПК в растворителях с повышенной основностью. В концентрированных растворах доля разрушенных ассоциатов ПК мала, что приводит к понижению степени метанолиза.
Образование доступных для метанолиза центров в ПК может оказаться стадией, определяющей скорость реакции. В данном случае исчезает чувствительность превращений к изменению концентрации катализатора.
Энтальпии активации метанолиза малы, что приводит к малой зависимости скоростей метанолиза ПК от температуры.
Список литературы
Fink J.K. Polymer Waste Management. Hoboken: Wiley, 2018.
Fukuoka Sh., Fukawa I.T., Fujita H., Sugiyama N., Sawa T. // Org. Process Res. Dev. 2019. V. 23. № 2. P. 145.
Kumar A., Gupta K., Tomer V., Kaur A., Kumar V. // Toxicol. Int. 2018. V. 25. № 1. P. 78.
Singh Sh., Li S.Sh.-L. // Int. J. Mol. Sci. 2012. V. 13. № 8. P. 10143.
Ullah A., Pirzada M., Jahan S., Ullah H., Khan M.J. // Toxicol. Ind. Health. 2019. V. 35. № 4. P. 78.
Okan M., Aydin H.M., Barsbay M. // J. Chem. Technol. Biotechnol. 2018. V. 94. № 1. P. 8.
Pérez J.M., Vilas J.L., Lazaa J.M., Arnáizb S., Mijangosa F., Bilbaoc E., Rodrígueza M., León L. M. // J. Mater. Proc. Techn. 2010. V. 210. № 5. P. 727.
Ronkay F. // Acta Polytech. Hung. 2013. V. 10. № 1. P. 209.
Zhang J., Panwar A., Bello D., Isaacs J.A., Jozokos T., Mead J. // Polym. Eng. Sci. 2018. V. 29. № 6. P. 1547.
Delva L., Hubo S., Cardon L., Ragaert K. // Waste Manag. 2018. V. 82. P. 198.
Montaudo G., Carroccio S., Puglisi C. // J. Anal. Appl. Pyrolysis. 2002. V. 64. № 2. P. 229.
Jang B.N., Wilkie C.A. // Thermochim Acta. 2005. V. 426. № 1–2. P. 73.
Siddiqui N.M., Halim S., Redhwi H.H., Antonakou E.V., Achilias D.S. // J. Anal. Appl. Pyrolysis. 2018. V. 132. P. 123.
Feng Y., Li Z., Wang Y., Chen W., Wang B., Liu Ch., Shen Ch. // Macromol. Mater. Eng. 2019. V. 304. № 4. P. 1800667-1-8.
Chiu S.T., Chen S.H., Tsai C.T. // Waste Manag. 2006. V. 26. № 3. P. 252.
Siddiqui N.M., Antonakou E.V., Redhwi H.H., Achi-lias D.S. // Thermochim. Acta. 2019. V. 675. P. 69.
Akiya N., Savage Ph.E. // Chem. Rev. 2002. V. 102. № 8. P. 2725.
Brunner G. Hydrothermal and Supercritical Water Processes. Amsterdam: Elsevier, 2014.
Tagaya H., Katoh K., Kadokawa J., Chiba K. // Polym. Degrad. Stab. 1999. V. 64. № 2. P. 289.
Huang Y., Liu Sh., Pan Z. // Polym. Degrad. Stab. 2011. V. 96. № 8. P. 1405.
Environmentally Friendly Syntheses Using Ionic Liquids / Ed. by J. Dupont, T. Itoh, P. Lozano, S.V. Malhotra. Boca Raton: CRC Press, 2015.
MacFarlane D.R., Kar M., Pringle J.M. Fundamentals of Ionic Liquids. From Chemistry to Applications. Weinheim: Wiley-VCH, 2017.
Song X., Liu F., Li L., Yang X., Yu Sh, Ge X. // J. Hazar. Mater. 2013. V. 244–245. P. 204.
Liu M., Guo J., Gu Y., Gao J., Liu F. // Polym. Degrad. Stab. 2018. V. 157. P. 9.
Guo J., Liu M., Gu Y., Wang Y., Gao J., Liu F. // Ind. Eng. Chem. Res. 2018. V. 57. № 32. P. 10915.
Liu M., Guo J., Gu Y., Gao J., Liu F., Yu Sh. // ACS Sustainable Chem. Eng. 2018. V. 6. № 10. P. 13114.
Taguchi M., Ishikawa Y., Kataoka Sh., Naka T., Funazukuri T. // Catal. Commun. 2016. V. 84. P. 93.
Liu F., Xiao Y., Sun X., Qin G., Song X., Liu Y. // Chem. Eng. J. 2019. V. 369. P. 205.
Hu L., Oku A., Yamada E. // Polymer. 1998. V. 39. № 16. P. 3841.
Kim D., Kim B., Cho Y., Han M., Kim B.-S. // Ind. Eng. Chem. Res. 2009. V. 48. № 14. P. 6591.
Samuilov A.Y., Samuilov Y.D. // Theor. Chem. Acc. 2019. V. 138. № 2. P. 24.
The Chemistry of the Hydroxyl Group / Ed. by S. Patai. London: Wiley, 1971.
Granjo J., Oliveira N. // Ind. Eng. Chem. Res. 2016. V. 55. № 1. P. 156.
CRC Handbook of Chemistry and Physics / Ed. by W.H. Haynes. Boca Raton: CRC Press, 2017.
Liu F., Li L., Yu S., Li Z., Ge X. // J. Hazar. Mater. 2011. V. 189. № 1–2. P. 249.
Liu F., Li Z., Yu S., Ge X. // J. Hazard. Mater. 2010. V. 174. № 1–3. P. 872.
Puglisi C., Samperi F., Carroccio S., Montaudo G. // Rapid Commun. Mass Spectr. 1999. V. 13. № 22. P. 2268.
King J.A., Bryant G.L. // Acta Crystal., C: Struct. Chem. 1993. V. 49. № 3. P. 550.
Solomos M.A., Bertke J.A., Swift J.A. // Acta Crysal., Crystal. Commun. 2017. V. 73. № 12. P. 1942.
Cabaleiro-Lago E.M., Rodríguez-Otero J. // ACS Omega. 2018. V. 3. № 8. P. 9348.
Podeszwa R., Bukowski R., Szalewic K. // J. Phys. Chem. A. 2006. V. 110. № 34. P. 10345.
Gilbert M. Brydson’s Plastic Materials. Amsterdam: Elsevier. 2017.
Hunter E.P., Lias S.G. // J. Phys. Chem. Ref. Data. 1998. V. 27. № 3. P.413.
Dybal J., Schmidt P., Baldrian J., Kratochvı J. // Macromolecules. 1998. V. 31. № 19. P. 6611.
Murzin D.Yu., Salmi T. Catalytic Kinetics. Chemistry and Engineering, Amsterdam: Elsevier, 2016.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)