

Высокомолекулярные соединения (серия Б), 2020, T. 62, № 5, стр. 323-347
СОВРЕМЕННЫЕ ДОСТИЖЕНИЯ В ОБЛАСТИ ПОЛУЧЕНИЯ И ИСПОЛЬЗОВАНИЯ ТЕРМОПЛАСТИЧНЫХ ЧАСТИЧНО КРИСТАЛЛИЧЕСКИХ ПОЛИУРЕТАНОВ С ЭФФЕКТОМ ПАМЯТИ ФОРМЫ
М. А. Горбунова a, *, Д. В. Анохин a, Э. Р. Бадамшина a
a Институт проблем химической физики Российской академии наук
142432 Черноголовка, Московской обл., проспект Академика Семенова, 1, Россия
* E-mail: zav@icp.ac.ru
Поступила в редакцию 22.01.2020
После доработки 05.03.2020
Принята к публикации 22.05.2020
Аннотация
Проанализировано современное состояние материаловедения в области получения перспективных функциональных термоэластопластов на основе полиуретанов и полиуретанмочевин. Интерес к данным материалам остается высоким в течение последних двадцати лет благодаря простоте получения, биосовместимости, хорошим термическим и механическим характеристикам, а также возможности настраивать свойства для конкретного приложения. Основной акцент сделан на описании частично кристаллических полиуретанов, в которых кристаллические домены играют роль временной физической сетки, что открывает возможность создания адаптивных материалов с эффектом памяти формы. Описываемые материалы разделены по типам внешних воздействий, являющихся переключателем механических свойств. Рассмотрено влияние природы и соотношения компонентов термочувствительных полиуретанов на механические свойства и основные характеристики эффекта памяти формы. Обсуждены последние достижения в области синтеза, модификации и характеризации полиуретановых материалов, а также приведены наиболее интересные примеры их применения в медицине.
ВВЕДЕНИЕ
Полимеры с эффектом памяти формы (ЭПФ) и их композиты привлекают большое внимание исследователей благодаря способности принимать одну или несколько временных форм и восстанавливать свою первоначальную форму (или другие временные формы) под действием внешних факторов. По типу внешнего воздействия полимеры с ЭПФ разделяют на термочувствительные (воздействие на материал температуры), электрочувствительные (электричества), магниточувствительные (магнитного поля), рН-чувствительные (изменение рН среды), светочувствительные (воздействие светового излучения). Такие полимеры относятся к классу адаптивных или “умных” материалов. Важно отметить, что в большинстве случаев полимеры с ЭПФ являются термочувствительными или термически активированными (индуцированными), т.е. возвращают свою первоначальную форму при локальном нагреве.
Областью применения с большим потенциалом роста полимеров с ЭПФ является медицинская промышленность (рис. 1) [1]. К преимуществам таких материалов относится их биосовместимость, биоразлагаемость и способность к изменению формы при температуре человеческого тела.

Среди полимеров с ЭПФ особый интерес представляют частично кристаллические сегментированные термопластичные полиуретаны и полиуретанмочевины, что подтверждается ростом обзоров и статей за последние два десятилетия [2–6]. Серьезные исследования в области термопластичных полиуретанов с ЭПФ начались в начале 2000-х годов после того, как A. Lendlein с соавторами сформулировали концепцию создания биоразлагаемых термочувствительных полиуретанов [7–11]. С тех пор многие исследователи пытаются создать биосовместимые и биоразлагаемые термопластичные полиуретаны и полиуретанмочевины для разного применения в медицине, в частности для имплантируемых устройств и инвазивной хирургии без повреждения окружающих тканей/органов. Важный фактор для развития адаптивных материалов − большие объемы производства полиуретанов, а также широкие возможности для улучшения их функциональных свойств [12–15].
С точки зрения фундаментальных исследований данные полимеры являются интереснейшим примером формирования структуры на различных иерархических уровнях и служат модельной системой для изучения влияния микроструктуры на макроскопические свойства. Прежде всего это связано с нетривиальной природой взаимовлияния двух конкурирующих процессов, определяющих структуру и физико-механические свойства материала – микрофазового разделения термодинамически несовместимых мягких и жестких блоков и кристаллизации гибких фрагментов полиэфиров. Целенаправленное регулирование указанных процессов открывает новые возможности для практического создания “умных” материалов с тонко настраиваемыми свойствами.
В настоящее время большинство обзоров и статей посвящено общим классам полимеров c ЭПФ [16], способам активации формы полимеров [17, 18], свойствам адаптивных полиуретанов [17] и различным областям их применения [19–22]. Однако нет обобщенных данных о взаимосвязи структура–свойства термопластичных полиуретанов и полиуретанмочевин, содержащих кристаллизующиеся гибкие фрагменты и способах управления их структурой, позволяющих целенаправленно регулировать механические свойства и ЭПФ. Именно этот класс полимеров представляет наибольший интерес для использования в различных областях, в том числе и медицине.
В обзоре обсуждаются последние достижения в области получения частично кристаллических термопластичных полиуретанов и полиуретанмочевин с ЭПФ, способы регулирования, методы активации формы, а также в области использования этих полимеров в медицине.
ОБЩИЕ ПРЕДСТАВЛЕНИЯ ОБ ЭФФЕКТЕ ПАМЯТИ ФОРМЫ. СТРОЕНИЕ ТЕРМОПЛАСТИЧНЫХ ПОЛИУРЕТАНОВ И ПОЛИУРЕТАНМОЧЕВИН С ЭФФЕКТОМ ПАМЯТИ ФОРМЫ
Полимеры с ЭПФ изменяют свою форму под действием внешних воздействий. Их свойства определяются сочетанием структуры полимера с технологией обработки и программирования временной формы. Строение адаптивных полимеров схематически показано на рис. 2 [23].

Полимер с ЭПФ представляет собой фазово-разделенную систему, формирующую две независимые сетки: постоянную (стабильную) и временную. Узлы стабильной сетки образованы химическими или водородными связями, либо являются результатом запутывания цепей. Временную сетку образуют кристаллические, жидкокристаллические или аморфные домены мягкого блока, ионные комплексы, комплексы включения и т.д. При изготовлении образца ему придают постоянную форму, при этом формируются обе сетки. На стадии программирования временная сетка под внешним воздействием (нагревание, облучение, магнитное поле, изменение pH и другие) разрушается, после чего образец переходит в эластичное состояние, в котором происходит деформация и придание временной формы. Завершается программирование фиксацией временной формы, при которой формируется новая временная сетка. На заключительном этапе постоянная форма восстанавливается за счет разрушения временной физической сетки повторным внешним воздействием. Описанный процесс называется двустадийным. При повторном программировании можно получить новую временную форму, отличающуюся от первой.
Один из наиболее интересных классов адаптивных полимеров как с фундаментальной, так и с практической точки зрения – полиуретаны и композиты на их основе. Термопластичные полиуретаны и полиуретанмочевины являются, по сути, линейными блок-сополимерами, образованными мягкими и жесткими блоками. Наиболее простые по строению – диблочные сополимеры, состоящие из двух линейных гомополимеров, соединенных ковалентной связью. В результате термодинамической несовместимости жестких и мягких блоков в полимере наблюдается разделение фаз (микросегрегация). Жесткие блоки объединяются в домены главным образом за счет водородных связей и создают в полимере так называемую физическую сетку, мягкие блоки обеспечивают высокоэластическую деформируемость материала. Благодаря своей фазово-разделенной структуре, полиуретан (или полиуретанмочевина) проявляет ЭПФ, т.е. восстанавливает свою первоначальную форму при внешнем воздействии, чаще всего при нагревании [24–29]. Таким образом, жесткий блок отвечает за прочность материала и фиксацию временной формы, в то время как мягкий блок – за эластичность и способность принимать временную форму [29].
Термопластичные полиуретаны со структурой, благоприятной для реализации ЭПФ, впервые были получены сотрудниками фирмы “Mitsubishi Heavy Industries (Япония)” [30, 31]. Синтез осуществляли двустадийным способом через стадию формирования макродиизоцианата из 4,4'-дифенилметандиизоцианата и полиэфирдиола с использованием удлинителя цепи 1,4-бутандиола. Такими образом, мягким блоком является полиэфирдиол, жестким блоком – уретандиоловые фрагменты, образованные при взаимодействии 4,4'-дифенилметандиизоцианата с 1,4-бутандиолом. Классическая схема синтеза термопластичного полиуретанового блок-сополимера через стадию формирования макродиизоцианата приведена ниже.
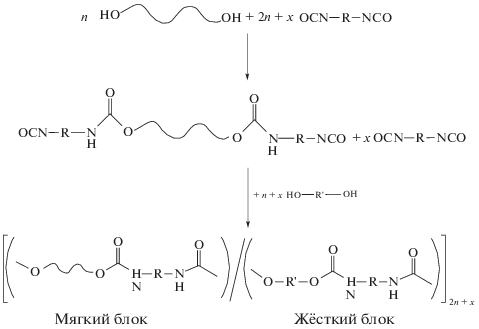
Здесь R и R' – фрагмент олефиновой цепи.
ТЕРМОЧУВСТВИТЕЛЬНЫЕ ПОЛИУРЕТАНЫ И ПОЛИУРЕТАНМОЧЕВИНЫ
Общая схема реализации эффекта памяти формы в термочувствительных полиуретанах и полиуретанмочевинах
Термическое воздействие является одним из наиболее распространенных прямых способов активации ЭПФ и основано на передаче тепловой энергии от внешнего источника к полимеру. В литературе описаны примеры изменения формы в термочувствительных полимерах за счет формирования сетки водородных связей, при этом скорость восстановления формы сильно зависит от температуры, поскольку слабые водородные взаимодействия чувствительны к нагреванию [32].
Ниже показана схема реализации ЭПФ и сопровождающей ее эволюции структуры в доменах мягкого и жесткого блока термочувствительного полиуретана.
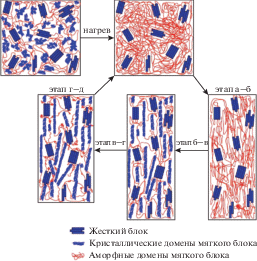
Этап а–б–в–г иллюстрирует процесс получения и фиксации временной формы за счет кристаллизации мягкого блока. Этап г–д показывает плавление кристаллической фазы мягкого блока и восстановление постоянной формы [33].
В исходных образцах вследствие фазового разделения (благодаря несовместимости мягкого и жесткого блока) оба типа блоков способны формировать изотропно распределенные домены. В данном случае при нагревании выше температуры переключения Ttrans происходит разрушение временной физической сетки, увеличение подвижности цепей в доменах мягкого блока и переход образца в эластичное состояние, в котором он способен восстанавливать свою исходную форму даже при высоких степенях вытяжки. Однако если вытянутые образцы охладить до исходной температуры T0 в изомерных условиях (при сохранении формы), появится возможность зафиксировать форму образца в термопластичном состоянии вследствие снижения подвижности цепей в мягких блоках. При повторном нагревании происходит обратный переход из термопласта в эластомер и восстановление постоянной формы образцов.
Для исследования ЭПФ проводят классические испытания на растяжение при фиксированной температуре, которые позволяют изменять приложенную деформацию ε или напряжение σ, а также скорость деформации. В результате из кривых напряжение–деформация можно получить такие значения, как модуль Юнга E, удлинение при разрыве εr и напряжение при разрыве σr.
Схематическое изображение типичных циклических зависимостей напряжение–деформация для термочувствительных полиуретанов и полиуретанмочевин с двустадийным способом формирования эффекта памяти формы (полимер, содержащий один тип временной сетки) приведено на рис. 3.
Рис. 3.
Схематическое изображение результатов циклических термомеханических исследований для двух различных тестов. а: схема напряжение σ–деформация ε, где 1 – растяжение образца до εm при Т > Ttrans, 2 – охлаждение до Т0 при постоянной εm, 3 – зажимное расстояние снято до исходного состояния, 4 – при ε = 0% нагревание до Т > Ttrans, 5 – начало второго цикла; б: схема ε–T–σ, где 1 – растяжение до εm при Т > Ttrans, 2 – охлаждение до Т0 со скоростью k = dT/dt при σ = 0 МПа, 3 – зажимное расстояние уменьшается (снятие нагрузки) до σ = 0 МПа, 4 – нагревание до Тtrans со скоростью k = dT/dt при σ = 0 МПа, 5 – начало второго цикла [7].

Важно отметить, что процесс программирования может быть выполнен в условиях контроля напряжения или деформации. Когда контролируется деформация ε, появляется возможность измерять эволюцию напряжения σ в образце при определенных условиях температурного отклика на деформацию и наоборот – можно регистрировать эволюцию деформации ε при контроле напряжения σ. Таким образом, циклические термомеханические испытания позволяют описать одновременно стадии программирования и восстановления [34].
Типичный тест с контролем деформации состоит из трех этапов [34]: 1) нагревание образца до температуры выше температуры переключения (Т > Тtrans), деформирование до εm; 2) охлаждение до исходной температуры T0 с определенной скоростью охлаждения при постоянной деформации εm для фиксации временной формы; 3) разгрузка образца до нулевого напряжения при T0, что приводит к некоторой усадке образца до εu. После установления временной формы процесс восстановления может быть произведен на шаге 4 при нагревании до Т > Тtrans. Цикл N (шаг 5) повторяется несколько раз, так как первый цикл обычно применяется для стирания термомеханической истории образца, а результаты циклов N ≥ 2 используются для количественного определения свойств памяти формы, о которых речь пойдет ниже.
При циклических испытаниях с контролем напряжения (рис. 3б) этапы программирования 1 и 3 остаются неизменными, а на этапе 2 после фиксированного времени выдержки при εm процесс продолжается до максимального напряжения σm, который затем поддерживается постоянным в процессе охлаждения. Таким образом, контролируется изменение удлинения образца при охлаждении.
Для количественной характеристики термочувствительных полиуретанов и полиуретанмочевин используют следующие показатели, предложенные A. Lendlein и S. Kelch [7]: температура переключения Ttrans, соответствующая температуре стеклования или температуре плавления, доля фиксированной деформации Rf, доля остаточной деформации Rr, максимальная деформация εm и максимальное напряжение при восстановлении σm.
Доля остаточной деформации или коэффициент восстановления формы Rr характеризует способность материала к запоминанию своей постоянной формы и является мерой восстановления после деформации:
где εm − максимальная деформация при механическом воздействии, εp − остаточная деформация после восстановления формы.Доля фиксированной деформации или коэффициент фиксации формы Rf описывает способность материала фиксировать механическую деформацию, т.е. способность запоминать временную форму в результате механического воздействия. Коэффициент фиксации формы Rf определяют как отношение деформации εu, остающейся после снятия напряжений, к величине максимальной деформации εm по формуле
Величины Rr и Rf при проведении N циклов испытаний рассчитываются следующим образом:
Механические свойства термочувствительных полиуретанов и полиуретанмочевин с эффектом памяти формы с температурой стеклования в качестве температуры переключения
Термочувствительные полиуретаны и полиуретанмочевины делятся на два типа. Для первого типа изменение механических свойств происходит при расстекловывании аморфной фазы мягкого блока (Ttrans = Tg) при нагревании в широком температурном диапазоне, для второго типа переключение связано с плавлением кристаллитов мягкого блока (Ttrans = Tm) и наблюдается в узком температурном интервале.
В качестве примеров полимера первого типа, в котором температурой переключения является температура стеклования Тg, в качестве аморфной фазы мягкого блока используют такие олигомеры, как полиэтиленадипинат (Тg = –45…–48°С) [35], полипропиленоксид (Тg = –5…–48°С) [36], а также уретановые фрагменты на основе 4,4'-дифенилметандиизоцианата и 1,6-гександиола [37]. Изменением длины мягкого аморфного блока можно управлять Тg, как температурой переключения и соответствующими механическими свойствами. При изучении теплофизических, прочностных свойств и ЭПФ полиуретанового блок-сополимера первого типа с мягкими аморфными блоками на основе 4,4'-дифенилметандиизоцианата и 1,6-гександиола и жесткими кристаллическими блоками на основе 1,6-гексаметилендиизоцианата и 1,2-бутандиола авторами [38] было установлено влияние длины мягкого блока на Тg и гистерезис в цикле нагрузка–разгрузка: с уменьшением длины блоков ухудшается фазовое разделение между мягкими и жесткими блоками, что приводит к уменьшению гистерезиса. М. Kayalvizhi с соавторами [39] синтезировали полимер на основе аморфного мягкого полистирольного блока и жесткого блока, сформированного с участием 1,6-гексаметилендиизоцианата и удлинителей цепи (этилендиамина, бутандиамина и гексаметилендиамина), и исследовали влияние строения последних на температуру стеклования в качестве Ttrans. Установлено, что температура стеклования мягкого блока снижается при переходе от этилендиамина к гексаметилендиамину с 89 до 86°C. Температура плавления жесткого блока также зависит от типа удлинителя цепи и снижается с увеличением его молекулярной массы за счет снижения степени кристалличности блока, формируемого с участием удлинителя цепи: 266, 217 и 157°C для этилендиамина, бутандиамина и гексаметилендиамина соответственно. Таким образом, уменьшение степени кристалличности жесткого сегмента с ростом длины удлинителя цепи приводит к снижению температуры плавления, термической стабильности термопластичного полиуретана и повышению температуры стеклования, определяющей Ttrans для данного типа полимеров с аморфными мягкими блоками.
Механические свойства термочувствительных полиуретанов и полиуретанмочевин с эффектом памяти формы с температурой плавления в качестве температуры переключения
Следует отметить, что наиболее часто на практике используются термопластичные полиуретаны и полиуретанмочевины с ЭПФ второго типа, т.е. с температурой плавления в качестве температуры переключения. В данном случае мягким блоком являются макрогликоли на основе простых или сложных полиэфирдиолов, способные к кристаллизации (рис. 4).
Рис. 4.
Фазовое разделение в термопластичных полиуретанах c кристаллизующимся мягким блоком: свободная (а, в) и связанная (б, г) водородными связями карбонильная группа жесткого (а, б) и мягкого блока (в, г) [39].

В результате кристаллизации мягких аморфных блоков формируется дополнительная сетка физических узлов. Это существенно влияет на поведение материала при деформации, что сказывается на силе фиксирования формы и на механических свойствах полиуретанов и полиуретанмочевин. Временная форма образуется за счет способности аморфного мягкого блока кристаллизоваться при деформации или за счет выдержки в деформированном состоянии. Под внешним воздействием происходит разрушение дополнительной физической сетки и обратный переход из термопластичного полимера в эластомерный, после чего становится возможным восстановление исходной формы. Если внешним воздействием является нагрев [26–28, 40–42], то температура переключения формы соответствует плавлению кристаллов полиэфирного мягкого блока Тm. Варьируя тип и молекулярную массу полиэфирдиолов, можно управлять температурой переключения, механическими свойствам и ЭПФ полиуретанов и полиуретанмочевин.
В качестве кристаллизующихся макрогликолей при синтезе термочувствительных полиуретанов и полиуретанмочевин используют в основном алифатические полиэфирдиолы, такие как поли-ε-капролактон диол [43], полиэтиленгликоль [44], полигидроксибутират [45], поли-L-лактид [46], полигликолид (полигликолевая кислота), поли(1,4-бутиленгликоль)адипинат [47], поли-δ-валеролактон [48] и другие. Более подробно типы олигомеров и термочувствительные полиуретаны и полиуретанмочевины на их основе описаны в обзоре [49].
Исследования показали, что структура и свойства адаптивных полиуретанов и полиуретанмочевин во многом определяются выбором исходных компонентов, способом синтеза, степенью фазового разделения, морфологией, степенью кристалличности и наличием водородных связей в полимере. В связи с этим понимание механизма взаимодействия жестких и мягких блоков имеет важное значение для получения материалов с памятью формы для конкретных применений.
Одним из важных факторов, определяющих механические свойства адаптивных материалов, является молекулярная масса мягкого блока. В.К. Kim с сотрудниками [50, 51] исследовали влияние молекулярной массы поли-ε-капролактон диола в диапазоне (2–8) × 103 на механические свойства и параметры ЭПФ при нагревании полиуретана, полученного двустадийным способом из поли-ε-капролактон диола, 4,4'-дифенилметандиизоцианата и 1,4-бутандиола в расплаве при 60°С. Обнаружено, что доля фиксированной деформации Rf повышается с ростом молекулярной массы поли-ε-капролактон диола с 60 до 90%. При исследовании серии образцов полиуретанов, отличающихся молекулярной массой поли-ε-капролактон диола в интервале (5 × 102)–(10 × 103), авторы [52] обнаружили, что за счет изменения молекулярной массы мягкого блока можно варьировать температуру переключения в диапазоне 37–42°С. Нижний предел молекулярной массы поли-ε-капролактон диола, способного кристаллизоваться при комнатной температуре, по данным авторов, является 2 × 103 [52]. На основании многочисленных экспериментов установлено, что для получения термопластичных полиуретанов с оптимальными механическими свойствами молекулярная масса полиэфирных блоков должна лежать в области (2–5) × 103 [53]. Низкомолекулярные полиэфирдиолы (менее 2 × 103) препятствуют способности мягкого блока кристаллизоваться и ухудшают кристаллизацию жестких блоков. Высокомолекулярные полиэфирдиолы (более 5 × 103) создают трудности при синтезе и переработке из-за большой вязкости, однако обладают более высоким Rf за счет лучшей кристалличности и эффективного фазового разделения. Таким образом, механические свойства адаптивных полиуретанов с кристаллизующимися полиэфирными блоками определяются строением кристаллов мягкого блока. Кристаллизация мягкого блока в фазово-разделенной системе ограничена в количестве кристаллической фазы, размере и форме кристаллов [54–57]. В этом случае возможны два типа геометрических ограничений: мягкие и жесткие [58, 59]. Последний тип соответствует системам, в которых кристаллизация в доменах мягкого блока не вызывает разрушения основной фазово-разделенной морфологии. Если же движущая сила фазового разделения оказывается слабее процесса кристаллизации, то протекает так называемая разрывная кристаллизация и морфология блок-сополимера изменяется [60–62]. В большинстве исследований фазово-разделенных блок-сополимеров рассматривается случай кристаллизации жесткого блока в мягкой аморфной матрице, т.е. случай разрывной кристаллизации.
На механические свойства и эффект памяти формы аналогично термочувствительным полиуретанам и полиуретанмочевинам первого типа оказывает влияние тип и строение удлинителей цепи. Так, в работе [63] исследовали влияние удлинителя цепи на механические свойства и эффект памяти формы термочувствительным полиуретанам второго типа на основе поликарбонатдиола с М = 2 × 103, 4,4'-дифенилметандиизоцианата и серии удлинителей цепи различного строения (этиленгликоль, 1,4-бутандиол, 1,6-гександиол, триметилолпропан). Установлено, что природа удлинителя цепи позволяет регулировать механические свойства и эффект памяти формы термочувствительных полиуретанов. Так, наилучшими прочностными свойствами обладает полиуретан на основе 1,6-гександиола (прочность при разрыве составляет 56.5 МПа и удлинение при разрыве 645%). При этом полиуретан, полученный с использованием триметилолпропан, имеет самые низкие прочностные свойства (41.1 МПа, 475%). Все полиуретаны восстанавливают свою первоначальную форму при температуре, близкой к температуре человеческого тела, за время менее 20 с со значениями Rr = 100%, Rf = = 94%. Иногда для образования мочевинных групп, придающих дополнителную прочность полиуретана, наряду с традиционным удлинителем цепи 1,4-бутандиола вводят 2-аминоэтанол [64].
При выборе полиэфиров в качестве мягкого блока при синтезе термопластичных полиуретанов и полиуретанмочевин важно учитывать не только его молекулярную массу, но и полидисперсность и наличие функциональных групп. Влияние полидисперсности мягких блоков было исследовано в различных полиуретанах на основе политетраметиленоксидас М = (6.5 × 102)–(1.5 × 103), 4,4'-дифенилметандиизоцианата и удлинителей цепи 1,4-бутандиола или диэтиленгликоля [65]. Так, для полиуретанов на основе политетраметиленоксидас М = 1546, полученого смешением политетраметиленоксида (М = 10 × 103) и политетраметиленоксида (М = 2 × 103) или политетраметиленоксида (М = 650) и политетраметиленоксида (М = 2 × 103), 4,4'-дифенилметандиизоцианата и 1,4-бутандиола увеличение степени полидисперсности от 1.47 до 1.68 приводит к повышению прочности и низким значениям остаточного удлинения (до 140%) и сопровождается снижением степени кристалличности жесткого блока от 0.148 до 0.131. В случае низкой степени полидисперсности мягкого блока (1.22) увеличивались степень кристалличности жесткого блока до 0.167 и значения остаточного удлинения до 190%.
Водородная связь между группами N–H и C=O полиуретанов является еще одним фактором, способствующим улучшению фазового разделения, влияющего на их механические свойства и эффект памяти формы [66]. Авторы работы [67] проанализировали микрофазовую структуру и образование водородных связей в термопластичных полиуретанах на основе 1,6-гексаметилендиизоцианата, поли-ε-капролактон диола (М = = 2 × 103) и удлинителей цепи различного строения – 1,4-бутандиола и триметилолпропана методом ИК-спектроскопии. При введении второго удлинителя цепи разветвленного строения интенсивность полосы поглощения, соответствующей валентным колебаниям карбонильной группы, связанной водородной связью (1693 см–1), увеличивается с 20.6 до 26.9%, что приводит к повышению интенсификации агрегации доменов жесткого блока и, как следствие, к улучшению фазового разделения между мягкими и жесткими блоками. Возможная причина заключается, по мнению авторов, в том, что разветвленная структура триметилолпропана создает стерические препятствия для смешивания мягких и жестких блоков и тем самым увеличивает фазовое разделение. Это вызывает агрегацию доменов жестких блоков и интенсификацию образования водородных связей между группами N–H и C=O уретана. Интересно, что несвязанные водородной связью группы N–H на границе раздела могут легко образовать водородную связь с группой C=O полиэфира мягкого блока, несмотря на хорошее фазовое разделение. Это проявляется в повышении интенсивности валентных колебаний карбонильных групп поли-ε-капролактон диола, связанных водородной связью при 1721 см–1. Авторы объясняют подобное явление увеличением пространства для укладки жестких сегментов за счет удлинителя цепи разветвленного строения. Установлено, что при замене 1,4-бутандиола на триметилолпропан повышается модуль Юнга с 48.9 МПа в отсутствие триметилолпропана до 159.8 МПа при содержании триметилолпропана 100 мас. % за счет увеличения степени кристалличности и образования водородных связей.
Как было показано в работе [68], термическая обработка от 80 до 140°С, проведенная после синтеза полиуретана на основе сополимера тетрагидрофурана с азидсодержащим 3,3'-бис-(азидометил)оксетаном с М = 6 × 103 в качестве мягкого блока, 2,4-толуилендиизоцианата и удлинителя цепи диэтиленгликоля, также влияет на процесс фазового разделения за счет изменения прочности водородных связей жесткого блока. Авторы с использованием метода ИК-спектроскопии по изменению интенсивности полос поглощения групп N–H и C=O, связанных водородной связью, показали, что термическая обработка вносит значительные изменения в прочность водородных связей жестких блоков. Степень фазового разделения снижается при 80°С и затем постепенно увеличивается с ростом температуры обработки до 110–140°С. Начальное снижение фазового разделения авторы объясняют растворением жестких сегментов и уменьшением прочности водородных связей. Постепенное повышение температуры обработки приводит к фазовому разделению из-за перегруппировки сегментов жесткого блока. При увеличении подвижности жесткого блока и его реорганизации возрастают прочность с 1 до 6 МПа и максимальное удлинение с 484 до 808% после термической обработки. Однако стоит заметить, что изменение микрофазового разделения не согласуется с механическими свойствами. Это указывает на то, что микрофазовое разделение является неединственным способом улучшения механических свойств адаптивных полиуретанов.
Таким образом, процессы фазового разделения и кристаллизации мягкого блока оказывают существенное влияние на конечную структуру полимера и его механические и термические свойства [69, 70].
Строение и длина жесткого блока также влияют на механические свойства и эффект памяти формы результирующего полимера. В качестве примера можно привести исследование влияния строения жестких блоков на основе 4,4´-дифенилметандиизоцианата и 1,4-бутандиола [71, 72], 1,6-гексаметилендиизоцианата и 1,4-бутандиола [73], 4,4′-дициклогексилметандиизоцианата и 1,4-бутандиола [74], а также смеси 4,4'-дифенилметандиизоцианата и 2,4'-дифенилметандиизоцианата с 1,4-бутандиолом [75] на свойства термочувствительных полиуретанов. Авторы работы [76] методом ДСК на примере модельных уретанов на основе диизоцианатов различного строения (1,6-гексаметилендиизоцианат, 4,4′-дициклогексилметандиизоцианата и смеси 4,4'-дифенилметандиизоцианата и 2,4'-дифенилметандиизоцианата)и бутанола исследовали влияние природы диизоцианатов на скорость кристаллизации. Наибольшей скоростью кристаллизации обладает уретан на основе 1,6-гексаметилендиизоцианата линейного по сравнению с уретаном на основе диизоцианатов циклоалифатического и ароматического строения. Степень кристалличности изменяется в ряду 1,6-гексаметилендиизоцианат > 4,4′-дициклогексилметандиизоцианата > 4,4'-дифенилметандиизоцианат > > смесь 4,4'-дифенилметандиизоцианата и 2,4'-дифенилметандиизоцианата с 100 до 77, 72 и 65% соответственно. Полиуретан, полученный авторами [77] с использованием алифатического 1,6-гексаметилендиизоцианата, демонстрирует оптимальные физико-механические параметры – модуль Юнга составляет около 39 МПа, разрывная прочность (отнесенная к поперечному сечению недеформированного образца) – 33 МПа, удлинение при разрыве εr достигает 1000%. Превосходные прочностные характеристики полиуретана на основе 1,6-гексаметилендиизоцианата обусловлены наличием уретановых групп и сильными межмолекулярными взаимодействиями посредством водородных связей. При замене алифатического диизоцианата на 2,4-толуилендиизоцианата резко снижаются значения прочности при разрыве и удлинения при разрыве, что, вероятно, связано с уменьшением концентрации водородных связей между объемными ароматическими заместителями. В работе [78] изучали строение и термомеханические свойства термопластичных полиуретанов на основе политетраметиленгликоля, 4,4'-дифенилметандиизоцианата и 1,4-бутандиола. Оптимальное восстановление формы наблюдается при содержании жесткого блока 30–45%.
Еще один способ улучшения механических свойств термопластичных полиуретанов и полиуретанмочевин – увеличение числа блоков в цепи, т.е. переход к мультиблочным сополимерам, содержащим в своей цепи отличающиеся по строению два типа кристаллизующихся полиэфирных фрагмента, которые формируют две независимые временные физические сетки – поли-ε-капролактон диол и поли(1,4-бутиленгликоль)адипинат [77]. Использование второго кристаллизующего компонента и поли(1,4-бутиленгликоль)адипината позволяет контролировать содержание и размеры кристаллических доменов в полиуретане и, как следствие, управлять механическими свойствами материалов, температурой переключения и скоростью фиксации формы от нескольких минут до нескольких месяцев. Так, полиуретанмочевинный сополимер на основе поли-ε-капролактон диола демонстрирует эластичное состояние (удлинение при разрыве εr = 1445%, модуль Юнга Е = 7 МПа, прочность при разрыве σr = 14 МПа), а полиуретанмочевинный сополимер на основе поли(1,4-бутиленгликоль)адипината – жесткое термопластичное состояние (εr = = 807%, Е = 199 МПа, σr = 21 МПа). Полимер, содержащий смесь этих олигодиолов, является эластомером и характеризуется модулем Юнга 59 МПа, удлинением при разрыве εr = 1300%) и прочностью при разрыве σr = 14 МПа [78].
Для использования в медицине особенно важно, чтобы температура переключения полиуретанов была адаптирована к температуре тела человека. Для этих целей в качестве мягкого блока вводят олигомеры с низкой температурой плавления кристаллитов, например поли(1,4-бутиленгликоль)адипинат или поли-ε-капролактон диол или оба этих олигомера вместе. Так, авторы [79] показали, что низкую температуру переключения можно легко достичь путем формирования мультиблочных полиуретанмочевинных сополимеров на основе двух кристаллизующихся олигомеров поли(1,4-бутиленгликоль)адипината и поли-ε-капролактон диола со средними молекулярными массами. Введение второго кристаллизующегося компонента вызывает нуклеацию и тем самым заметное повышение температуры кристаллизации переключающего элемента до температуры 37°C, а также значительное увеличение прочности и модуля упругости. Таким образом, разница в механическом поведении мультиблочных полиуретанмочевинах, отличающихся составом кристаллизующегося мягкого и строением жесткого блока, демонстрирует возможность регулирования свойств полимеров при синтезе без изменения их состава.
Процессы фазового разделения и зародышеобразования при кристаллизации можно контролировать путем введения нанодобавок – нуклеирующих агентов, одними из которых являются углеродные нанонаполнители [80–89].
Опубликованные результаты исследований свидетельствуют о том, что некоторые нанодобавки не только влияют на кинетику кристаллизации частично кристаллических полимеров, но и улучшают фазовое разделение жестких и мягких доменов, оказывая влияние на механические свойства [90]. Так, N.G. Sahoo с соавторами [91] показали, что энтальпия кристаллизации и степень кристалличности полиуретанового нанокомпозита на основе поли-ε-капролактон диола в присутствии 2.5 мас. % многостенных углеродных нанотрубок выше, чем для исходного образца. Однако в работе [92] наблюдали уменьшение энтальпии кристаллизации и степени кристалличности полиуретанового композита на основе поли-ε-капролактон диола в присутствии многостенных углеродных нанотрубок. Таким образом, влияние углеродных нанотрубок на кинетику кристаллизации зависит от многих факторов, и, в общем случае, невозможно утверждать, что углеродные нанотрубки являются нуклеирующими добавками или топологическими дефектами для определенного типа матрицы.
Важно отметить, что для повышения механических свойств и эффекта памяти формы термопластичных полиуретанов и полиуретанмочевин большую роль играет способ введения наночастиц, их концентрация и отсутствие агломерации в матрице полимера. Для предотвращения последнего эффекта часто проводят функционализацию нанонаполнителей. Так, Y.C. Jung [93] предложил использовать многостенные углеродные нанотрубки, содержащие гидроксильные группы ОН и карбоксильные группы COOH (функционализированные углеродные нанотрубки получены обработкой смесью HNO3 и H2SO4) для дальнейшего образования полиуретановых композитов различными методами: полимеризация in situ, физическое смешение (классический способ) и химическая сшивка (рис. 5). Композит, полученный химическим сшиванием за счет образования ковалентных связей между ОН-группами углеродных нанотрубок и NCO-группами 4,4'-дифенилметандиизоцианата обладает лучшими механическими свойствами и свойствами памяти формы по сравнению с композитами, полученными физическим смешением углеродных нанотрубок с полиуретаном и химическим сшиванием функционализированных углеродных нанотрубок с 4,4'-дифенилметандиизоцианатом.
Рис. 5.
Влияние способа получения термопластичных полиуретановых композитов с окисленной формой многостенных углеродных нанотрубок (МУНТ) (а), кривые напряжение–деформация (удлинение 200% при 50°С) (б), а также восстановление формы и сохранение формы для полиуретановых композитов, приготовленных классически способом физического смешения, in situ и химическим сшиванием (в) [93].
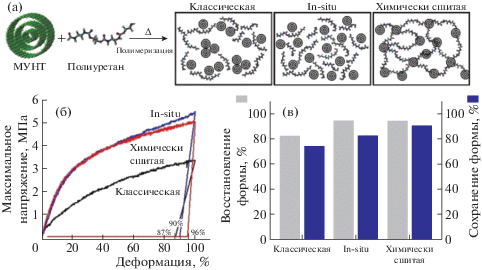
В другом подобном исследовании Y.C. Jung с соавторами [94] модифицировал поверхность многостенных углеродных нанотрубок с использованием УФ-облучения. Наличие гидроксильных и карбоксильных функциональных групп обеспечивает реакционную способность многостенных углеродных нанотрубок для образования ковалентной связи при синтезе термопластичных полиуретанов.
Введение нанокристаллической целлюлозы ускоряет кристаллизацию блока поли(1,4-бутиленгликоль)адипината и позволяет повысить температуру переключения с 10 до 25°С. Температура переключения может быть дополнительно увеличена путем смешивания полиуретана на основе поли(1,4-бутиленгликоль)адипината с олигомером поли-ε-капролактоном, что дает возможность повысить коэффициент фиксации формы до 98% [95]. В работе [95] показано, что введение 15 мас. % фосфорилированной нанокристаллической целлюлозы в полиуретан на основе поли(1,4-бутиленгликоль)адипината, 4,4'-дифенилметандиизоцианата и 1,4-бутандиола позволило увеличить модуль упругости со 150 МПа (исходный полиуретан) до 572 МПа (полиуретановый нанокомпозит) за счет ускорения кристаллизации полиэфирных блоков, улучшить Rf с 47 до 75% и повысить температуру фиксации с 10 до 25°С. Эти свойства могут быть полезны для использования в качестве жестких материалов для изготовления механических приводов или спортивного инвентаря [96]. Повышение температуры кристаллизации до температуры тела человека может быть особенно полезным для биомедицины [97].
Расширение спектра областей применения термочувствительных полиуретанов в медицине может быть достигнуто за счет полимеров со сложным откликом на изменение температуры. Подобные материалы с трехстадийным эффектом памяти формы, требующие двухэтапного программирования формы и наличия двух температур переключения, были описаны в работах [98, 99]. Молекулярная структура таких систем позволяет формировать два различных по химической природе домена с сильно различающимися температурами плавления и/или стеклования, обеспечивающих формирование двух независимых временных физических сеток. Различные комбинации отклика на температурные воздействия можно получить путем создания специфических температурных программ, предполагающих нагревание и растяжение образцов при заданных температурах. Одним из примеров адаптивных полиуретанов, принимающих более чем одну форму в широком температурном диапазоне, являются зведообразные полиуретаны с кристаллизующимися олигомерами [99]. Звездообразные блоки формируют постоянную сетку, а частично кристаллические сегменты поли-ε-капролактон диола – временную. Температурный интервал звездообразного полиуретана зависит от строения разветвленного звена поликапролактонового блока и от длины полимерной цепи. Временная форма задается механической деформацией выше температуры плавления поли-ε-капролактон диола. Подробные исследования механизмов формирования структуры и свойств термопластичных полиуретанов с многостадийным откликом выходят за рамки настоящего обзора.
Таким образом, можно заключить, что механические свойства, а также свойства памяти формы (доля остаточной деформации и доля фиксированных деформаций) термочувствительных полиуретанов и полиуретанмочевин зависят от строения фазово-разделенной структуры, т.е. от типа олигомерного мягкого блока и строения жесткого блока, варьирование которых позволяет регулировать силу межмолекулярного взаимодействия и, как следствие, доменную структуру и эксплуатационные характеристики адаптивных полиуретанов.
ЭЛЕКТРОЧУВСТВИТЕЛЬНЫЕ И МАГНИТОЧУВСТВИТЕЛЬНЫЕ ТЕРМОПЛАСТИЧНЫЕ ПОЛИУРЕТАНЫ И ПОЛИУРЕТАНМОЧЕВИНЫ
В случае, когда температура переключения немного выше температуры человеческого тела и внешний нагрев неприемлем, температуру переключения можно контролировать косвенным генерированием тепла внутри материала за счет электрического или магнитного поля. Такой нагрев может быть реализован путем введения в полимерную матрицу функциональных металлических нанонаполнителей с целью превращения электрической или магнитной энергии в тепловую.
Активируемые электрическим или магнитным излучением полиуретаны и полиуретанмочевины относятся к косвенным термочувствительным полимерам. Электрическое и магнитное поле как бесконтактный способ воздействия могут дистанционно управлять формой полимера in vitro в имплантируемых устройствах [100].
Важными характеристиками композитов для применения в качестве имплантируемых устройств, меняющих свою форму под действием локального нагрева, как и в случае термочувствительных полиуретанов, являются температура переключения и скорость восстановления формы [101]. Повышение температуры переключения до 40–45°С и не слишком высокая скорость восстановления формы позволяет имплантируемому устройству не деформировать ткани в процессе имплантации [102]. Важно отметить, что добавление магнитных и электропроводящих наполнителей в полиуретановую матрицу улучшает механические и формообразующие свойства памяти формы, не оказывая заметного влияния на термическое поведение [103].
Для переключения формы адаптивных полиуретанов электрическим полем перспективным представляется введение углеродных нанотрубок. Благодаря хорошим прочностным свойствам [104, 105], высокой тепло- [106, 107] и электропроводности [108] углеродные нанотрубки заметно улучшают эксплуатационные характеристики полимерных композитов при условии их равномерного распределения в матрице [109, 110], поскольку из-за высокого аспектного отношения углеродные нанотрубки образуют агрегаты, препятствующие полноценной реализации их свойств, что ограничивает широкое использование этих наноматериалов [111]. Для предотвращения агломерации углеродных нанотрубок применяют ультразвуковое диспергирование либо их функционализацию.
В работе [112] синтезировали электрочувствительный полиуретан на основе поли-ε-капролактон диола, 4,4'-дифенилметандиизоцианата и 1,4-бутандиола путем добавления 5 мас. % многостенных углеродных нанотрубок (диаметр 10–20 нм, длина 20 мкм), предварительно обработанных смесью азотной и серной кислот при 140°С в течение 10 мин с последующим диспергированием ультразвуком. Полимер способен восстанавливать свою первоначальную форму после деформации за 10 с при прикладывании напряжения 40 В. Исследования влияния функционализированных многостенных углеродных нанотрубок, содержащих гидроксильные и карбоксильные группы, на свойства смеси полиуретана (М = 333.6 × 103) и поли-L-лактида (М = 2.2 × 105) в соотношении 90 : 10 показали, что введение 4 мас. % [113] и 2–10 мас. % [114] многостенных углеродных нанотрубок обеспечивает способность реагировать на внешнее электрическое поле. Так, восстановление первоначальной формы полиуретанового композита, содержащего 10 мас. % многостенных углеродных нанотрубок, при 40 В происходит в течение 5 с до Rr = 95%. Для введения 10% многостенных углеродных нанотрубок смесь полиуретана с поли-L-лактидом смешивали с многостенными углеродными нанотрубками (60 об./мин в течение 15 мин при 190°С).
Для повышения скорости восстановления, смешиваемости компонентов полиуретановой матрицы и наполнителей, изменения термических и морфологических свойств композитов используют пластификаторы. Введение 5–10% полиэтиленгликоля в смесь коммерческого термопластичного полиуретана Desmopan 385E и поли-L-лактида, содержащую 2–4 мас. % одностенных углеродных нанотрубок, приводит не только к увеличению подвижности полимерных цепей, уменьшению фазового разделения и равномерному распределению одностенных углеродных нанотрубок, но и к росту температуры переключения и скорости восстановления первоначальной формы за счет повышения электропроводности композита [115]. Важно отметить, что в литературе встречается ограниченное число работ, описывающих влияние пластификаторов на электропроводящие свойства композитов.
Для активации памяти формы магнитным полем в полимерную матрицу, как правило, вводят магнитные наночастицы. Процесс переключения запускается индуктивным нагревом в переменном магнитном поле, при этом тепло генерируется внутри самого полимера, что вызывает быстрый рост температуры на 5–12°С [105]. Наночастицы могут выступать в качестве носителей лекарственных средств [116], при этом наиболее часто используются наноразмерные Ni, Fe2O3 и магнетит. Во многих случаях поверхность магнитных наночастиц функционализируют с целью адаптации свойств для конкретного применения, а их перемещение может контролироваться внешним магнитным полем. Особый интерес представляют наночастицы Fe3O4 из-за их хороших ферромагнитных свойств, низкой цитотоксичности и высокой биосовместимости [117].
Следует отметить, что большое значение при синтезе полимерных нанокопозитов с использованием наночастиц различной природы играет способ их введения в матрицу. Магнитные наночастицы – не исключение. Для их введения используют методы ex situ, т.е. диспергирование синтезированных наночастиц в полимерном растворе, и in situ, т.е. полимеризация мономеров в присутствии наночастиц [117] с предварительной обработкой наночастиц ультразвуком для предотвращения их агломерации.
Для достижения оптимальных свойств ЭПФ важно учитывать концентрацию вводимых наночастиц и режимы переменного магнитного поля. Так, введение от 10 до 30 мас. % наночастиц Fe2O3 методом in situ в термопластичный полиуретан на основе поли-ε-капролактон диола приводит к повышению температуры изменения формы полиуретана с 37 до 40–45°С, что позволит, по мнению авторов [102], в дальнейшим использовать данные композиты в качестве имплантирующих устройств. Наибольшей скоростью восстановления первоначальной формы (время восстановления формы 26 с, доля остаточных деформаций 97%) обладает композит, содержащий 30% Fe2O3, под действием магнитного поля (частота f = 45 кГц, амплитуда H = 29.7 и 36.7 кА/м) [102].
G. Shu-Ying [118] разработал полиуретановый биоразлагаемый композит на основе сополимера поли-L-лактида с поли-ε-капролактоном, 1,6-гексаметилендиизоцианата и удлинителя цепи политриметилолпропана с памятью формы в присутствии 3–9 мас. % Fe3O4 для использования в качестве саморасширяющихся сосудистых стентов. Стент может поддерживать свою временную форму при температуре тела в течение определенного времени, восстановление постоянной формы происходит при температуре немного выше температуры тела (около 40°C) как в воде, так и в переменном магнитном поле, обеспечивая достаточно высокую скорость переключения без внешнего нагревания. Нанокомпозиты имеют долю фиксированной деформации Rf > 99% и долю остаточной деформации Rr > 82% при температурах переключения 37 и 40°С.
Авторами статьи [103] установлен синергетический эффект электропроводящих и магнитных частиц на механические и термические свойства полиуретана на основе политетраметиленоксида (М = 2 × 103), 4,4'-дифенилметандиизоцианата и 1,4-бутандиола. Установлено, что при введении оптимального соотношения 1% графеновых нанопластин (размер частиц 2–10 нм) и 30% Fe2O3 в матрицу полимера прочность при разрыве повышается в 4.2 раза, разрывное удлинение – в 7.4 раза, а скорость восстановления формы снижается в 2 раза по сравнению с немодифицированным образцом.
Таким образом, выбор оптимальных концентраций, способов введения наночастиц в матрицу и режимов внешнего воздействия позволяет повышать температуру переключения и управлять скоростью восстановления формы.
СВЕТОЧУВСТВИТЕЛЬНЫЕ ТЕРМОПЛАСТИЧНЫЕ ПОЛИУРЕТАНЫ И ПОЛИУРЕТАНМОЧЕВИНЫ
Наряду с низкочастотным электромагнитным излучением, для управления эффектом памяти формы материала активно применяют лазеры в видимой области излучения (405–600 нм) [119–122], ближней ИК-области (600–800 нм) с высокой проникающей способностью и быстрым эффектом включения–выключения [123] или в УФ-области (200–400 нм) [124]. Преимуществом использования света в качестве источника активации является его доступность, безопасность, простота контроля интенсивности воздействия.
В литературе описывается в основном два механизма активации изменения формы светом – фототермический [125] и фотохимический [126].
Термопластичные полиуретаны и полиуретанмочевины с фототермической активацией эффекта памяти формы
Среди наполнителей термопластичных полиуретанов и полиуретанмочевин, индуцирующих тепло под действием электромагнитного излучения и обеспечивающих фототермический эффект, хорошо зарекомендовали себя наночастицы золота и серебра [125, 127–130]. Существует множество методов синтеза и областей применения таких наночастиц, что делает их наиболее эффективными для придания адаптивных свойств полимерам [131].
В фототермическом процессе резонансный свет возбуждает поверхностный плазмон наночастицы, а поглощенная энергия превращается в тепло из-за электрон-фотонных столкновений. Этот нагрев эффективен и сильно локализован в матрице полимера. Таким образом, под действием излучения в УФ-, видимой или ИК-области наблюдается повышение температуры образца на десятки градусов [132–134] и соответствующее изменение формы.
Н. Zhang с соавторами [135] ввели наночастицы золота от 0.1 до 1 мас. % в матрицу полиуретана на основе поли-ε-капролактон диола и 1,6-гексаметилендиизоцианата. Скорость восстановления формы контролировали, меняя интенсивность и продолжительность облучения светом в ближней ИК-области (700 нм). Установлено, что при оптимальной мощности лазера 0.57 В/см2 можно добиться увеличения температуры переключения на 25°С.
Отметим, эффект нагрева зависит от природы наночастиц, способа введения и концентрации наночастиц, а также от интенсивности и времени облучения [136]. В частности, показано, что равномерное распределение наночастиц серебра и золота в матрице полимера обеспечивает равномерную передачу тепла в нанокомпозитах и приводит к ускорению восстановления формы [131].
Описан фототермический эффект наночастиц серебра и золота, продуктивно использованный в наноструктурированном полиуретане для создания локализованного нагрева. Это позволило провести выборочную термическую обработку полимеров in situ для инициации процессов сшивки, коррекции дефектов материалов, управления термической активацией памяти формы полиуретана фирмы “Diaplex” [137], для улучшения теплофизических и механических свойств полиуретановых наноматериалов [138, 139]. Имеются немногочисленные сведения о влиянии наночастиц золота на термические, механические свойства и эффекта памяти формы полиуретанмочевины. Так, в работе [140] при введении различных концентраций наночастиц серебра в матрицу сверхразветвленного полиуретана на основе поли-ε-капролактон диола (М = 3 × 103), 2,4-толуилендиизоцианата и глицерола было показано, что с ростом концентрации наполнителя увеличиваются прочность на растяжение с 6.8 до 9.6 МПа и твердость по Шору с 72 до 76, а удлинение при разрыве и изгибе остается постоянным. Улучшение механических свойств обусловлено, по мнению авторов, хорошей диспергируемостью наночастиц в матрице полимера. Механические свойства композитов также можно настраивать, используя наночастицы разного размера. Крупные частицы в матрице приводят к снижению жесткости и более высоким значениям удлинения при разрыве по сравнению с композитами с меньшими частицами, которые демонстрируют повышенную жесткость и более низкое удлинение при разрыве.
Наряду с металлическими наночастицами серебра и золота в качестве фототермического наполнителя авторы работы [141] вводили 0.1–0.5% дикетопирролпиррола в матрицу полиуретана на основе поли-ε-капролактон диола, 1,6-гексаметилендиизоцианата и удлинителей цепи ди-трет-бутил-этилендиамина и диметилэтилендиамина. Восстановление формы происходило при лазерном облучении (доза облучения 0.5 Вт/см2 в течение 2 мин) в ближней ИК-области (808 нм). Полученные композиты имели хорошие антибактериальные и фоточувствительные свойства. Было установлено, что композит в присутствии 0.5 мас. % дикетопирролпиррола восстанавливает свою форму до Rr = 95% в течение 30 с из-за быстрого повышения температуры. При этом низкую эффективность восстановления формы (около 95%) нанокомпозита при облучении лазером авторы связывают с возможной агломерацией наночастиц в матрице полимера.
Для использования фоточувствительных материалов в медицине К.С. Hribar с сотрудниками смешивали биосовместимые золотые наностержни с полимером для фотопереключения формы и изменения температуры стеклования материала [142, 143]. Исследования показали отсутствие цитотоксичности композитов в присутствии золотых наностержней. Установлено, что воздействие лазера 770 нм мощностью 0.3 Вт приводит к повышению температуры стеклования композита от 40.7 до 53.3°С при концентрации наностержней 1.8 × 10–13 и 3.3 × 10–12 мас. % соответственно.
Необходимо отметить, что для активации формы под действием лазерного облучения в термопластичный полиуретан также вводят углеродные нанонаполнители (углеродные нанотрубки, сажа). В частности, одностенные углеродные нанотрубки улучшают механические свойства матрицы, а также имеют широкую полосу поглощения в ИК-области [144]. Под действием ИК-излучения происходит разогрев полимерной матрицы до плавления кристаллической фазы, что обеспечивает восстановление постоянной формы. Сопоставимые эффекты возникают при электрически индуцированном джоулевом нагреве при протекании тока через перколяционную сетку углеродных нанотрубок. Описывается новый способ получения гибридного полиуретанового композита на основе поли-ε-капролактон диолас М = = 4 × 103, 1,6-гексаметилендиизоцианата и гидроксиэтилакрилата под действием УФ-сшивания (365 нм, 8 Вт) с оксидом графена, функционализированным аллилизоцианатом. Показано, что гибридный полиуретановый композит в присутствии 1 мас. % оксида графена под действием света в ближней ИК-области восстанавливает свою форму на 90% в течение 1 мин, в то время как в отсутствие оксида графена – только на 10% в течение 6 мин. В качестве источника света авторы использовали Hg-Xe-лампу мощностью 25 мВт/см на расстоянии 20 см от образцов [145].
Области применения фототермически индуцируемых нанокомпозитов на основе термопластичных полиуретанов с эффектом памяти формы разнообразны. Они могут служить оптическими датчиками, микророботами, оптическими приводами и другими светочувствительными устройствами, в том числе медицинского назначения.
Композиты термопластичных полиуретанов и полиуретанмочевин с фотохимическим эффектом памяти формы
Фотохимический эффект основан на формировании или разрушении химических сшивок цепей термопластичных полиуретанов и полиуретанмочевин при облучении светом определенной длиной волны [146–149]. В качестве фотообратимых центров, вводимых в структуру полимера, используют производные коричной кислоты [150, 151], антрацен [152, 153], кумарин [154] или производные адаманта [155]. Накопление структурных изменений приводит к эволюции полимерной матрицы и даже к макроскопической деформации. В результате форма и механические свойства материала контролируемым образом меняются, в частности наблюдается эффект самозалечивания трещин и дефектов [151, 156].
Примером материала с фотохимической активацией формы может служить сополимер на основе полиэтиленгликоля, 4,4'-дигидроксиазобензола, 1,6-гексаметилендиизоцианата и 1,3,5,7-тетрагидроксиадамантана [157]. Введение производного адамантана существенно улучшает механические свойства и термостойкость полимера. Синтезированные образцы способны изменять форму под действием УФ-облучения, фиксировать форму в видимом свете и восстанавливать первоначальную форму в воде при комнатной температуре, т.е. являются одновременно термо-, УФ- и влагочувствительными материалами. Авторами продемонстрировано две временные формы материала, которые индуцировались при нагреве и под действием света. На первой стадии образцы деформировали при температуре выше плавления кристаллической фазы полиэтиленгликоля (80°С). При последующем охлаждении фиксировали первую временную форму. Под действием УФ-излучения полимер претерпевал деформацию скручивания и фиксировался во второй временной форме в видимом свете. Восстановление постоянной формы полимера происходило в течение 10 с в воде при 80°С. Хорошо известно, что восстановление формы в воде в основном объясняется пластификацией молекулами воды, а скорость восстановления зависит от скорости диффузии воды в полимерной матрице. Основная причина высокой скорости восстановления формы полиуретана, о котором говорится в работе [157], в воде заключается в том, что гидрофобные цепи полиэтиленгликоля довольно хорошо распределены в матрице и образуют сетчатую структуру при введении 1,3,5,7-тетрагидроксиадамантана, облегчающую диффузию воды.
Другой тип фоточувствительного полиуретана был создан на основе кумаринсодержащего кристаллизующегося поли-ε-капролактона [158]. При УФ-облучении с длиной волны больше 310 нм происходит димеризация кумарина до циклобутана, обратный процесс наблюдается при облучении с длиной волны меньше 260 нм. Таким образом, сложный эффект памяти формы со ступенчатым восстановлением первоначальной формы сначала при нагреве, а затем при УФ-облучении при λ = = 254 нм был продемонстрирован с помощью процедуры фиксации временной формы при деформации и УФ-облучении при λ =365 нм. Поли-ε-капролактон, содержащий кумарин, характеризуется высокой степенью кристалличности с температурой плавления выше комнатной. В этом случае соотношение коэффициентов восстановления первой и второй формы полиуретана может быть изменено путем подбора времени УФ-облучения и концентрации кумариновых звеньев.
Используя N,N-бис-(2-гидроксиэтил)циннамамид в качестве фоточувствительного мономера при синтезе полиуретана на основе 1,6-гексаметилендиизоцианата, поли-ε-капролактон диола и поли-L-лактида, авторы [159] показали, что боковые циннамамидные группы действуют как молекулярные переключатели под действием УФ-излучения в области 365–312 нм и обеспечивают полимеру обратимое [2+2] циклическое сшивание. При формировании дополнительной химической сетки увеличивались предел прочности от 10 до 20 МПа, модуль упругости от 20 до 230 МПа и удлинение при разрыве с 230 до 530%. Рост доли мягких сегментов поли-ε-капролактона также приводит к увеличению удлинения при разрыве. Долю фиксированных деформаций и обратимых деформаций можно улучшить, варьируя содержание N,N'-бис-(2-гидроксиэтил)циннамамида и поли-L-лактида соответственно. При содержании N,N'-бис-(2-гидроксиэтил)циннамамида 20 мас. % значение Rf достигает 50%, а максимальная величина Rr составляет более 95% при содержании поли-L-лактида 50 мас. %.
Авторы [160] разработали новый тип адаптивного полиуретана, состоящий из мягкого блока на основе N-метилдиэтаноламина и 1,6-гексаметилендиизоцианата и жесткого блока на основе 1,6-гексаметилендиизоцианата и 4,4'-азодибензойной кислоты. Благодаря своей структуре полимер способен принимать временную форму под действием УФ-облучения при 365 нм за счет цис-транс-фотоизомеризации 4,4'-азодибензойной кислоты и восстанавливать первоначальную форму при нагревании до 80°С. Такие полимеры, по мнению авторов, могут быть использованы в качестве интеллектуальных устройств захвата, искусственных мышц и роботизированных пальцев.
ВЛАГОЧУВСТВИТЕЛЬНЫЕ ТЕРМОПЛАСТИЧНЫЕ ПОЛИУРЕТАНЫ И ПОЛИУРЕТАНМОЧЕВИНЫ (ВОДА И PH)
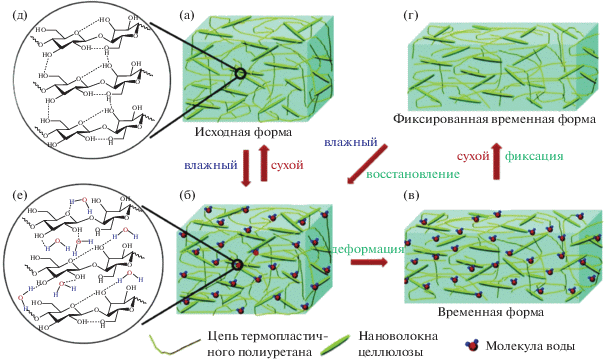
В литературе по исследованию влагочувствительных материалов, изменяющих свою форму под действием воды, упоминаются некоторые ограничения их использования в медицине вследствие низкой скорости восстановления формы. В связи с этим в настоящее время разрабатываются стратегии синтеза новых композиционных термопластичных полиуретанов и полиуретанмочевин на основе нановолокон целлюлозы [161], органоглины [162], оксида графена [163], которые могли бы улучшить механические свойства и биоразлагаемость, а также обеспечить высокую скорость восстановления в присутствии воды [164]. Так, в работе [165] предложен новый влагочувствительный композиционный полиуретан на основе политетраметиленгликоля, 4,4'-дифенилметандиизоцианата и 1,4-бутандиола в присутствии 2–40 мас. % нановолокон целлюлозы. Композит получен путем смешивания дисперсии нановолокон целлюлозы в диметилформамиде с набухшим полиуретаном. Установлено, что композит в присутствии 30% нановолокон целлюлозы обладает высоким коэффициентом восстановления формы (более 90%) при скорости восстановления менее 1 мин за счет разрушения в воде жесткой перколяционной сетки нановолокон в эластичной полиуретановой матрице (рис. 6). Высокая скорость отклика авторами объясняется хорошими эластомерными свойствами матрицы и улучшенной гидрофильности наполнителя. При набухании исходной сухой пленки (рис. 6а) в ней происходит формирование сетки водородных связей между нановолокнами и матрицей (рис. 6д). При набухании молекулы воды могут диффундировать в пленку (рис. 6б), при этом образуются конкурентные водородные связи между молекулами воды и гидроксильными группами нановолокон целлюлозы (рис. 6е), что приводит к разрушению временной сетки (рис. 6е). В этом состоянии пленка становится мягкой и легко деформируется под действием внешней силы (рис. 6в). После удаления воды происходит фиксация временной формы (рис. 6г) за счет восстановления водородных связей между нановолокнами и жесткой перколяционной сеткой, которая при повторном смачивании снова разрушается, и пленка восстанавливает свою первоначальную форму за счет хорошей эластичности полиуретановой матрицы.
Рис. 6.
Схема механизма изменения формы полиуретанового композита, активируемого водой [165]. Пояснения в тексте.
Такие влагочувствительные материалы могут претерпевать конформационные или фазовые изменения в ответ на внешние воздействия [166]. Данные материалы представляют собой одну из наиболее захватывающих развивающихся областей науки [167] в связи с перспективами их использования в тканевой инженерии [168] и системах доставки лекарств [169].
В работе [165] показано разнообразное использование композита из полиуретана на основе политетраметиленгликоля, 4,4'-дифенилметандиизоцианата и 1,4-бутандиола в присутствии 40% нановолокон целлюлозы (рис. 7). Так, пленка, принимающая развернутую временную форму из сложенной постоянной, может быть использована для захвата объекта в воде, а пленка с развернутой постоянной формой и спиральной или сложенной временной формой – для проектирования системы контролируемого высвобождения.
Рис. 7.
Восстановление развернутой формы водочувствительного полиуретанового композита из временной формы куба (а) и спирали (б), а также восстановление спиральной формы из временной кубической или плоской формы (в, г) [165].

Для применения в медицине отлично подходят материалы, изменяющие свою форму в физиологических растворах с заданным pH. Значения рН зависят от типа органов и тканей, кроме того, резкое изменение рН часто наблюдается в биологических системах при патологических состояниях [170].
На сегодняшний день опубликовано незначительное количество работ, посвященных реализации эффекта памяти формы через диссоциацию и ассоциацию водородных связей под действием изменения рН внешней среды. В одном из исследований [171] описан синтез полимера на основе полиэтиленгликоля, 4,4'-дифенилметандиизоцианата и N,N-бис-(2-гидроксиэтил)изоникотинамида, который проявляет высокую чувствительность к рН за счет ассоциации и диссоциации водородных связей между атомом водорода уретана и атомом азота пиридинового кольца (рис. 8). Механизм рН-активации заключается в образовании сетки водородных связей между атомами азота пиридинового кольца и группой Н–N уретана в нейтральных или щелочных средах. Данная связь разрушается в кислой среде за счет протонирования пиридинового кольца. ЭПФ проиллюстрирован на примере предварительного набухания полимера в кислоте (рН 1.3), фиксации формы в щелочи (рН 10) и последующего восстановления ее формы в кислоте при 20°С.
Рис. 8.
Схема осуществления эффекта памяти формы рН-чувствительного полимера на основе полиэтиленгликоля М 4 × 103, 4,4'-дифенилметандиизоцианата и N,N-бис-(2-гидроксиэтил)изоникотинамида [171].
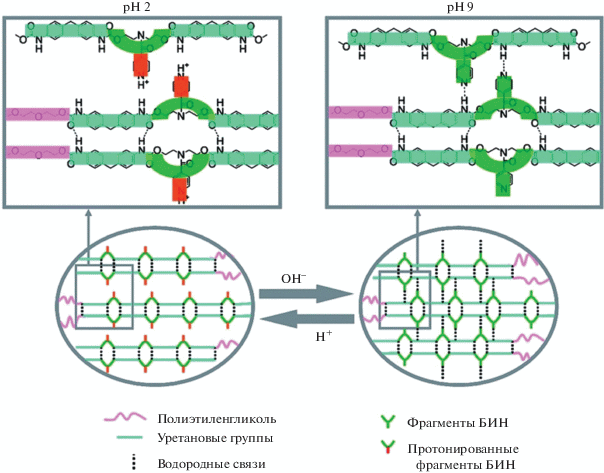
При замене уретановых групп на карбоксильные временная форма может быть зафиксирована при рН 2, а восстановление постоянной формы происходит при рН 9 [172].
В работе [173] сообщается о pH-чувствительном полиуретане на основе полиэтиленгликоля (с содержанием 20, 30 и 40%), 4,4'-дифенилметандиизоцианата и производного пиримидона – 2-уреидо-4[1H]-пиримидона. Этот материал в щелочной среде при рН 10 фиксирует временную форму за счет образования димеров пиримидона, в то время как при pH 1 в кислом растворе восстанавливает постоянную форму за счет диссоциации димерных групп. Важно отметить, что данный полимер является также термочувствительным и способным к восстановлению формы при нагревании. Авторы показали, что при содержании полиэтиленгликоля 30% в полиуретане наблюдается наилучший коэффициент восстановления формы (более 85%) под действием pH, а при содержании полиэтиленгликоля 20% – при нагревании выше 40°С.
В отличие от других систем, чувствительных к температуре, магнитному полю, свету и т.д., ключевую роль в управлении формой материала при изменении pH играет процесс диссоциации и ассоциации водородных связей. Полиуретан с таким типом переключения может использоваться в качестве систем доставки лекарств к тканям и органам, имеющим определенную физиологическую среду (pH). При попадании полимерной капсулы в кислую или щелочную среду за счет разрушения водородных связей полимерной матрицы происходит дозированное высвобождение лекарственного средства.
ПРИМЕНЕНИЕ ТЕРМОПЛАСТИЧНЫХ ПОЛИУРЕТАНОВ И ПОЛИУРЕТАНМОЧЕВИН В МЕДИЦИНЕ
Использование изделий из термопластичных полиуретанов и полиуретанмочевин в медицине связано с их доступностью, хорошими физико-механическими свойствами, цитотоксичностью, биосовместимостью, а также, в некоторых случаях, биоразлагаемостью [174–179]. Эти особенности полиуретанов позволяют заменить традиционно применяемые металлы, сплавы и керамику на полимерные материалы.
Биоразлагаемость адаптивных полиуретанов и полиуретанмочевин позволяет восстанавливать ткани с минимальным вмешательством без последующих хирургических процедур для извлечения имплантатов [180]. С целью исследования материалов на биоразложение и влагопоглощение используют в основном фосфатные буферные растворы и растворы 2 М HCl и 5 М NaOH. Для краткосрочного процесса биоразложения образцы погружают в соответствующие растворы (фосфатный буферный раствор, 2 М HCl или 5 М NaOH), а затем инкубируют при 37°C. Во время краткосрочного теста измеряют потерю массы образца через одну, две, три недели и 32 дня. В свою очередь при изучении долгосрочного процесса биоразложения с использованием фосфатного буферного раствора, вес образцов измеряют через неделю, один месяц и три месяца. Через определенные промежутки времени образцы промывают несколько раз в дистиллированной воде, затем сушат (три дня, 37°С) и снова взвешивают. Степень разложения определяют по проценту потери массы образца с течением времени [181]. Процесс биоразложения зависит от молекулярной массы, полидисперсности, кристалличности, гидрофильных и гидрофобных функциональных групп полиуретана. Период разложения настраивается с помощью выбора разлагаемых олигомеров и параметров полимеризации. Разложение мягкого полиэфирного блока полиуретана происходит через гидролиз сложноэфирной связи, и период разложения может составлять от недель до нескольких лет, в зависимости от типа полиэфира. Как правило, жесткий блок разлагается медленнее мягкого, поскольку уретановые связи гораздо менее подвержены гидролитической деградации. Использование диизоцианатов, низкомолекулярных диолов и диаминов приводит к образованию водородных связей между уретановыми группами, что обеспечивают полимеру дополнительную прочность и замедление гидролитической деградации [182].
Хорошая биосовместимость материалов на основе адаптивных полиуретанов подтверждается стандартными тестами на цитотоксичность и мутагенность [183–185]. Еще в 90-х годах М.С. Tanzi с сотрудниками [186] показали, что катализаторы, широко используемые в синтезе полиуретанов (такие как октоат олова, дибутилдилауринат олова, 1,4-диазабициклооктан, тетраметилбутанодиамин), повышают цитотоксичность полимера по отношению к эндотелиальным клеткам человека. Следовательно, синтез полиуретанов для медицинских целей необходимо осуществлять без использования указанных выше катализаторов, либо удалять их после синтеза [187, 188].
Биосовместимые и биоразлагаемые полиуретаны и полиуретанмочевины на основе поли-ε-капролактон диола, полипропиленгликоля, полиэтиленгликоля и поли-L-лактида [189–195] могут использоваться как эндоваскулярные устройства (устройства для окклюзии аневризмы и сосудистых стентов), в ортопедии, ортодонтии, эндоскопической хирургии, почечном диализе, покрытий для оборудования и т.д.
Адаптивные термочувствительные полиуретаны могут быть альтернативой традиционным материалам, применяемым для лечения искривлений зубов. Дуги из термопластичных полиуретановых сополимеров в ортодонтических брекетах более эстетичны, чем традиционный металлический арочный провод. Y.C. Jung и J.W. Cho [196] использовали экструдированный адаптивный провод, крепящийся к зубам в зубной модели при помощи кронштейнов из нержавеющей стали. Полиуретановую дугу получали традиционным двустадийным способом через стадию макродиизоцианата из циклоароматического 4,4'-дифенилметандиизоцианата, поли-ε-капролактон диола (М = 3 × 103) и 1,4-бутандиола в качестве удлинителя цепи. Показано, что при нагревании провода зубы медленно перемещались и выравнивались.
L.E. Aguilar с сотрудниками [197] создали термочувствительную полиуретановую матрицу, состоящую из нановолокон коммерческого полиуретана фирмы Esthane массой (М = 1 × 105) и сополимера поли(N-изопропилакриламид)-гидроксиметил-акриламида. Эту матрицу авторы использовали в качестве покрытия сосудистого нитинолового стента с ЭПФ для высокоэффективной гипертермической химиотерапии. Применяя переменное магнитное поле к стенту, покрытому термочувствительной матрицей и содержащему лекарственные средства, была достигнута эффективная нагревательная способность и контролируемое высвобождение лекарственных средств – 5-фторурацила и паклитаксела.
В работе [198] исследовали стабильность коммерческого полиуретана PurSil®AL20 на основе 4,4'-дициклогексилметандиизоцианата, 1,4-бутандиола, политетраметиленоксида и полидиметилсилоксана для использования в качестве средства доставки лекарства доцетаксела (полусинтетическое цитостатическое лекарственное средство растительного происхождения из группы таксанов, получаемое путем химического синтеза из природного сырья – из игл тиса европейского) через пищеводный лекарственный элюирующий стент. На высвобождение лекарственного средства влияет как рекристаллизация доцетаксела, так и температура хранения. Установлено, что оптимальными условиями для хранения стента, покрытого влагостойкой полиуретановой пленкой и находящегося в стенте доцетаксела, является комнатная температура 25°С. При этом низкая температура хранения (4°С) приводила к рекристаллизации доцетаксела и снижала скорость его высвобождения, а повышение температуры до 40°С – к увеличению размеров микроструктурных доменов и междоменных расстояний полиуретана (от 85 до 129 Å), что в дополнении к перекристаллизации доцетаксела препятствовало высвобождению лекарства.
Другой обширной областью применения биоразлагаемых адаптивных полиуретанов и полиуретанмочевин является неинвазивная тканевая инженерия. В этой технологии ткани можно выращивать на полиуретановых матриксах и неинвазивно вводить в организм (например, через катетер), обеспечивая лечение или реконструкцию тканей и органов. Имплантируемые эмболизирующие устройства и стенты на основе термопластичных полиуретанов продемонстрировали возможность их использования для эндоваскулярных процедур. Биоразлагаемые каркасы из адаптивных полиуретанов могут также применяться для реконструкции слизистой оболочки глотки, регенерации костей и восстановления органов. Исследовалась возможность использования термочувствительных полиуретанов в качестве внеклеточного матрикса для выращивания тканей in situ. D. Rickert с соавторами сообщили о росте клеток на биоразлагаемом полимере на основе гидроксилсодержащего поли-ε-капролактона [199]. Клетки глотки крысы выращивали на пористой и гладкой поверхности полимера для изучения перспективы реконструкции слизистой оболочки верхних дыхательных путей. В работе [200] исследовали рост мезенхимальных и мезотелиальных клеток человека, а также мезотелиальных клеток фибробластов L929 мышей на аналогичном полиуретане на основе поли-ε-капролактон диола. Было обнаружено, что мезотелиальные клетки создают антиадгезионный поверхностный слой, который может способствовать регенерации тканей, а также что использование тепла для переключения формы не влияло на большинство мезотелиальных клеток.
В работе [201] показана возможность применения в тканевой инженерии биосовместимых полиуретановых нановолокон на основе поли-ε-капролактон диола и L-лизиндиизоцианата в присутствии фиброина шелка, полученных электроформованием. Результаты клеточной пролиферации и морфологии клеток свидетельствуют о том, что нановолокна полиуретан/фиброин шелка способствуют повышению жизнеспособности клеток. Авторы установили, что нановолокнистые матрицы с пористой структурой (“sсaffold”) полиуретан/фиброин шелка в соотношении 40:60 соответствуют необходимым техническим требованиям для тканевой инженерии и могут быть использованы в качестве перспективной конструкции для клапанов сердца.
Обзор, сравнивающий методы электроформования и микрофлюидики для получения микро- и нановолокон полиуретана, вышел в 2017 г. [202]. В статье представлены примеры использования таких волокон для инженерии плотных тканей и генерации фибробластов связок человека. В частности, методом электроформования были получены тонкие волокна сегментированных термопластичных полиуретанов на основе полиэтиленгликоля, поли-L-лактида, поли(триметиленкарбоната) и алифатических диизоцианатов, из которых были изготовлены новые биоразлагаемые эластомерные высокопористые матрицы для инженерии мягких тканей [203].
При использовании адаптивных материалов в медицине важно обеспечить им бактерицидные свойства. В работе [204] сообщается о разработке нового покрытия из 3-(триметоксисилил)-пропилдиметилоктадециламмоний хлорида, полученного методом прививочной полимеризации акриловой кислоты из паровой фазы для коммерческих полиуретановых катетеров (Bioengineering Laboraties srl, Italy); покрытые катетеры проявили антибактериальную активность против кишечной палочки. В обзоре, посвященном последним достижениям в области противомикробных покрытий для мочевых катетеров [205], авторы описывают преимущества и недостатки различных полимеров, включая полиуретаны и полиуретанмочевины, для которых отмечается прочность, био- и гемосовместимость, а также простота нанесения. Пришивка катионных полиэтилениминных щеток к поверхности коммерческого полиуретанового стента (на основе алифатического полиэфира EG93A и 1,6-гексаметилендиизоцианата фирмы Tecoflex®, предотвращает воспаление мочеточника и закупорку тканей пациента, а пришивка алкильных цепей приводит к усилению антибактериального эффекта [206, 207]. Для получения внеклеточного матрикса для временной поддержки роста клеток и регенерации тканей использовали полиуретан Tecoflex EG-80A, полученный методом электроформования, с пропиткой из меда, виноградного масла и прополиса (смолистого вещества, произведенного пчелами) [208]. Полученные пленки демонстрировали антибактериальную активность и повышенную цитосовместимость, что позволяет использовать их для изготовления раневых повязок и тканей кожи. Методом электроформования были изготовлены нановолокона на основе полиуретана фирмы Estane® Skythane® X595A-11 с добавлением декстрана и эстрадиола для улучшения восстановления ранений кожи женщин в постклимактерическом периоде [209]. В обзоре приводится несколько примеров антибактериальных покрытий на основе полиуретана с различными характеристиками: высвобождающие салициловую кислоту [210], устойчивые к P. aeruginosa и E. coli, а также полиуретановое покрытие с большим сроком службы [211]. Для повышения антибактериальных свойств полиуретана вводят наночастицы металлов и оксидов металлов, таких как серебро или цинк. Известно, что наночастицы оксида цинка и серебра используются при лечении инфекционных заболеваний благодаря их антимикробным (антибактериальным) свойствам. Кроме того, стабильность наночастиц ZnO в жестких условиях обработки и относительно низкая токсичность в сочетании с сильными антимикробными свойствами усиливают их применение в качестве антимикробных препаратов [212]. Исследователи показали, что наночастицы ZnO обладают селективной токсичностью для бактерий и минимально воздействуют на клетки человека [213]. Наночастицы серебра способствуют заживлению ран, уменьшая воспалительный процесс. Следовательно, введение в полимерную матрицу наночастиц серебра позволит создавать нетоксичные и экологически безопасные материалы с антибактериальными свойствами [214–216]. Модифицированная термопластичными полиуретанами пленка на основе политетраметиленгликоля (М = 2 × 103), 4,4'-дифенилметандиизоцианата и 1,4-бутандиола наночастицами нитрата серебра и перфторалкила показала отличную антибактериальную активность против S. aureus и Escherichia сoli и может быть использована для предотвращения бактериального загрязнения внутрисосудистых катетер-ассоциированных инфекций [214]. Оксид цинка как наполнитель полиуретановой матрицы по сравнению с наночастицами серебра имеет более сильные межмолекулярные ван-дер-ваальсовы взаимодействия и может образовывать водородные связи с активным водородом уретановых (или мочевинных) групп, поэтому оксид цинка способен повышать механические свойства термопластичных полиуретанов и полиуретанмочевин. Для придания бактерицидных свойств полимеру на основе поли-ε-капролактон диола, 1,6-гексаметилендиизоцианата и 1,4-бутандиола, авторы [217] вводили наночастицы ZnO в количестве от 1 до 5 мас. %. Показано, что полиуретановые композиты проявляют антибактериальные свойства против стафилококков и кишечной палочки (Staphylococcus aureus, Micrococcus sp., Escherichia coli). Хорошо диспергированные наночастицы ZnO в полиуретановой матрице способствуют повышению гидрофобности полученных покрытий. В работе [218] описываются способы достижения антибактериальных свойств полиуретановых покрытий на основе политетраметиленгликоля, 4,4'-дифенилметандиизоцианата и удлинителя цепи 1,4-бутандиола путем введения наночастиц меди и серебра.
Адаптивные полиуретаны демонстрируют превосходные свойства по сравнению со многими другими полимерами в качестве основы адгезивов, чувствительных к давлению, они обеспечивают хорошую адгезию к коже и, следовательно, могут использоваться для хирургических раневых повязок и пластырей [219]. Полиуретановая раневая повязка может быть получена в виде полупроницаемой липкой пленки, перфорированной мембраны, гидроколлоида или пены [220]. В зависимости от формы (пленки, мембраны, пена и т.д.), химического состава и функционализации, они подходят для ран с разным количеством экссудата [219, 221]. Прозрачные полиуретановые пленки особенно подходят для защиты поверхностей и послеоперационных ран с отсутствием или слабым секретом экссудата. Через них не проникают бактерии и вода, но проникает воздух, таким образом позволяя ранам дышать. Полиуретановые покрытия могут быть единственным компонентом раневой повязки, но чаще используются в виде многокомпонентных материалов. В настоящее время на рынке представлены полиуретановые повязки с добавлением различных фармакологически активных веществ, антибактериальных (хлоргексидин (“Opsite CH”) и йод (“Tegaderm Plus”)), местных анестетиков, агентов с бактериостатическим эффектом и противогрибковых средств. Перевязочный материал для лечения язв на ногах содержит глицерин, цинк или коллаген [221].
За последние 10 лет, несмотря на большое число публикаций, касающихся полиуретанов, имеется лишь ограниченное число научных работ по тестированию биосовместимости медицинских полиуретановых перевязочных материалов. Одно из требований к этим материалам – наличие хороших механических свойств, способность поддерживать соответствующий уровень влажности раны и противомикробную защиту [222]. А. Yari с сотрудниками синтезировали новую полиуретановую мембрану на основе полиэтиленгликоля с М = (1–2) × 103, 1,6-гексаметилендиизоцианата и глицерола, предназначенную для ухода за раной. Мембрана способна поглощать экссудат и демонстрирует антимикробные свойства. Синтез проводили в присутствии эпоксидно-функционализированного четвертичного аммониевого соединения (глицидилтриэтиламмония хлорид). Применяя тест МТТ (колориметрический тест для оценки метаболической активности клеток) с использованием 1-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолия-бромида и метод прямого контакта с двумя клеточными линиями – фибропластами и эпидермальными кератиноцитами, была показана хорошая биосовместимость полученных мембран [223]. Современные исследования в области синтеза биосовместимых термопластичных полиуретанов для компонентов раневых повязок также сосредоточены на замене нефтехимического сырья полиолами природного происхождения, в частности натуральными маслами [224, 225]. M.U. Ozkaynak с соавторами показали возможность использования в качестве полиола льняного масла с целью получения полиуретановых мембран. Эти материалы демонстрируют хорошие механические свойства, газопроницаемость и кислотостойкость, в связи с чем было предложено их использование в качестве материала для медицинской раневой повязки [225, 226]. Проницаемость и адгезия материалов на основе термопластичного полиуретанового сополимера может быть улучшена за счет варьирования сшивающих агентов. В работе [227] описан синтез модифицированного полиуретанового гидрогеля, способного поглощать большое количество воды. Асимметричная мембрана из гидроксилсодержащего ПЭГ предотвращает быстрое обезвоживание раневой поверхности и проникновение бактерий, что помогает обеспечивать хорошее заживление ран. Тромбоциты притягиваются к поверхности термопластичных полиуретанов благодаря его гидрофильности, генерируя коагуляционный каскад [227]. C.V. da Silva с соавторами [228] сообщали об успешном добавлении масла огуречника (содержащего незаменимые жирные кислоты для заживления ран) и его высвобождении из перевязочных материалов на основе полиуретановой пены; авторы также рассматривают многие другие препараты, которые могут быть включены в такие повязки, например анальгетики, средства для рубцевания, антибиотики и противораковые препараты.
S. Zhu [229] разработал частичнокристаллический термопластичный полиуретан на основе поли-ε-капролактон диола, 4,4'-дифенилметандиизоцианата и 1,4-бутандиола с целью его использования в качестве мягкого активатора искусственной мышцы, способного выполнять два основных сокращения. Было показано, что степенью сокращения можно легко управлять путем изменения кристалличности мягкого блока. Авторы указывают на сенсорную способность синтезированных полимеров благодаря высокой чувствительности содержания кристаллической фазы к температуре. Данные материалы на основе одного полимера, по мнению авторов, расширяют функциональность термочувствительной мышцы без введения дополнительных компонентов и систем управления.
У адаптивных полиуретанов есть множество других интересных свойств, открывающих большие перспективы их использования в медицине: они просты в получении, обладают химической стабильностью к воде и органическим растворителям, устойчивы к воздействию солнечного света и характеризуются отличными эластичными свойствами, а также биосовместимостью.
ЗАКЛЮЧЕНИЕ
В настоящем обзоре представлены результаты последних достижений в области синтеза, модификации и применения частичнокристаллических термопластичных полиуретанов с эффектом памяти формы. Сделан акцент на взаимосвязи строения термопластичных полиуретанов и полиуретанмочевин с их адаптивными свойствами и способами активации памяти формы для возможного применения таких материалов в медицине. Показано, что на конечные свойства полиуретанов влияет ряд факторов: способ синтеза, состав, строение и относительное содержание мягкого и жесткого блоков, молекулярная масса и степень кристалличности мягкого полиэфирного блока. Природа и строение удлинителей цепи оказывает влияние на формирование постоянной сетки за счет образования водородных связей. Все это определяет механические свойства материала, условия переключения, степень фиксирования формы и скорость ее восстановления.
Важным преимуществом данного класса полимеров является возможная настройка свойств материалов для конкретного применения за счет оптимизации перечисленных параметров. Кроме того, введение разнообразных наполнителей в полиуретановую матрицу позволяет придавать композитам чувствительность к различным внешним воздействиям (температура, свет, влага, магнитное или электрическое поле, излучение), создавая материалы со сложным механическим откликом. Наряду с перечисленными достоинствами, адаптивным полиуретанам присущи и некоторые недостатки: низкая жесткость для многих составов, необходимость использования органических растворителей и т.п. Однако большой объем публикаций в данной области не оставляет сомнений, что исследователи смогут преодолеть эти недостатки, результатом чего будет широкое внедрение частичнокристаллических полиуретанов в различных высокотехнологичных областях.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (код проекта 19-53-15016) и по теме Государственного задания 0089-2019-0012 (номер государственной регистрации ААА-А19-119032690060-9).
Список литературы
Pulidindi K., Chakraborty S. // Global Market Insights. Competitive Market Share & Forecast, 2018.
Haryńska A., Kucinska-Lipka J., Sulowska A., Gubanska I., Kostrzewa M., Janik H. // Materials. 2019. V. 12. № 6. P. 887.
Menon A.V., Madras G., Bose S. // Polym. Chem. 2019. V. 10. P. 4370.
King X., Mi H.-Y., Turng L.-S. // J. Mech. Beh. Biomed. Mater. 2016. V. 60. P. 94.
Wang L. // Rev. Adv. Mater. Sci. 2013. V. 34. P. 123.
Chena S., Hua J.Y., C.-W., Chana L., Zhuo H. // Polym. Adv. Technol. 2010. V. 21. P. 377.
Lendlein A., Angew S.K. // Chem. Int. Ed. 2002. V. 41. P. 2034.
Lendlein A., Langer R. // Science. 2002. V. 296. № 5573. P. 1673.
Lendlein A., Jiang H., Jünger O., Langer R. // Nature. 2005. V. 434. I. 7035. P. 879.
Langer R.S., Lendlein. A. Pat. 6.388.043 USA. 2005.
Langer R.S., Lendlein A., Schmidt A., Grablowitz H. Pat. 6.160.084 USA. 2000.
Zhao W., Liu L., Zhang F., Leng J., Liu Y. // Mater. Sci. Eng. C. 2019. V. 97. P. 864.
Huang W.M. // Open Med. Dev. J. 2010. V. 2. P. 11.
Liu J.A.-C., Gillen J.H., Mishra S.R., Evans B.A., Tracy J.B. // Sci. Adv. 2019. V. 5. I. 8. P. eaaw2897.
Sáenz-Pérez M., Bashir T., Laza J.M., García-Barrasa J., Vilas J.L., Skrifvars M., León L.M. // Textile Res. J. 2018. V. 89. № 6. P. 1027.
Белошенко В.А // Успехи химии. 2005. V. 73. № 3. С. 285.
Liu T., Zhou T., Yao Y., Zhang F., Liu L., Liu Y., Leng J. // Composites. A. 2017. V. 100. P. 20.
Pilate F., Toncheva A., Dubois P., Raquez J.-M. // Eur. Polym. J. 2016. V. 80. P. 268.
Zhao W., Liu L., Zhang F., Leng J., Liu Y. // Mater. Sci. Eng. C. 2019. V. 97. P. 864.
Marzec M., Kucińska-Lipka J., Kalaszczyńska I., Janik H. // Mater. Sci. Eng. C. 2017. V. 80. P. 736.
Buffington. S.L., Paul J.E., Ali M.M., Macios M.M., Mather P.T., Henderson J.H. // Acta Biomater. 2019. V. 84. № 15. P. 88.
Fu Y.Q., Huang W.M., Luo J.K., Lu H. // Materials Today: Proceedings. 2018. Pt 1. V. 5. № 10. P. 21427.
Hu J.L., Chen S.J. // J. Mater. Chem. 2010. V. 20. P. 3346.
Kim B.K., Lee S.Y., Xu M. // Polymer. 1996. V. 37. P. 5781.
Ping P., Wang W., Chen X., Jing X. // J. Polym. Sci. 2007. V. 45. P. 557.
Li F., Hou J., Zhu W., Zhang X., Xu M., Luo X., Ma D., Kim B.K. // J. Appl. Polym. Sci. 1996. V. 62. P. 631.
Winey K., Thomas E., Fetters L. // Macromolecules. 1996. V. 24. P. 6182.
Shull J.R., Winey K., Thomas E., Kramer E. // Macromolecules. 1996. V. 24. P. 2748.
Nozaki S., Masuda S., Kamitani K., Kojio K., Takahara A., Kuwamura G., Hasegawa D., Moorthi K., Mita K., Yamasaki S. // Macromolecules. 2017. V. 50. P. 1008.
Ping P., Wang W., Chen X., Jing X. // J. Polym. Sci. 2007. V. 45. P. 557.
Tobushi H., Hayashi S., Kojima S. // JSME Int. J. 1992. V. 35. P. 296.
Hayashi S. // Int. Prog. Urethane. 1993. V. 6. P. 90.
Li J., Viveros J.A., Wrue M.H., Anthamatten M. // Adv. Mater. 2007. V. 19. P. 2851.
Hong S.J., Yu W.-R., Youk J.H. // Smart Mater. Struct. 2010. V. 19. P. 035022.
Wagermaier W., Kratz K., Heuchel M., Lendlein A. // Adv. Polym. Sci. 2010. V. 226. P. 97.
Takahashi T., Hayashi N., Hayashi S. // J. Appl. Polym. Sci. 1996. V. 60. P. 1061.
Caeter P., Goethals E.J., Gancheva V., Velichkova R. // Polym. Bull. 1997. V. 39. P. 589.
Kim B.K., Shin Y.J., Cho S.M., Jeong H.M. // J. Polym. Sci., Polym. Phys. 2000. V. 38. P. 2652.
Kayalvizhi M., Vakees E., Suresh J., Arun A. // Arab. J. Chem. 2015. V. 12. № 8. P. 2484.
Fernández-d’Arlas B., Rueda L., Fernández R., Khan U., Coleman J.N., Mondragon I., Eceiza A. // J. Soft Mater. 2010. V. 9. № 1. P. 79.
Kim B.K., Lee S.Y., Xu M. // Polymer. 1996. V. 37. № 26. P. 5781.
Li F., Hou J., Zhu W., Zhang X., Xu M., Luo X., Ma D., Kim B.K. // J. Appl. Polym. Sci. 1996. V. 62. № 4. P. 631.
Gupta A., Kim B.S. // Nanomater. 2019. V. 9. № 2. P. 225.
Ahmad M., Xu B., Purnawali H., Fu Y., Huang W., Miraftab M., Luo J. // Appl. Sci. 2012. V. 2. P. 535.
Gross R.A., Kalra B. // Science. 2002. V. 297. P. 803.
Chien Y.C., Chuang W.T., Jeng U.S., Hsu S.H. // ACS Appl. Mater. Interfaces. 2017. V. 15. № 6. P. 5419.
Both M., Emmerling F., Pretsch T. // Macromol. Chem. Phys. 2013. V. 214. P. 2683.
Huang Y., Chang R., Han L., Shan G., Bao Y., Pan P. // ACS Sustainable Chem.2016. V. 41. P. 121.
Chen. H.M. // Chinese J. Polym. Sci. 2018. V. 36. P. 905.
Li F., Zhang X., Hou J., Xu M., Luo X., Ma D., Kim B.K. // J. Appl. Polym. Sci. 1997. V. 64. № 8. P. 1511.
Kim B.K., Lee S.Y., Xu M. // Polymer. 1996. V. 37. № 26. P. 5781.
Ping P., Wang W.S., Chen X.S., Jing X.B. // Biomacromolecules. 2005. V. 6. № 2. P. 587.
Kalita H. Shape Memory Polymers: Theory and Application. Berlin; Boston: Walter de Gruyter GmbH, 2018.
Zhu L., Cheng S.Z.D., Calhoun B.H., Ge Q., Quirk R.P., Thomas E.L., Hsiao B.S., Yeh F., Lotz B. // J. Am. Chem. Soc. 2000. V. 122. P. 5957.
Zhu L., Mimnaugh B.R., Ge Q., Quirk R.P., Cheng S.Z., Thomas E.L., Lotz B., Hsiao B.S., Yeh F., Liu L. // Polymer. 2001. V. 42. P. 9121.
Sun Y.S., Chung T.M., Li Y.J., Ho R.M., Ko B.T., Jeng U.S., Lotz B. // Macromolecules. 2006. V. 39. P. 5782.
Chen H.-L., Wu C.-J., Lin T.-L., Lin J.S. // Macromolecules. 2001. V. 34. P. 6936.
Zhou Y., Ahn S.K., Lakhman R.K., Gopinadhan M., Osuji C.O., Kasi R.M. // Macromolecules. 2011. V. 44. P. 3924.
Nandan B., Hsu J., Chen H. // J. Macromol. Sci. C. 2006. V. 46. P. 143.
Yu-Su S.Y., Sheiko S.S., Lee H.L., Jakubowski W., Nese A., Matyjaszewski K., Anokhin D., Ivanov D. // Macromolecules. 2009. V. 42. P. 9008.
Xu J.T., Fairclough J.P., Mai S.M., Ryan A.J., Chaibundit C. // Macromolecules. 2002. V. 35. P. 6937.
Loo Y.L., Register R., Ryan A.J. // Macromolecules. 2002. V. 35. P. 2365.
Li S., Zhang J., Chen J., Yao M., Liu X., Jiang Z. // Appl. Polym. 2019. V. 59. № 2. P. E310.
Anokhin D.V., Gorbunova M.A., Estrin Ya.I., Komratova V.V., Badamshina E.R. // Phys. Chem. Chem. Phys. 2016. V. 18. № 46. P. 31769.
Prisacariu C., Scortanu E., Coseri S., Agapie B. // Ind. Eng. Chem. Res. 2013. V. 52. № 6. P. 2316.
Chen C.P., Dai S.A., Chang H.L., Su W.C., Wu T.M., Jeng R.J. // Polymer. 2005. V. 46. № 25. P. 11849.
Liu W.-K., Zhao Y., Wang R., Luo F., Li J.-S., Li J.-H., Tan H. // Chinese J. Polym. Sci. 2018. V. 36. P. 514.
Xiaoxia J., Yiwen H., Qilong Z. // J. Polym. Eng. 2016. V. 37. № 2. P. 169.
Sáenz-Pérez M., Laza J.M., García-Barrasa J., Vilas J.L., León L.M. // Polym. Eng. Sci. 2018. V. 58. № 2. P. 238.
Jiang L., Wu J., Nedolisa C., Saiani A., Assender H.E. // Macromolecules. 2015. V. 48. I. 15. P. 5358.
Chen S.J., Hu J.L., Zhuo H.T., Chen S.J. // J. Mater. Sci. 2011. V. 46. P. 5294.
Kumagai S., Motokucho S., Yabuki R., Anzai A., Kameda T., Watanabe A., Nakatani H., Yoshioka H. // Anal. Appl. Pyrolysis. 2017. V. 126. P. 337.
D'Arlas B.F., Rueda L., Caba K.D.L., Mondragon I., Eceiza A. // Polym. Eng. Sci. 2008. V. 48. P. 519.
Speckhard T.A., Hwang K.K.S., Cooper S.L., Chang V.S.C., Kennedy J.P. // Polymer. 1985. V. 26. P. 70.
Li W.J., Lin Y.L., Zheng Y.G. // Chem. Ind. Eng. Prog. 2011. V. 30. P. 1542.
Jiang L., Ren Z., Zhao W., Liu W., Liu H., Zhu C. // R. Soc. Open Sci. 2018. V. 5. P. 180536.
Gorbunova M.A., Shukhardin D.M., Lesnichaya V.A., Badamshina E.R., Anokhin D.V. // Key Eng. Mat. 2019. V. 816. P. 187.
Lee B.S., Chun B.C., Chung Y.C., Sul K.I.I., Cho J.W. // Macromolecules. 2001. V. 34. P. 6431.
Shirole A., Perotto C.U., Balog S., Weder C. // ACS Appl. Mater. Interfaces. 2018. V. 10. P. 29.
Fathilah B.A., Raja M. // Polym. Comp. 2010. V. 31. P. 1309.
Zeng R.T., Hu W., Wang M., Zhang S.D., Zeng J.B. // Polym. Test. 2016. V. 50. P. 182.
Yaricia T., Kodal M., Ozkoc G. // Polym. 2018. V. 146. P. 361.
Papageorgiou G.Z., Achilias D.S., Nanaki S., Beslikas T., Bikiaris D. // Thermochim. Acta. 2010. V. 511. P. 129.
Schawe J., Pötschke P., Alig I. // Polym. 2017. V. 116. P. 160.
Vaaben S.R., Aguilar A., Avalos F., Ramos-de Valle L.F. // J. Therm. Anal. Calorim. 2008. V. 93. P. 947.
Li J., Fang Z., Tong L., Gu A., Liu F. // Eur. Polym. J. 2006. V. 42. P. 3230.
Mitchell C.A., Krishnamoorti R. // Polymer. 2005. V. 46. P. 8796.
Zeng H., Gao C., Wang Y., Watts P.C.P., Kong H., Cui X., Yan D. // Polymer. 2006. V. 47. P. 113.
Grady B.P., Pompeo F., Shambaugh R.L., Resasco D.E // J. Phys. Chem. B. 2002. V. 106. P. 5852.
Xu J.Z., Zhong G.J., Hsiao B.S. // Prog. Polym. Sci. 2014. V. 39. P. 555.
Sahoo N.G., Jung Y.C., Yoo H.J., Cho J.W. // Compos. Sci. Technol. 2007. V. 67. P. 1920.
Jana R.N., Cho J.W. // Compos. A. 2010. V. 41. P. 1524.
Jung Y.C. // Carbon. 2010. V. 48. P. 1598.
Jung Y.C., Kim H.H., Kim Y.A., Kim J.H., Chom J.W., Mildred M.E., Dresselhaus S. // Macromolecules. 2010. V. 43. P. 6106.
Shirole A., Nicharat A., Perotto C.U., Weder C. // Macromolecules. 2018. V. 51. P. 1841.
Liu Y., Du H., Liu L., Leng J. // Smart Mater. Struct. 2014. V. 23. P. 023001.
Baer G., Wilson T., Matthews D.L., Maitland D. // J. Appl. Polym. Sci. 2007. V. 103. P. 3882.
Lai S.-M., You P.-Y., Chiu Y.T., Kuo C.W. // J. Polym. Res. 2017. V. 24. P. 161.
Yang X., Wang L., Wang W., Chen H., Yang G., Zhou S. // ACS Appl. Mater. Interfaces. 2014. V. 6. P. 6545.
Mohr R., Kratz K., Weigel T., Lucka-Gabor M., Moneke M., Lendlein A. // Proc. Natl. Acad. Sci. USA. 2006. V. 103. I. 10. P. 3540.
Hearon K., Gall K., Ware T., Maitland D.J., Bearinger J.P., Wilson T.S. // J. Appl. Polym. Sci. 2011. V. 121. P. 144.
Cai Y., Jiang J.-S., Zheng B., Xie M.-R. // J. Appl. Polym. Sci. 2013. V. 127. № 1. P. 49.
Urban M., Strankowski M. // Nanoplat. Mater. 2017. V. 10. № 9. P. 1083.
Gupta N., Gupta S.M., Sharma S.K. // Carbon Lett. 2019. V. 29. P. 419.
Maniecki T., Shtyka O., Mierczynski P., Ciesielski R., Czylkowska A., Leyko J., Mitukiewicz G., Dubkov S., Gromov D. // Fibre Chem. 2018. V. 50. P. 297.
Yang X., Zhang X., Cao B. // Nanomat. 2019. V. 9. I. 3. P. 477.
Kumanek B., Janas D. // J. Mater. Sci. 2019. V. 54. P. 7397.
Barnett C.J., Evans C., McCormack J.E., Gowenlock C.E., Dunstan P., Adams W., White A.O., Barron A.R. // Nano Lett. 2019. V. 19. № 8. P. 4861.
Ma P.-C., Siddiqui N.A., Marom G., Kim J.-K. // Appl. Sci. Manuf. A. 2010. V. 41. № 10. P. 1345.
Kalakonda P., Banne S., Kalakonda P.B. // Nanomat. Nanotech. 2019. V. 9. P. 1.
Rubel R.I., Ali Md.H., Jafor Md.A., Alam Md.M. // AIMS Mater. Sci. 2019. V. 6. № 5. P. 756.
Cho J.W., Kim J.W., Jung Y.C., Goo N.S. // Macromol. Rapid Commun. 2005. V. 26. P. 412.
Liu T., Huang R., Qi X., Dong P., Fu Q. // Polymer. 2017. V. 114. P. 28.
Raja M., Ryu S.H., Shanmugharaj A. // Eur. Polym. J. 2013. V. 49. P. 3492.
Sun Y.-C., Chu M., Huang M., Hegazi O., Naguib H.E. // Macromol. Mater. Eng. 2019. V. 34. № 10. P. 1900196.
Mahdavi M., Ahmad M.B., Haron Md.J., Namvar F., Nadi B., Zaki M., Rahman Ab., Amin J. // Molecules. 2013. V. 18. P. 7533.
Razzaq M.Y., Behla M., Lendlein A. // Nanoscale. 2012. V. 4. P. 6181.
Shu-Ying G. // J. Appl. Polym. Sci. 2018. V. 134. P. 45686.
Maity S., Bochinski J.R., Clarke L.I. // Adv. Funct. Mater. 2012. V. 2. P. 5259.
D’Agostino A., Taglietti A., Desando R., Bini M., Patrini M., Dacarro G., Cucca L., Pallavicini P., Grisoli P. // Nanomat. 2017. V. 7. P. 7.
Maity S., Downen L.N., Bochinski J.R., Clarke L.I. // Polymer. 2011. V. 52. P. 1674.
Alvarez-Lorenzo C., Bromberg L., Concheiro A. // Photochem. Photobiol. 2009. V. 85. P. 848.
Yung S.S., Chung T.M., Li Y.J., Ho R.M., Ko B.T., Jeng U.S., Lotz B. // Macromolecules. 2006. V. 39. P. 5782.
Deepen P., Sharmistha P., Nima R., Mark W., Pankaj V., Funct J. // Biomater. 2013. V. 4. P. 358.
Alkilany A.M., Thompson L.B., Boulos S.P., Sisco P.N., Murphy C.J. // Adv. Drug Deliv. Rev. 2012. V. 64. I. 2. P. 190.
Long K.N., Scott T.F., Qi H.J. // J. Mech. Phys. Solids. 2009. V. 57. № 7. P. 1103.
Lee S.H., Jun B.-H. // Int. J. Mol. Sci. 2019. V. 20. № 4. P. 865.
Kim M., Lee J.-H., Nam J.-M. // Adv. Sci. 2019. V. 6. № 17. P. 1900471.
Chugh H., Sood D., Chandra I., Tomar V., Dhawan G., Chandra R. // Artif. Cells, Blood Subs. 2018. V. 46. № 1. P. 1210.
Guo J., Rahme K., He Y., Li L.-L., Holmes J.D., O’Driscoll C.M. // Int. J. Nanomedicine. 2017. V. 12. P. 6131.
Advances in Polyurethane Biomaterials / Ed. by S.L. Cooper, J. Guan. Woodhead Publ.: Elsevier, 2016.
Zhu L., Cheng S.Z.D., Calhoun B.H., Ge Q., Quirk R.P., Thomas E.L., Hsiao B.S., Yeh F., Lotz B. // J. Am. Chem. Soc. 2000. V. 122. P. 5957.
Li S., Lin D., Zhou J., Zha L. // J. Phys. Chem. 2016. V. 120. P. 4902.
Zharov V.P., Mercer K.E., Galitovskaya E.N., Smeltzer M.S. // Biophys. J. 2006. V. 90. P. 619.
Zhang H., Xia H., Zhao Y. // J. Mater. Chem. 2011. V. 22. I. 3. P. 845.
Wang J., Yi G. // Nanoscale Res. Lett. 2019. V. 14. P. 292.
Mishra S.R., Tracy J.B. // ACS Appl. Nano Mater. 2018. V. 1. № 7. P. 3063.
Hsu S.-H., Chou C.-W. // Polym. Degrad. Stab. 2004. V. 85. I. 1. P. 675.
Hsu S.-H., Chou C.-W., Tseng S.-M. // Macromol. Mater. Eng. 2004. V. 289. I. 12. P. 1096.
Deka H., Karak N., Kalita R.D., Buragohain A.K. // Polym. Degrad. Stab. 2010. V. 95. P. 1509.
Zhang Y., Zhou S., Chong K.C., Wang S., Liu B. // Mater. Chem. Front. 2019. V. 3. P. 836.
Dickerson E.B., Dreaden E.C., Huang X., Elsayed I.H., Chu H., Pushpanketh S. // Cancer. Lett. 2008. V. 269. № 1. P. 57.
Hribar K.C., Metter R.B., Ifkovits J.L., Troxler T., Burdick J.A. // Small. 2009. V. 5. № 16. P. 1830.
Koerner H., Price G., Pearce N.A., Alexander M., Vaia R.A. // Nat. Mater. 2004. V. 3. I. 2. P. 115.
Park J.H., Kim B.K. // Smart. Mater. Struct. 2014. V. 23. P. 025038.
Fu S., Chen S. // Mater. Proc. 2019. V. 16. № 3. P. 1436.
Bellin I., Kelch S., Langer R., Lendlein A. // Proc. Natl. Acad. Sci. USA. 2006. V. 103. № 48. P. 18043.
Chatani S., Wang C., Podgorski M., Bowman C.N. // Macromolecules. 2014. V. 47. № 15. P. 4949.
Chen S., Yuan H., Chen S., Yang H., Ge Z., Zhuo H., Liu J. // J. Mater. Chem. A. 2014. V. 2. № 26. P. 10169.
Lendlein A., Jiang H., Jünger O., Langer R. // Nature. 2005. V. 434. № 7035. P. 879.
Yu Y., Nakano M., Ikeda T. // Nature. 2003. V. 425. P. 6954.
Sabzi M., Babaahmadi M., Rahnama M. // ACS Appl. Mater. Interfaces. 2017. V. 9. № 28. P. 24061.
Zhou L., Liu Q., Lv X., Gao L., Fang S., Yu H. // J. Mater. Chem. C. 2016. V. 4. № 42. P. 9993.
Wu Y., Huang H., Chen Y. // Mater. Today: Proceedings. 2019. V. 16. Pt. 3. P. 1507.
Fu S., Zhu J., Chen S. // Macromol. Res. 2018. V. 26. P. 1035.
Wu L., Jin C., Sun X. // Biomacromol. 2011. V. 12. № 1. P. 235.
Fua S., Chen S. // Mater. Today: Proceedings 2019. V. 16. Pt 3. P. 1436.
Wu Y., Huang H., Chen Y. // Mater. Today: Proceedings 2019. V. 16. Pt 3. P. 1507.
Wu L., Jin C., Sun X. // Biomacromolecules. 2011. V. 12. P. 235.
Ban J., Mu L., Yang J., Chen S. // J. Mater. Chem. A. 2017. V. 5. P. 14514.
Wang Y. // J. Mater. Chem. B. 2018. V. 6. P. 1668
Qi D., Yao X.L., Deng S., Zhou T.N., Fu Q. // J. Mater. Chem. A. 2014. V. 2. P. 2240.
Sedat X., Feina C., Sadhan C.J. // Polymer. 2008. V. 49. P. 2223.
Wang C., Chen S. // Mater. Today: Proceedings. 2019. V. 16. Pt 3. P. 1475.
Wang Y. // J. Mater. Chem. B. 2018. V. 6. P. 1668.
Alarcon D.H., Pennadam S., Alexander C. // Chem. Soc. Rev. 2005. V. 34. P. 276.
Liu F., Urban M.W. // Prog. Polym. Sci. 2010. V. 35. P. 3.
Hoffmann J., Plotner M., Kuckling D. and Fischer W.J. // Sens. Actuators, Part A. 1999. V. 77. P. 139.
Bajpai A.K., Shukla S.K., Bhanu S., Kankane S. // Prog. Polym. Sci. 2008. V. 33. P. 1088.
Fleigea E., Quadirb M.A., Haag R. // Adv. Drug Delivery Rev. 2012. V. 64. P. 866.
Chen H., Li Y., Liu Y., Gong T., Wang L., Zhou S. // Polym. Chem. 2014. V. 5. P. 5168.
Song Q., Chen H., Zhou S., Zhao K., Wang B., Hu P. // Polym. Chem. 2016. V. 7. № 9. P. 1739.
Song Q., Chen H., Yu W., Ni H., Hu P., Zhao K. // Mater. Today: Proceedings. 2019. V. 16. Pt 3. P. 1405.
Vroman I., Tighzert L. // Materials. 2009. V. 2. P. 307.
Dolynchuk O. // Macromolecules. 2017. V. 50. P. 3841.
Zhou B., Hu Y., Li J., Li B. // J. Bio. Macromol. 2014. V. 64. P.402.
Zhou X., Zhang T., Guo D., Gu N. // Coll. Surf. A. 2014. V. 441. P. 34.
Wang Y., Hong Q., Chen Y., Lian X., Xiong Y. // Coll. Surf. B. 2012. V. 100. P. 77.
Middleton J.C., Tipton A.J. // Biomater. 2000. V. 21. P. 2335.
Baker R.M., Tseng L.-F., Iannolo M.T., Oest M.E., Henderson J.H. // Biomater. 2015. V. 76. P. 388.
Kucińska-Lipka J., Malysheva K., Włodarczyk D., Korchynskyi O., Karczewski J., Kostrzewa M., Gubanska I., Janik H. // Polymers. 2017. V. 9. P. 329.
Aksoy E.A., Taskor G., Gultekinoglu M., Kara F., Ulubayram K. // App. Polym. 2017. V. 135. № 5. P. 45764.
Komez A., Buyuksungur S., Hasirci V., Hasirci N. // J. Bioact. Comp. Polym. 2018. V. 33. № 5. P. 479.
Gopinath A., Subaraj M., Nasar A.S. // Eur. Polym. J. 2018. V. 108. P. 517.
Arévalo F.R., Osorio S.A., Valcárcel N.A. // Polym. Ren. Res. 2018. V. 9. № 1. P. 23.
Tanzi M.C., Verderio P., Lampugnani M.G., Resnati M., Dejana E., Sturani E. // J. Mater. Sci. Mater. Med. 1994. V. 5. P. 393.
Heijkants R.G.J.C., Van Calck R.V., Van Tienen T.G., De Groot J.H., Buma P., Pennings A.J., Veth R.P.H., Schouten A.J. // Biomater. 2005. V. 26. P. 4219.
Barrioni B.R., De Carvalho S.M., Oréfice R.L., De Oliveira A.A.R., Pereira M.D.M. // Mater. Sci. Eng. C. 2015. V. 52. P. 22.
Ping P., Wang W., Zhang P., Chen X., Jing X. // Front. Chem. China. 2007. V. 4. P. 331.
Neuss S., Blomenkamp I., Stainforth R., Boltersdorf D., Jansen M., Butz N., Perez-Bouza A., Knüchel R. // Biomater. 2009. V. 30. P. 1697.
Yakacki C.M., Lyons M.B., Rech B., Gall K., Shandas R. // Biomed. Mater. 2008. V. 3. P. 015010.
Razzaq M.Y., Reinthaler M., Schröder M., Wischke C., Lendlein A. // Encyclop. Biomed. Eng. 2019. P. 394.
Balk M., Behl M., Wischke C., Zotzmann J., Lendlein A. // Adv. Drug Deliv. Rev. 2016. V. 107. № 15. P. 136.
Boia R., Dias P.A.N., Martins J.M., Galindo-Romero C., Aires I.D., Vidal-Sanz M., Agudo-Barriuso M., de Sous H.C., Ambrósio A.F., Brag M.E.M., Santiago A.R. // J. Contr. Rel. 2019. V. 316. P. 331.
Chien Y.C., Chuang W.T., Jeng U.S., Hsu S.H. // ACS Appl. Mater. Interfaces. 2017. V. 15. № 9. P. 5419.
Jung Y.C., Cho J.W. // J. Mater. Sci., Mater. Med. 2010. V. 21. P. 2881.
Aguilar L.E., Nejad A.G., Park C.H., Kim C.S. // Nanomed. Nanotechnol. Biol. Med. 2017. V. 13. P. 527.
Shaikh M., Choudhury N.R., Knott R., Kanwar J.R., Garg S. // Eur. J. Pharm. Biopharm. 2016. V. 101. P. 82.
Rickert D., Lendlein A., Peters I., Moses M., Franke R.-P. // Eur. Archiv. Oto-Rhino-Laryng. Head and Neck. 2006. V. 262. P. 215.
Neuss S., Blomenkamp I., Stainforth R., Boltersdorf D., Jansen M., Butz N., Perez-Bouza A., Knuchel R. // Biomater. 2009. V. 30. P. 1697.
Du J., Zhu T., Yu H., Zhu J., Sun C., Wang J., Chen S., Wang J., Gu X. // Appl. Surf. Sci. 2018. V. 447. I. 31. P. 269.
Cheng J., Jun Y., Qin J., Lee S.-H. // Biomater. 2017. V. 114. P.121.
Trinca R.B., Abraham G.A., Felisberti M.I. // Mater. Sci. Eng. C. 2015. V. 56. P. 511.
Zanini S., Polissi A., Maccagni E.A., Dell’Orto E.C., Liberatore C., Riccardi C. // J. Colloid. Interface. Sci. 2015. V. 451. P. 78.
Singha P., Locklin J., Handa H. // Acta Biomater. 2017. V. 50. P. 20.
Gultekinoglu M., Kurum B., Karahan S., Kart D., Sagiroglu M., Ertas N., Ozen A.H., Ulbubayram K. // Mater. Sci. Eng. C. 2017. V. 71. P. 1166.
Gultekinoglu M., Kurum B., Karahan S., Kart D., Sagiroglu M., Ozen H., Ulubayram K. // Tiss. Eng. A. 2018. V. 21. P. S19.
Chao C.Y., Mani M.P., Jaganatha S.K. // PLoS One. 2018. V. 13. № 10. P. e0205699.
Unnithan A.R., Sasikala A.R.K., Murugesan P., Gurusamy M., Wu D., Park C.H., Kim C.S. // Int. J. Biol. Macromol. 2015. V. 77. P. 1.
Nowatzki P.J., Koepsel R.R., Stoodley P., Min K., Harper A., Murata H., Donfack J., Hortelano E.R., Ehrlich G.D., Russell A.J. // Acta Biomater. 2012. V. 8. P. 1869.
Sun X., Cao Z., Porteous N., Sun Y. // Acta Biomater. 2012. V. 8. P. 1498.
Reddy K.M, Feris K., Bell J., Wingett D.G., Hanley C., Punnoose A. // Appl. Phys. Lett. A. 2007. V. 90. № 213902. P. 2139021.
Nanoparticles and their Potential Application as Antimicrobials. In Science Against Microbial Pathogen: Communicating Current Research and Technological Advances / Ed. by A. Méndez-Vilas. Badajoz: Formatex Research Center, 2011. P. 197.
Xu D., Su Y., Zhao L., Meng F., Liu C., Guan Y., Zhang J., Luo J. // J. Biomed. Mater. Res. A. 2017. V. 105. № 2. P. 531.
Król B., Pielichowska K., Sochacka-Piętal M., Król P. // Polym. Adv. Technol. 2018. V. 29. № 3. P. 1056.
Pant B., Park M. and Park S.-J. // Polymers. 2019. V. 11. P. 1185.
Seyed Dorraji M.S., Rasoulifard M.H., Shajeri M., Ashjari H.R., Azizi M., Rastgouy-Houjaghan M. // Polym. Bull. 2018. V. 75. P. 1519.
Savelyev Y., Gonchar A., Movchan B., Gornostay A., Vozianov S., Rudenko A., Rozhnova R., Travinskay T. // Mater. Today: Proceedings. 2017. V. 4. P. 87.
Singh A.K., Mehra D.S., Niyogi U.K., Sabharwal S., Singh G. // Radiat. Phys. Chem. 2014. V. 103. P. 75.
Vermette P., Griesser J., Laroche H., Guidoin R. Biomedical Applications of Polyurethanes (Tissue Engineering Intelligence Unit). Georgetown, Texas: Landes Biosci., Eurekah. com., 2001.
Triller C., Huljev D., Smrke D.M. // Acta Medica Croatica. 2012. V. 66. P. 65.
Yucedag F., Atalay-Oral C., Erkal S., Guner F.S. // J. Appl. Polm. Sci. 2010. V. 115. P. 1347.
Yary A., Yegneh H., Bakhshi H. // J. Mater. Sci., Mater. Med. 2012. V. 23. № 9. P. 2187.
Rogulska M., Podkoscielny W., Kultis A., Pikus S., Pozdzik E. // Eur. Polym. J. 2006. V. 42. P. 1786.
Ozkaynak M.U., Atalay-Oral C., Tantekim-Ersolmaz S.B., Guner S. // Macromol. Symp. 2005. V. 228. P. 177.
Mosmann T., Immunolog J. // Meth. 1983. V. 65. P. 55.
Morgado P.I., Aguiar-Ricardo A., Correia I.J. // J. Membr. Sci. 2015. V. 490. P. 139.
da Silva C.V., Pereira V.J., Rosa P.T.V., Cabral-Albuquerque E.C.M., Vieira de Melo S.A.B., Costa G.M.N., Dias A.M.A., de Sousa H.C., Bragac M. E.M. // J. Supercrit. Fluids. 2016. V. 115. P. 1.
Zhu S., Hu J. // Mater. Today: Proceedings. Pt 3. 2019. V. 16. P. 1456.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)