

Высокомолекулярные соединения (серия Б), 2020, T. 62, № 4, стр. 306-314
Синтез полимеров уретанового типа с полидиметилсилоксановыми блоками для изготовления волокнистых матриксов методом электроформования
И. К. Шундрина a, *, И. В. Олейник a, В. И. Пастухов a, Л. А. Шундрин a, В. С. Черноносова b, П. П. Лактионов b
a Новосибирский институт органической химии им. Н.Н. Ворожцова
Сибирского отделения Российской академии наук
630090 Новосибирск, пр. академика Лаврентьева, 9, Россия
b Институт химической биологии и фундаментальной медицины
Сибирского отделения Российской академии наук
630090 Новосибирск, пр. академика Лаврентьева, 8, Россия
* E-mail: ishund@nioch.nsc.ru
Поступила в редакцию 02.12.2019
После доработки 28.02.2020
Принята к публикации 12.03.2020
Аннотация
Cинтезирован ряд полиуретанов, полимочевин и полиуретанмочевин с гибкими блоками на основе α,ω-бис-(гидроксиалкил)полидиметилсилоксана и α,ω-бис-(аминопропил)полидиметилсилоксана с молекулярными массами (5.6 и 0.9) × 103 соответственно для получения искусственных сосудов методом электроформования. Структура и свойства полимеров охарактеризованы методами гель-проникающей хроматографии, дифференциальной сканирующей калориметрии, термогравиметрии и динамического механического анализа. Установлено, что полидиметилсилоксановые сегменты в структуре полимеров формируют свою фазу с температурой стеклования, характерной для полидиметилсилоксана. Исследованы особенности получения волокон из растворов синтезированных эластомеров методом электроформования. Показано, что на морфологию представленных волокон большое влияние оказывает модуль упругости полимеров.
Современные технологии изготовления трехмерных конструкций, такие как электроформование и 3D-печать [1–3], позволяют принципиальным образом изменить подходы к созданию биопротезов, которые должны соответствовать протезируемым тканям по механическим свойствам и быть биологически совместимыми. Электроформованием можно несложно и воспроизводимо получать матриксы из синтетических и природных полимеров с разными механическими свойствами, различной пористостью, легко формовать трубки – протезы сосудов, укладывая волокна на цилиндрический электрод-коллектор [4–7].
Протезы сосудов могут быть выполнены из биологически инертных или биодеградируемых материалов [8–10]. Для изготовления матриксов методом электроформования применяют разнообразные синтетические и природные полимеры: нейлон, полиоксиэтилен, поликапролактам, полилактиды, полигликолиды, полиуретаны, хитозаны, белки и другие [11–13]. Однако далеко не все существующие изделия отвечают предъявляемым к ним требованиям. В частности, протезы сосудов малого диаметра не обеспечивают длительной проходимости и требуют принципиального улучшения механических свойств, био- и гемосовместимости.
Наиболее перспективными для изготовления протезов сосудов малого диаметра являются полиуретановые эластомеры, обладающие достаточными прочностью, эластичностью и высокой биосовместимостью [14, 15]. Однако полиуретаны в условиях in vivo склонны к биодеградации из-за реакций гидролиза и окисления [16]. В последние десять–двадцать лет появился набор более стабильных к гидролизу полиуретанов [17], не содержащих в своей структуре сложноэфирных групп. Окислительная деградация этих полиуретанов в биологических средах связана с отрывом метиленового протона, находящегося в α-положении к атому кислорода в эфирном фрагменте –О–(CH2)n– [18, 19], различными окислителями (кислородом или переходными металлами), которые могут генерироваться клетками организма (например, супероксидным радикалом в макрофагах, пероксидными радикалами, пероксидазой или каталазой и т.д.) [20, 21]. В результате наблюдается деструкция цепей или их сшивка, что приводит к потере прочности или росту трещин на поверхности имплантата [22, 23]. Для повышения биостабильности полиуретанов были предприняты попытки замены чувствительного полиэфирного мягкого сегмента на более устойчивый полидиметилсилоксановый как один из наиболее широко используемых полимерных материалов для производства биомедицинских устройств из-за их превосходной биосовместимости, низкой токсичности, окислительной и термической стабильности [17]. Эти преимущества также дают возможность применять ПДМС в качестве мягкого сегмента для синтеза биостабильных полиуретанов. Было показано, что полидиметилсилоксановые блоки препятствуют адгезии к поверхности имплантов макрофагов, которые инициируют окисление полиуретанов за счет генерации супероксид-радикалов [24].
Полиуретановые эластомеры обычно проявляют двухфазную микроструктуру, возникающую из-за химической несовместимости между мягкими и жесткими блоками [25]. Жесткий блок в этой двухфазной микроструктуре может действовать как физическая сшивка или армирующий наполнитель, в то время как мягкий блок образует эластичную матрицу. Такое микрофазное разделение демонстрирует превосходные физические и механические свойства полиуретановых эластомеров, среди них прочность и большое относительное удлинение в области упругой деформации [26, 27]. Степень сегрегации зависит не только от соотношения твердых и мягких блоков, но и от типа удлинителя цепи, химической структуры, молекулярной массы мягких блоков, а также от плотности водородных связей. Низкая полярность диолов ПДМС по сравнению с полиэфирными или поликарбонатными приводит не только к выраженной фазовой сегрегации, но и к плохой межфазной адгезии, а следовательно, плохим механическим свойствам [28]. Механические свойства (модуль упругости, прочность) силоксановых полиуретанов могут быть улучшены, если в полимерную цепь ввести мочевинные группы, которые повышают жесткость цепи, увеличивая плотность межмолекулярных водородных связей [29]. Несмотря на ряд публикаций о синтезе и свойствах биостабильных полиуретанов с силоксановыми блоками [30–33], в литературе отсутствует информация об их применении для изготовления полимерных матриксов по технологии электроформования из растворов.
В настоящей работе описан синтез ряда биостабильных низкомодульных полиуретанов, полимочевин и полиуретанмочевин, содержащих мягкие ПДМС-блоки, которые могут быть задействованы в технологии электроформования для изготовления протезов сосудов малого диаметра, и исследованы закономерности получения из них волокон методом электроформования из раствора.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
4,4'-Дифенилметандиизоцианат (4,4'-MDI) производства “Acros” и α,ω-бис-(аминопропил)полидиметилдисилоксан с Mn = 0.9 × 103 (Si-900) производства “ABCRGmbH&Co.KG”, а также 4,4'-дициклогексилметандиизоцианат (H12MDI), катализаторы дибутилдилаурат олова (SnBu2-dilaurate) и 2-этилгексаноат олова(II) (Sn(Oct)2), 1,2-диаминоэтан, 1,4-диаминобутан и 4,4'-диаминодифениловый эфир –все фирмы “Aldrich” применяли без предварительной очистки. α,ω-Бис-(гидроксиалкил)полидиметилсилоксан сМn = = 5.6 × 103 (Si-5600, “Aldrich”) предварительно сушили под вакуумом (1 мм рт. ст.) в течение 12 ч; 1,3-бис-(гидроксибутил)тетраметилдисилоксан (Si-278, “ABCRGmbH&Co.KG”) сушили методом азеотропной отгонки воды толуолом. 1,4-Бутандиол (BDO, “Мerck”) перегоняли в вакууме, среднюю фракцию (Ткип = 119–120 град/10 мм рт. ст.) задействовали для синтеза. N,N-Диметилацетамид сушили над CaH2 в течение двух суток при комнатной температуре, затем перегоняли в вакууме (Ткип = 56–57 град/12 мм рт. ст.) и хранили над молекулярными ситами 3 Å. Толуол и тетрагидрофуран кипятили со сплавом Na–K, перегоняли и хранили над молекулярными ситами 3 Å. Гексафторизопропанол (ГФИП; “P&MInvest”) использовали без дополнительной очистки.
Синтез полиуретанов PU-1, PU-3, PU-4 и полиуретанмочевин PU-7–PU-14 проводили по двухстадийной схеме, полиуретан PU-2 получали одностадийным синтезом (растворитель, температура и время выдержки на первой и второй стадиях приведены в табл. 1). На первой стадии к 2 ммолям диизоцианата (4,4'-MDI или H12MDI) в токе сухого аргона добавляли 1 ммоль олигомерного кремнийорганического диола и катализатор (SnBu2-dilaurate или Sn(Oct)2). Реакционную массу перемешивали при нагревании. На второй стадии к аддукту добавляли раствор 1 ммоля удлинителя цепи (диола или диамина), перед чем аддукт охлаждали до комнатной температуры, а затем продолжали нагревание реакционной массы. Концентрация реагентов в растворе на первой и второй стадиях составляла 40–45 и 20–25% соответственно. Полученные полимеры высаживали в этанол, сушили на воздухе, а затем под вакуумом (1 мм рт. ст.) при комнатной температуре до постоянной массы.
Таблица 1.
Состав, условия синтеза и выход полиуретанов и полиуретанмочевин
| Поли-мер | Макродиол, макро- диамин | Диизоцианат | Удлинитель цепи | Катализатор | Растворитель | Т, °С | Время, ч | Выход полимера, % |
|---|---|---|---|---|---|---|---|---|
| PU-1 | Si-5600 | H12MDI | Si-278 | SnBu2-dilaurate | без растворителя | 60 | 6* | 72 |
| толуол | 80 | 8** | ||||||
| PU-2 | Si-5600 | H12MDI | Si-278 | SnBu2-dilaurate | толуол | 80 | 8 | 88 |
| PU-3 | Si-5600 | H12MDI | HO(СН2)4OH | SnBu2-dilaurate | без растворителя | 60 | 6* | 99 |
| толуол | 90 | 9** | ||||||
| PU-4 | Si-5600 | 4,4'-MDI | HO(СН2)4OH | SnBu2-dilaurate | толуол | 60 | 4* | 79 |
| толуол +ДМАА | 90 | 9** | ||||||
| PU-5 | Si-900 | 4,4'-MDI | – | – | ДМАА+толуол (1 : 3) | 25 70 | 2* 5** | 87 |
| PU-6 | Si-900 | H12MDI | – | – | ДМАА +толуол | 25 | 4* | 78 |
| 70 | 9** | |||||||
| PU-7 | Si-5600 | H12MDI | H2N(CH2)2NH2 | Sn(Oct)2 | без растворителя | 60 | 6* | 75 |
| толуол | 70 | 6** | ||||||
| PU-8 | Si-5600 | H12MDI | H2N(CH2)4NH2 | Sn(Oct)2 | без растворителя | 60 | 6* | 90 |
| толуол | 70 | 6** | ||||||
| PU-9 | Si-5600 | H12MDI | 4,4'-Диамино-дифениловый эфир | Sn(Oct)2 | без растворителя толуол | 60 70 | 6* 6** | 96 |
| PU-10 | Si-5600 | 4,4'-MDI | H2N(CH2)2NH2 | Sn(Oct)2 | толуол | 60 | 6* | 89 |
| толуол+ДМАА | 70 | 6** | ||||||
| PU-11 | Si-5600 | 4,4'-MDI | H2N(CH2)2NH2 | Sn(Oct)2 | ДМАА | 60 | 6* | 91 |
| ДМАА | 70 | 6** | ||||||
| PU-12 | Si-5600 | 4,4'-MDI | H2N(CH2)4NH2 | – | толуол | 60 | 6* | 92 |
| толуол+ДМАА | 70 | 5** | ||||||
| PU-13 | Si-5600 | 4,4'-MDI | 4,4'-Диамино-дифениловый эфир | – | ДМАА ДМАА | 60 70 | 6* 6** | 91 |
| PU-14 | Si-5600 | 4,4'-MDI | 4,4'-Диамино-дифениловый эфир | – | ДМАА ДМАА | 60 70 | 6* 6** | 87 |
Синтез полимочевин PU-5, PU-6 осуществляли одностадийным методом без использования катализаторов (табл. 1) при концентрации реагентов в растворе 30%. В раствор 1 ммоля олигомерного кремнийорганического диамина при перемешивании в токе сухого аргона прикапывали раствор 1 ммоля диизоцианата (4,4'-MDI или H12MDI) и перемешивали сначала при комнатной температуре в течение 2 ч, затем, повысив температуру до 60°С, до окончания реакции. Полимеры высаживали в этанол, сушили на воздухе, потом под вакуумом (1 мм рт. ст.) при комнатной температуре до постоянной массы.
Во всех опытах завершенность реакции контролировали методом ИК-спектроскопии по отсутствию полосы валентных колебаний групп NCO (при 2275 см–1).
Молекулярно-массовые характеристики сополимеров измеряли методом ГПХ на приборе “Agilent LC-1200”, снабженном высокоэффективной колонкой “PL 1110-6500-gel 5 μm Mixed-C”. В качестве элюента использовали ТГФ, скорость подачи равнялась 1 мл/мин, температура составляла 40°С. Калибровку осуществляли по ПС-стандартам “Waters”.
Логарифмическую приведенную вязкость ηln растворов полимеров в ТГФ определяли с помощью капиллярного вискозиметра Уббелоде при концентрации 0.5 г/дл при 25°С. Исследования методом ДСК осуществляли на приборе “Netzsch STA-409” при скорости нагревания 10 град/мин в потоке гелия 50 мл/мин. Калибровку температуры и теплового потока проводили в соответствии со стандартом ISO 11357-1 по температуре плавления и энтальпиям фазовых переходов стандартных веществ из калибровочного набора от фирмы “NETZSCH” (C6H5COOH – 99.5%, RbNO3 – 99.99%, In – 99.99%, Sn – 99.99%, Bi – 99.9995%, Zn – 99.999%). Полученные данные обрабатывали с использованием программного обеспечения “NETZSCH Proteus Thermal Analysis”.
Испытания методом динамического механического анализа (ДМА) вели на приборе “DMA-242 C NETZSCH” в режиме растяжения и интервале температуры от –150 до +120°C в токе аргона. Образцы для испытаний представляли собой пленки толщиной ~300 мкм. Полоски полимеров размером 10 × 3 мм сканировали при скорости нагревания 3 град/мин, при частоте 1 Гц и амплитуде осцилляций 30 мкм. Температуру стеклования Tc устанавливали по положению максимума модуля механических потерь.
ИК-спектры регистрировали в режиме пропускания с использованием ИК-спектрометра “Tensor 27” с фурье-преобразователем в диапазоне волновых чисел 4000–500 см–1 для высушенных пленок полимеров, полученных из растворов в ГФИП на поверхности дисков из КBr.
Для изготовления волокнистых матриксов использовали растворы синтезированных полимеров в ГФИП. Динамическую вязкость растворов определяли при 20°С на вискозиметре “Brookrfield LVDV-II+PRO”, шпиндель СP-40. Матриксы изготавливали методом электроформования c применением вращающегося электрод-коллектора (диаметр 27 мм, длина 45 мм, скорость вращения 300 об/мин) при напряжении 16–24 кВ, расстоянии между электродами 180–200 мм и скорости подачи раствора 0.8–1.5 мл/ч при температуре 23–25°С, влажности 25–30% на лабораторной установке для электроформования (NF-103; “MECC”, Япония). После изготовления матриксы снимали с коллектора, высушивали не менее 12 ч под вакуумом при остаточном давлении не более 10 Па и хранили запаянными в пленке при 4°С.
Структуру матриксов исследовали с помощью сканирующего электронного микроскопа (СЭМ) TM-1000 фирмы “Hitachi” (Япония): ускоряющее напряжение 15 кэВ в режиме низкого вакуума. Образец без предварительной обработки фиксировали на предметном столике с помощью токопроводящего скотча.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Синтез высокомолекулярных полиуретанов на основе кремнийорганических диолов представляет собой экспериментальную сложность из-за большой разницы в параметрах растворимости между неполярными ПДМС-блоками и высоко полярными уретановыми сегментами. Нежелательное макроскопическое разделение фаз часто происходит уже во время синтеза полимеров. Поэтому выбор растворителя для синтеза очень критичен [34, 35]. Растворитель должен обеспечивать совместимость силоксановых олигодиолов или диаминов с диизоцианатами и удлинителями цепи, а также хорошую растворимость самих полимеров. Выбранные растворители для синтеза полимеров приведены в табл. 1. Следует отметить, что полностью исключить фазовое разделение при синтезе полиуретанов, полимочевин, полиуретанмочевин не удалось. Подбор растворителя помогал проводить реакцию в гомогенных условиях на стадии получения аддукта и небольших степенях конверсии на второй стадии. Как правило, при увеличении степени конверсии наблюдалось помутнение реакционной смеси, а в некоторых случаях –образование геля.
Структуры полидиметилсилоксановых диолов и диамина, используемых в синтезе полимеров, приведены ниже:
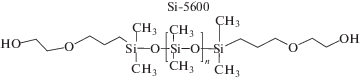
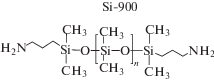
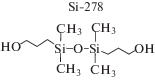
Для синтеза полиуретанов PU-1–PU-4 были использованы кремнийорганический олигодиол Si-5600 c Mn = 5.6 × 103, алифатический H12MDI или ароматический 4,4'-MDI диизоцианат (табл. 1). Удлинителем цепи служил низкомолекулярный 1,3-бис-(гидроксибутил)тетраметилдисилоксан (Si-278) или бутандиол. Олигодиол Si-5600 хорошо совместим с алифатическим диизоцианатом H12MDI и плохо – с ароматическим диизоцианатом 4,4'-MDI. Поэтому при синтезе полиуретанов по двухстадийной схеме с H12MDI (PU-1, PU-3) первую стадию можно проводить без растворителя. Оптимальным растворителем для второй стадии или одностадийного синтеза PU-2 являлся толуол. Полиуретан с ароматическим диизоцианатом 4,4'-MDI (PU-4) был получен на первой стадии в растворе толуола, на второй стадии в более полярном растворе толуол–ДМАА. Химическая природа удлинителя цепи оказывает влияние на выход полиуретанов на основе алифатического диизоцианата H12MDI. Наибольший выход продукта (99%) отмечен для PU-3 с бутандиолом в качестве удлинителя цепи по сравнению с PU-1 и PU-2, полученными с низкомолекулярным ПДМС-диолом Si-278. В последнем случае максимальный выход полимера PU-2 (88%) достигнут при использовании двухстадийной схемы синтеза. Несмотря на большую реакционную способность ароматических диизоцианатов по сравнению с алифатическими диизоцианатами, выход PU-4 оказался меньше выхода PU-3, что связано с бóльшей фазовой несовместимостью малополярных силоксановых блоков и более полярного ароматического диизоцианата.
Синтез полимочевин осуществляли в одну стадию без катализатора и удлинителя цепи при мольном соотношении α,ω-бис-(аминопропил)полидиметилдисилоксан с Mn = 0.9 × 103 : диизоцианат (1 : 1) в смеси растворителей ДМАА : : толуол. Полимочевины PU-5 и PU-6 получены с выходами 87 и 78% соответственно, которые коррелируют с различием в реакционной способности ароматических и алифатических диизоцианатов.
Синтез полиуретанмочевин проводили по двухстадийной схеме на основе кремнийорганического олигодиола Si-5600 c Mn = 5.6 × 103 и диизоцианатов H12MDI или 4,4'-MDI. В качестве удлинителя цепи на второй стадии применяли низкомолекулярные алифатические этилендиамин и 1,4-диаминобутан и ароматический 4,4'-диаминодифениловый эфир. PU-7–PU-9 на основе алифатического диизоцианата H12MDI получали на первой стадии без растворителя, на второй стадии использовали толуол. Полимеры имели высокий выход 87–96%. PU-10–PU-14 на основе ароматического диизоцианата получали в среде толуола или ДМАА. Выходы полимеров составили не менее 87%.
Структура синтезированных полиуретанов, полимочевин и полиуретанмочевин была подтверждена методом ИК-спектроскопии. Характерные для них ИК-спектры приведены на рис. 1. В ИК-спектрах наиболее интенсивные полосы поглощения относятся к колебаниям полиметилсилоксановых фрагментов: 1257 см–1 (деформационные колебания δ (С–Н) в Si–CH3); 1079, 1010 см–1 (валентные асимметричные νa и симметричные νs колебания Si–O–Si); 788 см–1 (маятниковые колебания ρ (С–Н) во фрагментах Si–CH3).
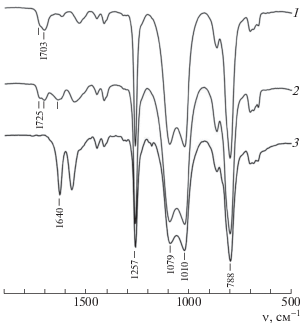
Валентные колебания свободных и связанных с водородом карбонилов С=О уретановых групп проявляются при 1725 и 1703 см–1, мочевинных групп – при 1640 см–1. Валентные колебания водородно-связанных групп NH имеют вид широкой полосы при 3326 см–1.
Хорошую растворимость полимеров в органических растворителях можно назвать важным условием для получения волокон методом электроформования. Для изготовления медицинских матриксов на основе полиуретанов предпочтение отдается ТГФ и ГФИП. Последний используется для волокон из смесей полиуретанов с биополимерами, например, с желатином [36]. Данные по растворимости синтезированных полимеров при комнатной температуре приведены в табл. 2. Все синтезированные полимеры за исключением полиуретанмочевин PU-11–PU-13 оказались хорошо растворимыми и в ТГФ, и в ГФИП. Причиной плохой растворимости полиуретанмочевин PU-11–PU-13, полученных с использованием ароматического диизоцианата, является сильная фазовая сегрегация в процессе синтеза полимеров. Так, PU-10 и PU-11 имеют одинаковый мономерный состав, хотя PU-10 хорошо растворим, а PU-11 только набухает. Синтез этих полимеров проходил в неодинаковых растворителях. Аналогично можно сравнить PU-13 и PU-14. Данные примеры указывают на важную роль растворителя в процессе синтеза полимеров.
Таблица 2.
Растворимость, вязкость и молекулярно-массовые характеристики полиуретанов, полимочевин и полиуретанмочевин
| Полимер | Растворимость | ηln, дл/г | Мn × 10–3 | Мw × 10–3 | Đ | |
|---|---|---|---|---|---|---|
| ТГФ | ГФИП | |||||
| PU-1 | + | + | 0.57 | 32 | 57 | 1.8 |
| PU-2 | + | + | 0.42 | 25 | 48 | 1.9 |
| PU-3 | + | + | 0.33 | 23 | 45 | 2.0 |
| PU-4 | + | + | 0.13 | 15 | 29 | 1.9 |
| PU-5 | + | + | 0.21 | 18 | 38 | 2.1 |
| PU-6 | + | + | 0.19 | 20 | 36 | 1.8 |
| PU-7 | + | + | 0.56 | 26 | 55 | 2.1 |
| PU-8 | + | + | 0.47 | 24 | 45 | 1.9 |
| PU-9 | + | + | 0.45 | 23 | 43 | 1.9 |
| PU-10 | + | + | 0.77 | 40 | 72 | 1.8 |
| PU-11 | _ | _ | – | – | – | – |
| PU-12 | _ | _ | – | – | – | – |
| PU-13 | Гель | Гель | – | – | – | – |
| PU-14 | + | + | 0.52 | 25 | 49 | 2.0 |
Молекулярно-массовые характеристики Mn и Mw, индекс полидисперсности Đ растворимых в ТГФ полимеров определены методом ГПХ (табл. 2). Для полиуретанмочевин PU-7–PU-10 и PU-14 среднечисленная молекулярная масса варьировалась в диапазоне (23–40) × 103, а среднемассовая – (43–72) × 103, при этом индекс полидисперсности не превышал 2.1. Для полимочевин PU-5 и PU-6 молекулярные массы меньше по сравнению с полиуретанмочевинами. Среднечисленная и среднемассовая молекулярные массы полиуретанов PU-1–PU-4 зависят от природы удлинителя цепи и его совместимости с мягким силоксановым блоком. Большие значения молекулярной массы получены при использовании более совместимого 1,3-бис-(гидроксибутил)тетраметилдисилоксана вместо 1,4-бутандиола (PU-1, PU-2). Значения приведенной вязкости растворов полимеров в ТГФ коррелируют со значениями молекулярной массы (табл. 2).
Следует отметить, что молекулярные массы синтезированных полимеров меньше по сравнению с коммерческими несилоксановыми полиуретанами TecoflexEG-80A и Pellethane 2363-80A, которые используются для получения медицинских матриксов методом электроформования. Значения Mw, определенные с помощью гель-хроматографии, составили (150 и 190) × 103 соответственно. Основная причина, по которой у этих полимеров более низкая молекулярная масса –фазовая несовместимость в процессе синтеза. Одним из вариантов решения данной проблемы является выбор структуры и длины силоксановых блоков. Существует достаточно широкий набор по молекулярным массам ПДМС-диолов с силанольным типом концевых гидроксильных групп. Однако высокая реакционная способность силанольных групп в реакциях с нуклеофильными реагентами даже в мягких условиях синтеза полиуретанов приводит к протеканию различных побочных реакций. Кроме того, связь Si–O–C крайне неустойчива к реакциям гидролиза. Этих недостатков лишены силоксановые олигомеры с концевыми алкилгидроксильными группами Si–R–OH. Кроме того, алкилгидроксильные группы в структуре ПДМС-олигомеров также могут иметь и другие преимущества перед силанольными Si–OH, среди них повышение совместимости с другими мономерами [37]. Однако выбор коммерчески доступных силоксановых олигомеров с концевыми алкилгидроксильными группами крайне ограничен.
Устойчивость синтезированных полимеров к термическому разложению определена методом термогравиметрии. Для характеристики термостойкости была выбрана температура, при которой полимер теряет 5% своей массы. Значения Т5% для синтезированных полимеров в сравнении с промышленным полиуретаном медицинского назначения Tecoflex-80A приведены в табл. 3. Все синтезированные полиуретаны термически устойчивы, Т5% > 290°С. Так, наиболее термостойким полимером оказалась полиуретанмочевина PU-14, полученная с использованием ароматического диизоцианата и ароматического удлинителя цепи 4,4'-диаминодифенилового эфира.
Таблица 3.
Термические свойства и модуль упругости полиуретанов, полимочевин и полиуретанмочевин
| Полимер | Температура начала разложения Т5%, °С | Тс, °С | Тпл, °С | Модуль упругости, Е ', МПа |
|---|---|---|---|---|
| Tecoflex-80A | 305 | –57 | 85 | 14.0 |
| PU-1 | 304 | –110 | – | 3.1 |
| PU-2 | 305 | –109 | – | 3.3 |
| PU-3 | 303 | –110 | – | 3.4 |
| PU-4 | 306 | –111 | – | 4.1 |
| PU-5 | 292 | –106 | 82 | 42 |
| PU-6 | 298 | –105 | 63 | 28 |
| PU-7 | 304 | –112 | – | 9.5 |
| PU-8 | 303 | –109 | – | 9.7 |
| PU-9 | 305 | –110 | – | 10.1 |
| PU-10 | 314 | –114 | – | 12.2 |
| PU-14 | 337 | –109 | – | 13.3 |
Температура стеклования и модуль упругости полимеров установлены методом ДМА, температура плавления – методом ДСК (табл. 3). Представленные характеристики определяют такие важные эксплуатационные свойства для протезов сосудов, как эластичность. Коммерческий Teco-flex-80A, полученный на основе поли(тетраметиленоксид)диола, H12MDI и 1,4-бутандиола, имеет температуру стеклования мягкого блока –57°С и температуру плавления +85°С. Мягкий блок в синтезированных полимерах образован ПДМС-фрагментами, которые выделяются в отдельную фазу с характерной для ПДМС низкой температурой стеклования (ниже –100°С). Значения температуры стеклования мягкого блока полимочевин PU-5 и PU-6, полученных с использованием более низкомолекулярного ПДМС-блока Si-900, несколько выше по сравнению с остальными полимерами, синтезированными на основе олигодиола Si-5600. Значения температуры плавления жестких блоков 82 и 63°С зафиксированы только для полимочевин PU-5 и PU-6 соответственно. Это может быть связано с разной массовой долей жестких блоков в структуре синтезированных полимеров, составляющей 21.2–21.7 и 9.2–12.6% для полиуретанов и полиуретанмочевин. Фазовое состояние синтезированных полимеров оказывает влияние на их модуль упругости (табл. 3). Самые высокие модули упругости имеют полимочевины PU-5 (42 МПа) и PU-6 (28 МПа), причем модуль упругости полимочевины на основе ароматического диизоцианата значительно выше. Модули упругости полиуретанмочевин варьируются в диапазоне 9.5–13.3 МПа и близки к Tecoflex-80A. Полидиметилсилоксановые полиуретаны (PU-1–PU-4) представляют собой эластичные материалы c низким модулем упругости 3.0–4.1 МПа.
Синтезированные полиуретаны, полиуретанмочевины и полимочевины, растворимые в ГФИП, были использованы для получения волокон методом электроформования. Концентрация полимеров в растворе ГФИП варьировалась от 3 до 40%. Физико-механические свойства и морфология волокнистого материала, изготовленного методом электроформования, зависят от свойств полимера, растворителя, условий среды и самого процесса [30, 31]. Особое влияние на морфологию матриксов оказывают концентрация полимера в растворе и его молекулярная масса, которые и определяют вязкость рабочего раствора. Оптимальной концентрацией рабочего раствора считалась концентрация, при которой формировались волокна без таких дефектов, как бусинки на волокне. Для коммерческого полиуретана Tecoflex-80A формирование бездефектных волокон происходит при концентрации раствора не менее 3%. Поскольку молекулярная масса синтезированных полимеров меньше, чем у Tecoflex-80A, то при такой концентрации раствора процесс электроформования протекает в режиме электрораспыления. Формирование бездефектных волокон из полимочевин и полиуретанмочевин происходит при концентрации раствора 16–20%. Динамическая вязкость растворов при указанных значениях концентрации варьировалась в диапазоне 160–300 сП. С ростом концентрации раствора увеличивается толщина синтезируемых волокон. Так, из растворов полиуретанмочевины PU-10 c концентрацией 16, 18, 20% получены матриксы со средним диаметром волокон 600, 800 и 1000 нм соответственно. Процесс электроформования волокон из растворов синтезированных полиуретанов значительно отличался от полимочевин и полиуретанмочевин (рис. 2).
Рис. 2.
Микрофотографии волокон, полученных электроформованием из растворов в ГФИП, полиуретана PU-1 с концентрацией 25 (а) и 40% (б), а также полимочевины PU-6 (в) и полиуретанмочевины PU-10 (г) с концентрацией 20%.

Для синтезированных полиуретанов не наблюдалось формирования волокон правильной формы. Полимер осаждался на подложку в виде плоских лент вне зависимости от концентрации растворов. Известно, что весьма значительное влияние на морфологию волокон оказывает макромолекулярная природа полимера. Методом электроформования достаточно легко получают волокна из растворов кристаллизующихся полимеров, таких как полилактид, поликарбонат, поликапролактам, полиакрилонитрил, полиамиды [38–41]. Волокна таких полимеров формируются при испарении растворителя и кристаллизации ориентированных макромолекулярных цепей. Получение волокон из аморфных эластомеров имеет ряд ограничений, связанных с релаксационными явлениями в полимере. Успешный опыт получения волокон из эластомеров методом электроформования описан только для термоэластопластов и сегментированных полиуретанов [42–45], в которых жесткие блоки играют роль физических узлов сетки. Синтезированные в данной работе полиуретаны представляют собой высокоэластичные низкомодульные эластомеры с низкой долей жестких блоков. После испарения растворителя в процессе электроформования волокна из таких полиуретанов сильно деформируются и теряют форму. Введение в структуру силоксановых полиуретанов мочевинных фрагментов или увеличение доли жестких блоков приводит к росту их модуля упругости и оказывает положительное влияние на формирование волокнистых матриксов.
Авторы выражают благодарность Химическому исследовательскому центру коллективного пользования СО РАН за проведение спектральных и аналитических измерений.
Работа выполнена при финансовой поддержке комплексной программы фундаментальных исследований Сибирского отделения Российской академии наук (коды проектов 0302-2018-0009 и 0309-2018-0014).
Список литературы
Hollister S.J. // Nature Mater. 2005. V. 4. № 7. P. 518.
Jia A.J., Ratima S.C. // Engineering. 2015. V. 1. № 2. P. 261.
Ingavle G.C., Leach J.K. // Tissue Eng. B. 2014. V. 20. № 4. P. 277.
Stitzel J.D., Liu J., Lee S.J., Komura M., Berry J., Soker S., Lim G., Dyke M.V., Czerw R., Yoo J.J., Atala A. // Biomaterials. 2006. V. 27. № 7. P. 1088.
Chernonosova V.S., Gostev A.A., Gao Yun, Chesalov Y.A., Shutov A.V., Pokushalov E.A., Karpenko A.A., Laktionov P.P. // BioMed. Res. Int. 2018. V. 2018. Art. ID 1380606.
Firoozi S., Derakhshan M.A., Karimi R. // Nanomed. Res. J. 2017. V. 2. № 2. P. 131.
Kennedy K.M., Bhaw-Luximon A., Jhurry D. // Acta Biomater. 2017. V. 50. P. 41.
Hasan A., Memic A., Annabi N., Hossain M., Paul A., Dokmeci M.R., Dehghani F., Khademhosseini A. // Acta Biomater. 2014. V. 10. № 1. P. 11.
Gostev A.A., Karpenko A.A., Laktionov P.P. // Polym. Bull. 2018. V. 75. № 9. P. 4311.
Aldana A.A., Abraham G.A. // Int. J. Pharmaceutics. 2017. V. 523. № 2. P. 441.
Wenguo C., Zhou Y., Chang J. // Sci. Technol. Adv. Mater. 2010. V. 11. № 1. P. 014108.
Abdoluosefi H.E., Honarasa G. // Mater. Res. Express. 2017. V. 4. № 10. P. 105308.
Pashneh-Tala S., MacNeil S., Claeyssens F. // Tissue Eng. B. 2016. V. 22. № 1. P. 68.
Nezarati R.M., Eifert M.B., Dempsey D.K., Cosgriff-Hernandez E. // J. Biomed. Mater. Res. B. 2015. V. 103. № 2. P. 313.
Kucinska-Lipka J., Gubanska I., Janik H., Sienkiewicz M. // Mater. Sci. Eng. C. 2015. V. 46. P. 166.
Lamba N.M.K., Woodhouse K.A., Cooper S.L., Lelah M.D. // Polyurethanes in Biomedical Applications. Boca Raton: CRC Press, 2012.
Gunatillake A.P., Martin D.J., Meijs G.F., McCarthy S.J., Adhikari R. // Aust. J. Chem. 2003. V. 56. P. 545.
Takanahara A., Coury A.J., Herdenrother R.W., Cooper S.L. // J. Biomed. Mater. Res. 1991. V. 25. № 3. P. 341.
Szycher M., Reed A.M., Arthur A., Siciliano A.A. // J. Biomater. Appl. 1991. V. 6. № 2. P. 110.
Mathur A.B., Collier T.O., Kao W.J., Wiggins M., Schubert M.A., Hiltner A., Anderson J.M. // J. Biomed. Mater. Res. 1997. V. 36. № 2. P. 246.
Schubert M.A., Wiggins M.J., Anderson J.M., Hiltner A. // J. Biomed. Mater. Res. 1997. V. 34. № 4. P.493.
Thomas V., Jayabalan M. // J. Biomed. Mater. Res. A. 2009. V. 89. № 1. P. 192.
Pinchuk L. // J. Biomater. Sci., Polym. Ed. 1995. V. 6. № 3. P. 225.
Jones J.A., Dadsetan M., Collier T.O., Ebert M., Stokes K.S., Ward R.S., Hiltner P.A., Anderson J.M. // J. Biomater. Sci., Polym. Ed. 2004. V. 15. № 5. P. 567.
Ojha U., Kulkarni P., Faust R. // Polymer. 2009. V. 50. № 15. P. 3448.
Blackwell J., Nagarajan M.R., Hoitnik T.B. // Polymer. 1982. V. 23. № 7. P. 950.
Assinik R.A., Wilkies G.L. // J. Appl. Polym. Sci. 1981. V. 26. № 11. P. 3689.
Speckhard T.A., Cooper S.L. // Rubber Chem. Technol. 1986. V. 59. № 3. P. 405.
Yilgor E., Burgaz E., Yurtsever E., Yilgor I. // Polymer. 2000. V. 41. № 3. P. 849.
Zia K.M., Ahmad A., Anjum S., Zuber M., Anjum M.N. // J. Elast. Plast. 2015. V. 47. № 7. P. 625.
Adhikari R., Gunatillake P.A., Mccarthy S.J., Meijs G.F. // J. Appl. Polym. Sci. 1999. V. 74. № 12. P. 2979.
Adhikari R., Gunatillake P.A., Bown M. // J. Appl. Polym. Sci. 2003. V. 90. № 6. P. 1565.
Yang J., Gao Y., Li J., Ding M., Chen F., Tan H., Fua Q. // RSC Adv. 2013. V. 3. № 22. P. 8291.
Balaban M., Antic V., Pergal M., Francolini I., Martinelli A., Djonlagic J. // J. Serb. Chem. Soc. 2012. V. 77. № 10. P. 1457.
Yilgör I., Wilkes G.L., McGrath J.E. // Polym. Prep. 1983. V. 24. № 2. P. 80.
Kim S.E., Heo D.N., Lee J.B., Kim J.R., Park S.H., Jeon S.H., Kwon K. // Biomed. Mater. 2009. V. 4. P. 044106.
Tyagi D., McGrath J.E., Wilkes G.L. // Polym. Eng. Sci. 1986. V. 26. № 20. P. 1371.
Korycka P., Mirek A., Kramek-Romanowska K., Grzeczkowiczand M., Lewińska D. // Beilstein J. Nanotechnol. 2018. V. 9. P. 2466.
Pham Q.P., Sharma U., Mikos A.G. // Tissue Eng. 2006. V. 12. № 5. P. 1197.
Luu Y.K., Kim K., Hsiao B.S., Chu B., Hadjiargyrou M. // J. Controlled Release. 2003. V. 89. № 2. P. 341.
Yoshimoto H., Shin Y.M., Terai H., Vacanti J.P. // Biomaterials. 2003. V. 24. № 12. P. 2077.
Hao X., Zhang X. // Mater. Lett. 2007. V. 61. № 6. P. 1319.
Chuangchote S., Sirivat A., Supaphol P. // Polym. J. 2006. V. 38. № 9. P. 961.
Demir M.M., Yilgor I., Yilgor E., Erman B. // Polymer. 2002. V. 43. № 11. P. 3303.
Zhuo H., Hu J., Chen S., Yeung L. // J. Appl. Polym. Sci. 2008. V. 109. № 1. P. 406.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)