

Высокомолекулярные соединения (серия Б), 2020, T. 62, № 5, стр. 348-355
РАЗЛИЧНАЯ РЕАКЦИОННАЯ СПОСОБНОСТЬ ЦИКЛОКАРБОНАТСОДЕРЖАЩИХ ЦЕПЕЙ ТРИГЛИЦЕРИДОВ РАСТИТЕЛЬНЫХ МАСЕЛ КАК ПРИЧИНА АНОМАЛЬНОЙ КИНЕТИКИ УРЕТАНООБРАЗОВАНИЯ С ИХ УЧАСТИЕМ
М. В. Забалов a, *, М. А. Левина a, Р. П. Тигер a
a Федеральный исследовательский центр химической физики им. Н.Н. Семенова
Российской академии наук (ФИЦ ХФ РАН)
119991 Москва, ул. Косыгина, 4, Россия
* E-mail: zabalov@chph.ras.ru
Поступила в редакцию 31.03.2020
После доработки 13.04.2020
Принята к публикации 12.05.2020
Аннотация
Изучена сравнительная реакционная способность олеиновых, линолевых и линоленовых фрагментов цепей триглицеридов растительных масел в процессах их превращения в соответствующие гидроксиуретаны. По данным квантово-химических расчетов методом функционала плотности наибольшую реакционную способность демонстрируют линоленовые фрагменты цепей триглицеридов. Активационные барьеры реакций с участием второй циклокарбонатной группы линолевого фрагмента, второй и третьей групп линоленового фрагмента ниже, чем барьеры превращения первых циклокарбонатных групп соответствующих моделей, что объясняется каталитическим содействием групп ОН гидроксиуретанов, образующихся в первой стадии аминолиза, акту переноса протона.
ВВЕДЕНИЕ
Зеленая химия полиуретанов, основанная на реакциях первичных аминов с циклокарбонатами, предполагает не только исключение высокотоксичных изоцианатов из процесса синтеза полимеров, но и возможность использования возобновляемого растительного сырья [1–9]. В этом отношении определенные перспективы имеют циклокарбонилированные производные растительных масел, которые получают путем окисления последних до эпоксидсодержащих триглицеридов с последующей каталитической карбонизацией под действием СО2. Такого типа олигомеры из соевого и подсолнечного масел подробно нами изучены методом масс-спектрометрии с целью установления их молекулярного состава и распределения по функциональным группам в цепях [10, 11]. Они представляют собой триглицериды циклокарбонилированных производных некоторых ненасыщенных (линолевой − L, олеиновой − О, линоленовой − Ln) кислот и насыщенных (стеариновой − S, пальмитиновой − P) различного состава. Так, в продуктах из соевого масла около 25 таких триглицеридов с функциональностью по циклокарбонатным группам от 0 до 6: PSS (0), SSS (0), PPL (2), POO (2), PLS (2), PLO (3), OOO (3), OLS (3), SLnS (3), PLL (4), PLnO (4), OLO (4), LLS (4), OLnS (4), LLO (5), OLnO (5), PLnL (5), LnLnO (5), LLL (6), LLnL (5), LLnO (6). В продуктах из подсолнечного масла – 16 типов триглицеридов, и главным отличием олигомеров на основе соевого и подсолнечного масел является отсутствие в последних Ln-фрагментов.
При изучении кинетики аминолиза циклокарбонатсодержащих олигомеров из растительных масел в избытке амина в растворах были обнаружены отклонения кинетических кривых от закона первого порядка [11–15]. Такие отклонения не наблюдались при изучении кинетики уретанообразования с участием простых модельных соединений – этиленкарбоната и его замещенных, а также олигомеров нефтяного происхождения [12–14]. Скорее всего, аномальная кинетика аминолиза триглицеридов может быть связана с особенностями их строения и агрегации в растворе. Недавно А. Cornille с соавторами [16] объясняли неполную конверсию в реакции некоторых 5- и 6-членных циклокарбонатов с олигомерным диамином замедлением диффузии реагентов за счет накопления в растворе гидроксильных групп гидроксиуретана, образующих водородные связи с циклокарбонатными и уретановыми группами, а также с молекулами амина. Это действительно может способствовать замедлению реакции, особенно в массе или при участии в реакции диаминов, однако в нашем случае речь идет об обычной органической реакции в разбавленном растворе, и ее поведение не должно отличаться от модельной реакции аминолиза этиленкарбоната. Более того, как следует из работ [14, 17, 18], образование гидроксильных групп должно наоборот способствовать ускорению реакции за счет сольватации ими реакционного центра и экранированию избыточной электронной плотности, возникающей в переходном состоянии (TS), а то и за счет встраивания гидроксильной группы в циклы переноса протона в TS (эффект каталитического содействия).
В настоящей работе дается интерпретация аномальной кинетики уретанообразования в реакции с участием триглицеридов растительных масел, основанная на квантово-химических данных о разной реакционной способности циклокарбонатсодержащих цепей природного олигомера.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ И МЕТОДИКА КВАНТОВО-ХИМИЧЕСКИХ РАСЧЕТОВ
В дополнение к опубликованным ранее данным [11, 13, 15] в настоящей работе изучена кинетика уретанообразования при взаимодействии циклокарбонилированных триглицеридов соевого и подсолнечного масел с н-бутиламином в ДМСО при 55°С. В избытке амина по сравнению с концентрацией циклокарбонатных групп реакция протекала в режиме псевдопервого порядка. Кинетику изучали методом ИК-фурье-спектроскопии по изменению во времени оптической плотности на частоте колебаний карбонильных групп циклокарбоната по методике, подробно описанной ранее в работах [13, 14, 17]. Реакция протекала непосредственно в термостатируемой кювете спектрометра, и закон Ламберта–Бэра соблюдался во всем интервале изменения оптических плотностей раствора.
Расчет поверхностей потенциальной энергии исследуемых реакций проводили в рамках теории функционала плотности с использованием неэмпирически обобщенного градиентного приближения и функционала PBE [19, 20] в базисе TZ2P по программе PRIRODA [21, 22]. Моделями служили реакции метиламина с упрощенными молекулами замещенных циклокарбонатов, имитирующими структуру олеинового, линолевого и линоленового фрагментов цепей производных триглицеридов растительных масел (см. ниже). Для всех исходных реагентов, интермедиатов и переходных состояний проводили оптимизацию геометрии. Характер найденных стационарных точек (минимум или седловая точка на поверхности потенциальной энергии) определяли вычислением собственных значений матрицы вторых производных энергии по координатам ядер. Принадлежность переходных состояний данному превращению устанавливали с помощью расчетов координаты реакции (IRC). Энергию активации рассчитывали как разность электронных энергий переходных состояний (TS) и изомера предреакционного комплекса (RC) с минимальной энергией, найденного на данном типе поверхностей потенциальной энергии.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
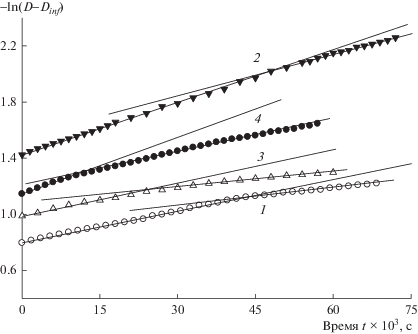
На рис. 1 приведены типичные анаморфозы кинетических кривых н-бутиламинолиза циклокарбонилированных производных растительных масел в координатах уравнения первого порядка. В исследуемых временных интервалах, как видно на рис. 1, наблюдаются отклонения от линейного характера анаморфоз, по начальным участкам которых в работах [13, 15] мы рассчитывали наблюдаемые константы скорости kobs уретанообразо-вания. Интервал степеней превращения α, при которых происходят переломы анаморфоз кинетических кривых, разный для производных соевого и подсолнечного масел и зависит от концентрации амина. В случае подсолнечного масла (кривые 1, 2) α = 40–50%, в случае соевого (кривые 3, 4) α = 15–25%.
Рис. 1.
Типичные кинетические анаморфозы реакции аминолиза олигомеров из подсолнечного (1, 2) и соевого (3, 4) масел в полулогарифмических координатах. Концентрации амина в растворе [BuNH2] = 0.56 (1), 0.7 (2), 0.5 (3), 0.65 моль/л (4), циклокарбонатных групп (1.5−5.0) × 10–3 моль/л. Анаморфоза (3) из статьи [13].
Наличие в исходных олигомерах циклокарбонатсодержащих цепей разного типа может оказывать влияние на наблюдаемые кинетические закономерности изучаемых реакций. Ранее было показано [13, 15, 23, 24], что заместители в этиленкарбонате, обладающие индуктивным −I эффектом, активируют карбонильную группу циклокарбоната за счет оттягивания электронной плотности и увеличения положительного заряда на карбонильном атоме углерода, способствуя тем самым раскрытию цикла при взаимодействии с амином. Согласно нашим квантово-химическим расчетам, (2-оксо-1,3-диоксолан-4-ил)метильная группа (модель второго цикла в линолевом фрагменте) обладает слабым –I эффектом, и наличие такого заместителя при циклокарбонате способствует ускорению реакции аминолиза по сравнению с алкилзамещенными циклокарбонатными группами [13]. В настоящей работе вопросы реакционной способности различных типов циклокарбонатных групп, встречающихся в циклокарбонилированных растительных маслах, рассмотрены более детально и на количественном уровне. При этом представлялось важным правильно выбрать модельные системы для расчетов, максимально полно отражающие строение циклокарбонатных группировок в углеводородных цепях триглицеридов. Для моделирования олеинового фрагмента триглицерида растительного масла использовали 3,4-диметил-замещенный циклокарбонат (a), линолевого фрагмента – 3-(2-оксо-1,3-диоксолан-4-ил)метил-4-метил-замещенный циклокарбонат (b), линоленового фрагмента – b и 3,4-ди((2-оксо-1,3-диоксолан-4-ил)метил)-замещенный циклокарбонат (c).
Основные типы циклокарбонилированных кислотных остатков растительных масел и соответствующие модельные соединения a, b, c, используемые для теоретических расчетов реакций аминолиза, приведены ниже.

В наших кинетических исследованиях [12, 13, 17], выполненных на модельных объектах, было показано, что аминолиз циклокарбонатов в растворе протекает параллельно по двум каналам с участием одной и двух молекул амина (или его димера). Во всех случаях даже при низких концентрациях вклад канала с участием двух молекул амина в суммарную скорость некаталитической реакции является основным. По этой причине в настоящей работе при проведении квантово-химических расчетов использовали реакции модельных циклокарбонатов с двумя молекулами метиламина.
Потенциальные кривые превращения модельных циклокарбонатов a, b и c в гидроксиуретаны в реакциях с двумя молекулами амина приведены на рис. 2, а наиболее характерные структуры предреакционных комплексов RC и переходных состояний TS – на рис. 3. Поскольку возможно образование различных изомеров и конформеров, энергии на рис. 2 представлены с интервалом значений. Сравнение активационных параметров ведется по минимальным значениям энергий, соответствующим наиболее выгодным структурам TS.
Рис. 2.
Поверхности потенциальной энергии превращения циклокарбонатов а–с в комплексы из продуктов реакции (PC), представляющие собой сольватированные молекулой амина гидроксиуретаны. Звездочкой отмечены данные работы [13].
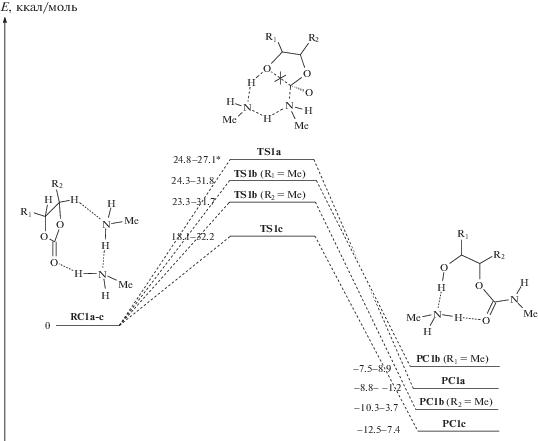
Рис. 3.
Наиболее характерные структуры RC и TS, найденные на поверхностях потенциальной энергии реакций аминолиза соединений b и c. В скобках указана энергия активации в ккал/моль.

С усложнением типа модельного соединения растет и число изомеров/конформеров TS. Так, для TSa было найдено всего 4 изомера/конформера, для соединения TSb – уже 32, а для TSс – 426! При этом уменьшить число изомеров для соединений a–c удалось за счет учета Z-стереоизомерии исходных природных соединений, т.е. растительных масел, и за счет того, что эпоксидирование и циклокарбонилирование не меняет эту конфигурацию заместителей.
В качестве уровня отсчета для энергий активации выбирался изомер RC с минимальной энергией. Разброс энергий изомеров RC составил 0.5 (RCa), 8.7 (RCb), 11.6 ккал/моль (RCc), что довольно существенно, и могло бы внести большую погрешность в расчеты, если бы отсчет энергии активации каждого TS велся от соответствующего ему по координате реакции RC, как это обычно принято. Мы ведем отсчет энергий активации на каждой поверхности потенциальной энергии от энергии одного комплекса RC, соответствующего структуре с минимальной энергией, поэтому такой ошибки не возникает (см. работы [18, 25]). Во всех случаях изомеры RC с минимальной энергией представляют собой одинаковые по структуре соединения с водородными связями между карбонильным атомом O и протоном аминогруппы первой молекулы амина и атомом N первой молекулы амина с протоном аминогруппы второй молекулы амина (рис. 2). Так как строение RC одинаково, сравнение энергий активации, рассчитанных на разных поверхностях потенциальной энергии, корректно [18, 25].
Как видно на рис. 2, минимальные активационные барьеры превращения циклокарбонатов а и b (R1 = Me, R2 = Me) близки между собой. Они расположились на диаграмме в интервале 1.5 ккал/моль в соответствии с индуктивным эффектом заместителя. Наибольший барьер превращения имеет диметилзамещенный циклокарбонат a. Для соединения b возможно два пути раскрытия цикла, приводящие к разным продуктам. При раскрытии цикла по соседству с метильным заместителем (R1 = Me) энергия активации выше и приближается к значению для циклокарбоната a. Иными словами, влияние (2-оксо-1,3-диоксолан-4-ил)метильного заместителя в данном случае минимально. Если же связь C–O разрывается около (2-оксо-1,3-диоксолан-4-ил)метильного заместителя (R2 = Me), то энергия активации оказывается ниже на 1 ккал/моль.
На снижение активационного барьера влияет слабый –I эффект (2-оксо-1,3-диоксолан-4-ил)метильной группы, но не только. Дополнительное влияние оказывает самосольватация переходного состояния соседней циклокарбонатной группой, как например, в TS1b (R2 = Me, Ea = = 23.3) на рис. 3. Ранее мы оценили влияние этих эффектов на схожем модельном соединении как ∼1 ккал/моль для эффекта сольватации и ∼2 ккал/моль для индуктивного эффекта [13]. Близкие значения энергий эффектов найдены и на основании анализа структур TS1b.
Наименьшая энергия активации, как следует из рис. 2, наблюдается при аминолизе соединения с, что свидетельствует о наибольшей реакционной способности линоленовых фрагментов цепей циклокарбонилированных триглицеридов. Среди множества изомеров TSc можно выделить несколько, характеризующих его структурные и электронные особенности (рис. 3). Как и для TSb, на стабильность структур TSс оказывает влияние индуктивный эффект двух (2-оксо-1,3-диоксолан-4-ил)метильных заместителей, а также эффект самосольватации соседней циклокарбонатной группы. Однако в данном случае влияние этих двух эффектов сильнее. Так, энергия структуры TS1c с самосольватацией (TS1c с Ea = 18.1 на рис. 3) на 6.9 ккал/моль ниже, чем энергия TS1c с влиянием только индуктивного эффекта (TS1c с Ea = 25.0 на рис. 3). Дополнительное сильное влияние на Ea может оказывать стерическое отталкивание двух (2-оксо-1,3-диоксолан-4-ил)метильных заместителей, приводящее к росту Ea до 32.2 ккал/моль для TS1c (рис. 3).
Таким образом, в соответствии с приведенными выше результатами расчетов основной вклад в превращение на первой стадии вносит реакция с участием центральных циклокарбонатных групп линоленовых фрагментов. В то же время данное обстоятельство не может оказывать сильное влияние на кинетику превращения, поскольку линоленовых фрагментов в соевом масле всего 7%, а в подсолнечном масле их нет вообще. Из результатов об энергиях активации следует, что линолевые и олеиновые фрагменты реагируют с амином с меньшей и примерно одинаковой скоростью. Это означает, что перелом анаморфоз не связан только с первой стадией реакции, а вклад в увеличение скорости на начальном этапе вносит и вторая стадия, при которой во взаимодействие с амином вступает циклокарбонатная группа, соседствующая с карбаматной (уретановой). В работе [15] мы показали, что вторая стадия аминолиза протекает со значительно меньшим активационным барьером из-за возможности сольватации реакционного центра гидроксильной группой. В дополнение к предыдущим исследованиям в настоящей работе изучена еще одна возможность содействия гидроксильной группы протеканию реакции – ее встраивание в цикл переноса протона в переходном состоянии.
Для упрощения модели и корректного сравнения результатов с предыдущими расчетами рассматривались две стадии аминолиза соединения d (R1 = (2-оксо-1,3-диоксолан-4-ил)метил, R2 = H) по концертному механизму с участием на каждой стадии по одной молекуле амина (рис. 4). На этом рисунке приведен только один путь с разрывом связи C–O в цикле по соседству с (2-оксо-1,3-диоксолан-4-ил)метильным заместителем, так как именно данный путь более выгоден из-за влияния –I эффекта заместителя [13].
Рис. 4.
Поверхности потенциальной энергии реакций последовательного раскрытия циклокарбонатных групп под действием молекул метиламина в модельном соединении d. Звездочкой на диаграмме отмечены данные работы [15].
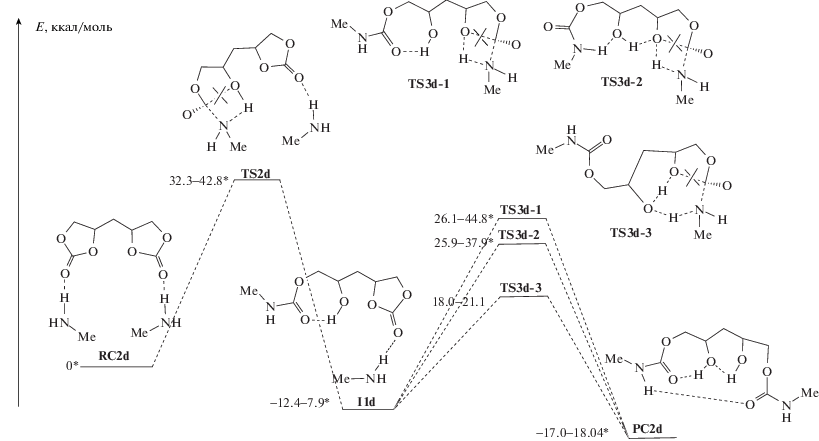
Для бициклокарбоната d исследование двух стадий аминолиза проводили на единой поверхности потенциальной энергии с участием сразу двух молекул амина. В качестве уровня отсчета относительных энергий был выбран изомер RC2d с минимальной энергией, в котором отсутствовали взаимодействия между двумя молекулами амина, и каждая молекула амина образовывала по одной водородной связи с атомами O разных карбонильных групп, т.е. при таком способе расчета две стадии аминолиза протекали независимо друг от друга.
Присоединение первой молекулы амина на первой стадии реакции происходит без каких-либо особенностей через TS2d с образованием интермедиата I1d. Стабильность этого интермедиата на поверхности потенциальной энергии определяется возможностью образования водородных связей и циклов. Наиболее устойчивый изомер I1d приведен на рис. 4. Он включает образование 7-членного цикла с водородной связью между карбонильным атомом O и протоном гидроксильной группы.
Следующая реакция аминолиза – присоединение второй молекулы амина ко второй циклокарбонатной группе, протекает через TS3d. Мы разделили изомеры TS3d на три группы (рис. 4), наиболее полно характеризующие структурные особенности этого переходного состояния (TS3d-1, TS3d-2, TS3d-3).
В TS3d-1 нет никакого содействия гидроксильной группы протеканию реакции. Гидроксильная группа или вовлечена в различные циклы с водородной связью, как показано на рис. 4, или вообще свободна. Относительно высокая стабильность TS3d-1 достигается только за счет очень устойчивого 7-членного цикла с водородной связью, другие типы структур значительно менее устойчивы.
Во вторую группу мы выделили изомеры с сольватацией реакционного центра гидроксильной группой (TS3d-2), т.е. со слабым влиянием последней на реакцию аминолиза второй циклокарбонатной группы. Как следует из рис. 4, изомеры TS3d-2 практически не отличаются по энергии от TS3d-1, если сравнивать изомеры с минимальной энергией в каждой группе.
Третья группа (TS3d-3), впервые рассчитанная в настоящей работе, включает структуры с встраиванием гидроксильной группы в цикл переноса протона, т.е. с сильным влиянием ее на процесс. При этом образуется менее напряженный 6-членный цикл переноса протона и значительно снижается барьер реакции до 18.0 ккал/моль. Никаких стерических или структурных препятствий для образования таких структур TS3d-3 нет (рис. 5).
Важно подчеркнуть, что значительное снижение барьера реакции аминолиза второй циклокарбонатной группы в линоленовых и линолевых фрагментах вносит вклад в общее ускорение реакции в совокупности с несколько большей реакционной способностью указанных фрагментов по сравнению с олеиновыми группировками, что и проявляется экспериментально в виде отклонения анаморфоз кинетических кривых от линейного закона.
Работа выполнена в рамках Госзадания 0082-2019-0003 (№ АААА-А20-120021090129-9).
Список литературы
Guan J., Song Y., Lin Y., Yin X., Zuo M., Zhao Y., Tao X., Zheng Q. // Ind. Eng. Chem. Res. 2011. V. 50. № 11. P. 6517.
Figovsky O., Shapovalov L., Leykin A., Birukova O., Potashnikova R. // PU Magazine. 2013. V. 10. № 4. P. 1.
Nohra B., Candy L., Blanco J.-F., Guerin C., Raoul Y., Mouloungui Z. // Macromolecules. 2013. V. 46. № 10. P. 3771.
Blattmann H., Fleischer M., Bähr M., Mülhaupt R. // Macromol. Rapid Commun. 2014. V. 35. № 14. P. 1238.
Rokicki G., Parzuchowski P.G., Mazurek M. // Polym. Adv. Technol. 2015. V. 26. № 7. P. 707.
Maisonneuve L., Lamarzelle O., Rix E., Grau E., Cramail H. // Chem. Rev. 2015. V. 115. № 22. P. 12407.
Cornille A., Auvergne R., Figovsky O., Boutevin B., Caillol S. // Eur. Polym. J. 2017. V. 87. P. 535.
Błażek K., Datta J. // Critical Revs Environmental Sci.Technol. 2019. V. 49. № 3. P. 173.
Carré C., Ecochard Y., Caillol S., Avérous L. // ChemSusChem. 2019. V. 12. № 15. P. 3410.
Levina M.A., Miloslavskii D.G., Pridatchenko M.L., Gorshkov A.V., Shashkova V.T., Gotlib E.M., Tiger R.P. // Polymer Science B. 2015. V. 57. № 6. P. 584.
Levina M.A., Miloslavskii D.G., Zabalov M.V., Pridatchenko M.L., Gorshkov A.V., Shashkova V.T., Krasheninnikov V.L., Tiger R.P. // Polymer Science B. 2019. V. 61. № 5. P. 540.
Levina M.A., Zabalov M.V., Krasheninnikov V.L., Tiger R.P. // Polymer Science B. 2017. V. 59. № 5. P. 497.
Levina M.A., Zabalov M.V., Krasheninnikov V.L., Tiger R.P. // Polymer Science B. 2018. V. 60. № 5. P. 563.
Zabalov M.V., Levina M.A., Tiger R.P. // Russ. J. Phys. Chem. B. 2019. V. 13. № 5. P. 778.
Levina M.A., Zabalov M.V., Krasheninnikov V.G., Tiger R.P. // React. Kinet. Mech. Cat. 2020. V. 129. № 1. P. 65.
Cornille A., Blain M., Auvergne R., Andrioletti B., Boutevina B., Caillol S. // Polym. Chem. 2017. V. 8. № 3. P. 592.
Levina M.A., Krasheninnikov V.G., Zabalov M.V., Tiger R.P. // Polymer Science B. 2014. V. 56. № 2. P. 139.
Zabalov M.V., Tiger R.P. // Theoret. Chem. Accounts. 2017. V. 136. Article 95.
Perdew J.P., Burke K., Ernzerhoff M. // Phys. Rev. Lett. 1996. V. 77. № 18. P. 3865.
Ernzerhoff M., Scuseria G.E. // J. Chem. Phys. 1999. V. 110. № 11. P. 5029.
Laikov D.N. // Chem. Phys. Lett. 1997. V. 281. P. 151.
Laikov D.N., Ustiniuk Yu.A. // Russ. Chem. Bull. Int. Ed. 2005. V. 54. № 3. P. 820.
Garipov R.M., Sysoev V.A., Mikheev V.V., Zagidullin A.I., Deberdeev R.Y., Irzhak V.I., Berlin A.A. // Dokl. Phys. Chem. 2003. V. 393. № 1. P. 289.
Lamarzelle O., Durand P.-L., Wirotius A.-L., Chollet G., Grau E., Cramail H. // Polym. Chem. 2016. V. 7. № 7. P. 1439.
Zabalov M.V., Tiger R.P. // Russ. Chem. Bull. Int. Ed. 2016. V. 65. № 3. P. 631.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)