

Высокомолекулярные соединения (серия Б), 2020, T. 62, № 6, стр. 465-480
ПОЛУЧЕНИЕ IN SITU ПОЛИМЕРНЫХ НАНОКОМПОЗИТОВ НА ОСНОВЕ ЗОЛЕЙ ПОВЕРХНОСТНО-МОДИФИЦИРОВАННЫХ ДЕТОНАЦИОННЫХ НАНОАЛМАЗОВ МЕТОДАМИ КЛАССИЧЕСКОЙ И КОНТРОЛИРУЕМОЙ РАДИКАЛЬНОЙ ПОЛИМЕРИЗАЦИИ
Е. В. Сивцов a, *, А. В. Калинин b, А. И. Гостев a, А. В. Смирнов c, Л. В. Агибалова b, Ф. А. Шумилов b
a Санкт-Петербургский государственный технологический институт (технический университет)
190013 Санкт-Петербург, Московский пр., 26, Россия
b Научно-исследовательский институт синтетического каучука имени С.В. Лебедева
198035 Санкт-Петербург, ул. Гапсальская, 1, Россия
c Санкт-Петербургский национальный исследовательский университет
информационных технологий, механики и оптики
197101 Санкт-Петербург, Кронверкский пр., 49, Россия
* E-mail: pjeka@yahoo.fr
Поступила в редакцию 07.04.2020
После доработки 28.04.2020
Принята к публикации 15.05.2020
Аннотация
Рассмотрена возможность поверхностной модификации детонационных наноалмазов для придания им диспергируемости в метилметакрилате. Классической радикальной полимеризацией и полимеризацией с обратимой передачей цепи на основе образующихся устойчивых золей детонационных наноалмазов в мономере получены in situ полимерные композиты. Приведены доказательства диспергирования наноалмазов в полимерной матрице до размера частиц порядка единиц нанометров. Изучено влияние детонационных наноалмазов на молекулярно-массовые характеристики и температуру стеклования полиметилметакрилата, полученного в их присутствии.
ВВЕДЕНИЕ
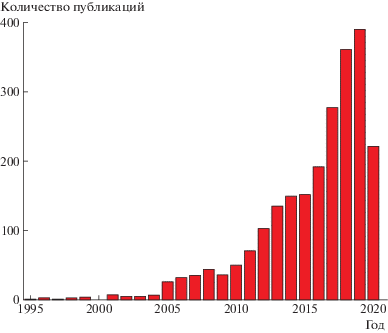
Интерес к применению детонационных наноалмазов (ДНА) в составе полимерных композиций неуклонно растет в течение последних двух десятилетий. На рис. 1 показана статистика по количеству публикаций, отвечающих условиям поиска по ключевым словам “ДНА” и “полимер” в базе данных www.sciencedirect.com. После появления первых же сведений о синтезе ДНА они сразу стали таким же многообещающим наноуглеродным наполнителем для полимеров, как фуллерены и нанотрубки [1–13]. Хорошие предпосылки к совмещению с полимерами различного строения дает присутствие на поверхности ДНА функциональных гидроксильных, карбоксильных, ангидридных, кетонных и лактонных групп, наличие и концентрация которых сильно зависят от процедур очистки и последующей обработки ДНА после детонационного синтеза [14]. Принципиальной проблемой при создании полимерных нанокомпозитов с ДНА является необходимость максимально полного диспергирования ДНА в матрице, желательно до первичных частиц размера порядка 5 нм. Только в этом случае в полной мере может проявиться эффект от их введения, связанный с высокой удельной поверхностью ДНА. Однако отличающая ДНА от других наноуглеродных наполнителей остро выраженная склонность к агрегированию является серьезным препятствием на этом пути. Традиционные методы, основанные на влиянии сдвиговых деформаций и воздействии ультразвука, обычно разбивают агрегаты на фрагменты микронного масштаба, которые в полимерной матрице проявляются просто дефектами структуры [15]. Именно по этой причине реальный эффект от введения ДНА в типичных концентрациях (максимум до нескольких мас. %) обычно более чем скромен, не редко и отрицателен [16].
Рис. 1.
Распределение по годам выпуска количества публикаций, посвященный ДНА в составе полимерных композиций.
Получение устойчивых дисперсий ДНА включает два аспекта: разрушение агломератов и агрегатов первичных частиц (в агломератах частицы связаны друг с другом слабыми взаимодействиями, рассматриваемыми в теории Дерягина–Ландау–Фервея–Овербека (ДЛФО), а в агрегатах – химическим взаимодействием поверхностных функциональных групп) и стабилизация полученной коллоидной системы. Первое достигается, как правило, обработкой ДНА ультразвуком, несмотря на существующее мнение, что ультразвуковой обработки недостаточно для разрушения очень прочных агрегатов размером 100–200 нм [17, 18]. Стандартным решением проблемы придания устойчивости дисперсным системам является их стабилизация с помощью ПАВ или полимеров. В работе [19] частицы ДНА средним размером 53 нм были успешно диспергированы в минеральном масле с помощью различных ПАВ и их синергической смеси. Адсорбция на поверхности ДНА происходила за счет как химических взаимодействий (благодаря группам –NH2, –COOH, –SO3H, –OH, имеющимся в составе ПАВ), так и образования водородных связей. Гидрофобные радикалы ПАВ обеспечивали стерический механизм стабилизации полученных дисперсий. Аналогично диспергируемость ДНА в растворе акрилового полимера была улучшена с помощью олеиламина [20].
Стабилизация гидрозолей ДНА также успешно осуществлена в работе [21] с помощью водорастворимого поли-N-винилпирролидона. Размер частиц составил 30–35 нм. В работе [22] концентрированные (30 мас. %), устойчивые гидрозоли ДНА получены стабилизацией блок-сополимерами поли(аммониевая соль метакриловой кислоты–блок–2-феноксиэтилакрилат). Размер агрегатов при этом уменьшился с 250 до 55 нм.
При получении полимерных композитов, содержащих ДНА, необходим анализ трех практических аспектов: во-первых, способа введения ДНА в полимерную матрицу; во-вторых, возможности модификации поверхности ДНА для лучшей диспергируемости и совмещения с полимером; в-третьих, ожидаемого эффекта от введения ДНА.
Способ введения ДНА в полимерную матрицу признается определяющим. Получение полимерных композитов, содержащих ДНА, возможно через расплав полимера, раствор или синтезом полимера непосредственно в присутствии ДНА.
Например, через расплав полимера в двухшнековом экструдере при 180°С получены композиты биомедицинского назначения на основе полимолочной кислоты, содержащие 0.1–5.0 мас. % ДНА [23]. Неудивительно, что такой способ смешения приводит к тому, что ДНА в полимере оказываются в виде крупных агрегатов, которые не разрушаются под действием сдвиговых нагрузок в экструдере и имеют тем бόльший размер, чем выше содержание ДНА в композите. Хорошее совмещение с полимером обеспечивается в данном случае взаимодействием полярных групп матрицы и наполнителя. Похожим способом предварительно фосфорилированные ДНА вводились в сополимер этилена с винилацетатом литьем под давлением смеси полимера с наполнителем при 165°С [24]. Распределение и размер частиц ДНА в конечном изделии не контролировались. В сополимер этилена с 1-октеном ДНА вводились ультразвуковым диспергированием в толуольном растворе полимера с последующим осаждением ацетоном [25]. Пленки получали из высушенного композита литьем под давлением при 150–160°С.
Примером получения композитов через раствор является введение ДНА с помощью ультразвука в жидкую эпоксидную смолу диспергированием их в растворе смолы в ацетоне с последующим удалением растворителя [26]. Авторы указанной работы не приводят надежных доказательств, что ДНА удалось диспергировать до наноразмерного уровня при их концентрации в матрице 0.3 мас. %. Зато при концентрации 1 мас. % отчетливо наблюдались агрегаты до десятков мкм. В работах [15, 27] ДНА (в частности, в нетипично большом количестве (25 об. %) [15]) вводили в эпоксидную матрицу через раствор в ТГФ под действием ультразвука; затем растворитель отгоняли, а из полученной пасты формировали изделия горячим прессованием, повышая температуру с 80 до 165°С. В работах [28, 29] ДНА добавляли в эпоксидную смолу без разбавления растворителем и подвергали действию ультразвука. Авторы работы [28] не отмечали образование агрегатов в отвержденной матрице. В работе [30] ДНА в количестве 0.2–1.5 мас. % вводили в состав тонких пленок, образованных смесью хитозана и поливинилового спирта, предварительным смешением водных растворов компонентов и под действием ультразвука. Несмотря на то что авторам удалось достичь равномерного распределения ДНА в матрице, они находились в ней в виде крупных агрегатов. Аналогично добавляли ДНА в количестве 0.6 мас. % в чистый поливиниловый спирт для последующего формирования пленок [31]. Редкий пример получения композита непосредственно в ходе синтеза полимера описан в работе [32]. Волокна полианилина осаждали полимеризацией из водного раствора анилина, содержащего ДНА и ПАВ для их лучшей диспергируемости.
Модификация поверхности ДНА способна влиять на их склонность к агрегированию, кардинально улучшать диспергируемость и совместимость с полимерной матрицей. Помимо традиционного отжига в кислородной и инертной атмосфере, обработки смесью сильных минеральных кислот [33, 34] модификация поверхности может осуществляться хлорированием, фторированием, этерификацией ацилхлоридом, силилированием, арилированием, присоединением эпоксидного олигомера на основе бисфенола А, фуллереном, взаимодействием с хлорангидридами органических кислот, путем захвата поверхностью ДНА радикалов, образующихся при распаде диазосоединений, взаимодействием поверхностных карбоксильных групп с аминами и другими способами [6, 35–50]. Интересный способ модификации поверхности предложен для подготовки ДНА к прививке биологически активных соединений [51]. Он состоит из стадий унификации функционального состава поверхности восстановлением всех групп до атомов H и групп –OH, последующего их хлорирования (или фторирования) и прививки к ним этилен- или гексаметилендиамина.
При получении полимерных композитов, содержащих ДНА, всегда подразумевается их диспергирование в органических средах. Было показано, что с увеличением полярности среды улучшается и диспергируемость частиц ДНА [52, 53]. Это позволило сделать вывод об увеличении устойчивости дисперсий ДНА в органических жидкостях при модификации поверхности в сторону ее гидрофобизации. Используя данный принцип, авторы достигли удовлетворительной диспергируемости предварительно модифицированных ДНА в толуоле, причем размер частиц составил порядка 18 нм, что близко к нативному размеру.
Принципы “click chemistry” использованы при модификации ДНА, на поверхности которых предварительно были созданы азидные группы, ацетиленами с заместителями различной природы [54], позволив тем самым получить устойчивые дисперсии и уменьшить размер агрегатов по сравнению с немодифицированными исходными ДНА, содержащими на поверхности аминогруппы. В работе [55] “click” реакцией тиол-содержащего полиэтиленгликоля с винильными группами на поверхности ДНА проведена их функционализация. Судя по всему, “тиол-ен”-взаимодействие может служить универсальным средством прививки на поверхность ДНА тиолсодержащих соединений.
Известны несколько способов успешной модификации поверхности ДНА прививкой полимеров и олигомеров различной природы. Так, в работе [56] описана прививка полиметилметакрилата, поли-N-диметиламиноэтилметакрилата, поли-2-(метакрилоилокси)этил триметиламмоний хлорида, полистирола и полиакриловой кислоты к графитизированной поверхности ДНА радикальной полимеризацией соответствующих мономеров, инициированной распадом пероксида бензоила под действием ультразвука и микроволнового излучения. В результате стало возможным получение устойчивых дисперсий ДНА в разных по природе средах. Используя поверхностные гидроксильные группы, к ДНА был привит адамантановый фрагмент этерификацией с 1-адамантанкарбонил хлоридом [57]. Поверхностный слой использован далее для проведения взаимодействий типа “гость–хозяин” с β-циклодекстрином, содержащим гиперразветвленный полиглицерол. Полученные трехслойные частицы имеют высокий потенциал как средство доставки лекарств. Полимеризацией с раскрытием цикла к поверхности ДНА, предварительно функционализованной с образованием аминогрупп, были привиты цепи полиаминокислот [58]. Результатом стало улучшение диспергируемости ДНА в воде и уменьшение размера агрегатов. Этим же способом были привиты поли(L-молочная кислота) и поли(ε-капролактон) [59] к поверхности ДНА, на которой предварительно созданы гидроксильные группы. Такая прививка улучшила диспергируемость ДНА в органических растворителях, а размер агрегатов уменьшился от микрометрового до 70 нм.
Особый интерес представляет модификация поверхности ДНА прививкой полимеров посредством проведения контролируемой радикальной полимеризации, используя предварительно созданные на поверхности функциональные группы. Так, полимеризация с переносом атома (ATRP – Atom Transfer Radical Polymerization в англ. литературе) была осуществлена в присутствии последовательно обработанных тионилхлоридом и гидроксиэтил-2-бромизобутиратом ДНА. Привиты цепи поли-изо-бутилметакрилата и поли-трет-бутилметакрилата с плотностью прививки пять полимерных цепей на 100 нм2 [60]. Еще один пример функционализации с помощью ATRP–прививка поли-трет-бутилметакрилата к ДНА, на поверхности которого были предварительно созданы группы –PhCH(Br)CH3, являющиеся катализаторами ATRP [61]. Последующий гидролиз трансформировал гидрофобные цепи поли-трет-бутилметакрилата в гидрофильные полиакриловой кислоты. Аналогично взаимодействием гидроксильных групп на поверхности ДНА с 2-бромоизобутирил бромидом были синтезированы поверхностные группы –C(O)–C(CH3)2Br, на которых затем проведена ATRP 2-метакрилоил-оксиэтил фосфорилхолина [62].
Техника контролируемой радикальной полимеризации с обратимой передачей цепи (ОПЦ) была использована для прививки на ДНА флуоресцентных сополимеров 2-метакрилоилоксиэтилфосфорилхолина с диакрилоил флуоресцеином [63]. Для этого на поверхности ДНА были созданы тритиокарбонатные группы. Таким образом ДНА играл роль агента обратимой передачи цепи.
Интерпретация данных по влиянию ДНА на свойства полимерных композитов почти всегда противоречива. Например, признавать ли очень скромный эффект или его отсутствие как принципиально индифферентное действие ДНА, либо относить на счет плохой диспергируемости или неравномерного распределения ДНА в полимерной матрице. Ниже приведены несколько типичных примеров.
Так, введение ДНА в полимолочную кислоту в количестве 0.1–5.0 мас. % не влияет на температуру стеклования полимерной матрицы, но несколько увеличивает степень кристалличности полимера. Прочностные характеристики и термостабильность незначительно улучшаются [23]. Наиболее изучено влияние ДНА на свойства отвержденных эпоксидных смол. Заметное увеличение твердости и прочности отвержденной эпоксидной смолы было достигнуто при введении 0.3 мас. % ДНА [26]. Авторы указанной работы объясняют достигнутый результат химическим взаимодействием эпоксидной смолы с функциональными группами на поверхности ДНА. Важно, что при данной концентрации наблюдался максимум всех физико-механических характеристик, что хорошо коррелирует с качеством диспергирования ДНА: при концентрации выше 0.3 мас. % прослеживается сильное агрегирование, и свойства ухудшаются. Значит, достигаемый эффект напрямую зависит от размера частиц ДНА, которого удается достичь при диспергировании. В работе [28] 0.2 мас. % ДНА в составе отвержденной эпоксидной смолы позволили несколько увеличить твердость и прочностные характеристики композита. Сильный эффект наблюдался от введения в эпоксидную матрицу ДНА в концентрации 25 об. % [15]. При этом модуль Юнга увеличивался на 470%, твердость на 300%, значительно увеличивалась устойчивость к царапанью и термопроводность. Последнее объясняется тем, что при такой концентрации ДНА не приходится говорить о дисперсии, а скорее, о сплошной структуре из частиц ДНА, пронизывающей весь объем полимера. Аналогичное изменение физико-механических свойств отвержденной эпоксидной смолы описали авторы работы [29]; но в этом случае максимальное значение модуля Юнга было при содержании ДНА 30 мас. %, прочности при растяжении – при 1 мас. %. Повышение трибологических и прочностных свойств отвержденной эпоксидной смолы наблюдали при содержании ДНА 0.1%, причем при дальнейшем увеличении концентрации ДНА характеристики либо изменялись незначительно, либо ухудшались [64]. Авторы объясняют это явление резко возрастающей тенденцией к агрегированию при увеличении содержания ДНА сверх указанного значения. Наиболее оригинальный способ модификации свойств эпоксидной смолы ДНА основан на использовании аминированных ДНА в качестве отвердителя эпоксидной смолы в эквивалентном соотношении по функциональным группам [27]. Модуль Юнга отвержденной композиции в данном случае увеличивался на 700%.
Устойчивость к царапанью акриловых покрытий была улучшена с помощью введения 1.5 мас. % ДНА [20, 65]. Добавление 1 мас. % ДНА в тонкие пленки, образованные смесью хитозана с поливиниловым спиртом, позволило в несколько раз повысить их адсорбционную емкость по отношению к ионам Pb(II). Увеличение количества ДНА не привело к усилению эффекта. Содержание 0.6% ДНА в пленках поливинилового спирта несколько улучшило их твердость и прочность [31]. Введение ДНА в количестве до 1.44 об. % в сополимер этилена с 1-октеном влечет повышение степени кристалличности, модуля Юнга и прочности [25]. Прочностные и термомеханические характеристики полиэтилена низкой плотности были улучшены добавлением 1–11 мас. % ДНА с поверхностью, модифицированной алкильными радикалами различной длины [66]. Трибологические свойства пленок из политетрафторэтилена были улучшены добавлением 2 мас. % ДНА [67]. Однако о совмещении ДНА с полимерной матрицей в данном случае речи не идет: ДНА покрывали частицы полимера в водной суспензии, из которых потом формировалась пленка.
Таким образом, понятно, что в использовании таких наполнителей, как ДНА, до сих пор нет готовых рецептов: ни в том, что касается типа модификации поверхности (а она для получения полимерных композитов необходима), ни в количестве вводимых ДНА, где не действует принцип “чем больше, тем лучше”. Приведенные примеры демонстрируют, что увеличение концентрации ДНА выше десятых долей процента создает риск их агрегации. По существу, все работы, посвященные наполнению полимеров ДНА, являются поисковыми, а их практический результат чаще всего разочаровывающим. Известны, конечно, исключения, например в цитируемой выше работе [27], где модифицированные аминогруппами ДНА были использованы как своеобразный наполнитель – отвердитель эпоксидных смол.
В настоящей работе проведена новая, ранее не использовавшаяся функционализация поверхности ДНА для создания на их основе полимерных композитов. Выбор функциональных групп вполне объясним – особые свойства перфторалкильных радикалов и силоксанов общеизвестны. В модификации ими поверхности ДНА ожидалось выявить новые перспективные эффекты, но прежде всего – предотвращение агрегации первичных частиц ДНА и их легкой диспергируемости в модельном мономере. Эффект, достигаемый от введения ДНА в полимерную матрицу, напрямую зависит от степени диспергирования ДНА. Она, в свою очередь, определяется способом введения ДНА в композит и природой их поверхности. Показано, что наименее изучены и использованы возможности изготовления полимерных композитов in-situ непосредственно в процессе синтеза полимера, хотя именно такой способ представляется наиболее перспективным. Поэтому целью работы было изучить возможность поверхностной модификации ДНА таким образом, чтобы они могли образовать устойчивые золи в модельном мономере – метилметакрилате, полимеризацией которого затем получить композиты с различным содержанием ДНА и исследовать влияние ДНА на молекулярно-массовые характеристики образующегося полимера и его температуру стеклования.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В данной работе использовали детонационные наноалмазы марки “ТАН”, предоставленные Специальным конструкторско-технологическим бюро “Технолог” (Россия), суммарное содержание гидроксильных и карбоксильных групп 18 ммоль/г. Метилметакрилат (ММА) дважды перегоняли (Ткип = 101°С) и хранили в холодильнике. Чистоту контролировали по отсутствию сигналов примесей в спектрах ЯМР 1H. ДАК дважды перекристаллизовывали из этанола при 50 (±2)°C, и сушили в вакууме при 20°C (Tпл = 104°C). Дибензилтритиокарбонат (БТК) синтезировали по методике [68] с некоторыми модификациями [69].
Поверхностная модификация ДНА типа I
К ДНА (3 г), которые поместили в 100 мл трехгорлую колбу, оснащенную мешалкой, обратным холодильником с хлоркальциевой трубкой и термометром, добавляли 50 мл тионилхлорида, затем реакционную массу кипятили при постоянном перемешивании в течение 8 ч. Далее содержимое колбы подвергали воздействию ультразвука частотой 44 кГц в течение 5 мин, и кипячение содержимого колбы продолжали 16 ч с обратным холодильником при интенсивном перемешивании. После этого оставшийся тионилхлорид отгоняли. Отдельно готовили раствор аллилата натрия в аллиловом спирте: 0.46 г натрия растворяли в 50 мл безводного аллилового спирта. Его при перемешивании приливали к сухому остатку ДНА, содержимое колбы кипятили с обратным холодильником в течение 8 ч. Полученную суспензию ДНА в аллиловом спирте центрифугировали (скорость вращения 6000 об./мин), отделяли ДНА, промывали этилацетатом и вновь центрифугировали. Затем обработанные таким образом ДНА распределяли путем воздействия ультразвука в 30 мл гептаметилтрисилоксана. К полученной суспензии прибавляли 0.1 мл катализатора Карстеда, при перемешивании нагревали до 110°С в течение 2 ч. После этого избыток гептаметилтрисилоксана отгоняли под вакуумом водоструйного насоса (30–35 мм рт. ст.), ДНА промывали этилацетатом и осаждали из образовавшейся суспензии путем центрифугирования (6000 об./мин).
Поверхностная модификация ДНА типа II
Навеску ДНА (2.5 г) помещали в трехгорлую круглодонную колбу объемом 100 мл, снабженную мешалкой, термометром, обратным холодильником с хлоркальциевой трубкой. В колбу вносили 50 мл триметилхлорсилана, в течение 4 ч осуществляли кипячение под обратным холодильником. Затем содержимое колбы подвергали действию ультразвука частотой 44 кГц, время воздействия 5 мин, после чего избыток триметилхлорсилана отгоняли.
Поверхностная модификация ДНА типа III
Тщательно осушенную навеску ДНА кипятили в среде тионилхлорида в течение 8 ч в 100 мл трехгорлой круглодонной колбе, снабженной мешалкой, обратным холодильником с хлоркальциевой трубкой и термометром. Полученную взвесь ДНА в тионилхлориде подвергали воздействию ультразвука с частотой 44 кГц в течение 5 мин, после чего кипячение под обратным холодильником продолжали. Отдельно готовили образец натриевого алкоголята 2,2',3,3',4,4',5,5',5''-нонафторамилового спирта: навеску металлического натрия (0.23 г) растворяли в 50 мл перегнанного обезвоженного нонафторамилового спирта. Затем тионилхлорид досуха отгоняли из колбы, содержащей навеску ДНА, и туда добавляли раствор нонафторамилата натрия в избытке нонафтор-амилового спирта. Содержимое колбы перемешивали и кипятили с обратным холодильником в течение 4 ч, затем подвергали воздействию ультразвука с частотой 44 КГц в течение 5 мин, после чего кипячение продолжали еще 4 ч. В результате содержимое колбы представляло собой однородную суспензию серо-коричневого цвета, не оседающую в течение 8 ч. Далее нонафторамиловый спирт отгоняли, оставшиеся ДНА заливали 70 см3 этилацетата (для отмывки избыточного алкоголята), центрифугировали при 6000 об./мин, и вновь промывали этилацетатом. Контроль полноты отмывки от избыточного алкоголята осуществляли полоской индикаторной бумаги, смоченной дистиллированной водой. Модифицированные таким образом ДНА сушили при температуре +60°С.
Определение функциональных групп, содержащих подвижный водород, по методу Чугаева–Церевитинова
В условиях инертного газа и полного отсутствия паров воды ДНА обрабатывали раствором этилмагнийиодида в дибутиловом эфире. При этом количественно [70] происходила реакция
Объем выделившегося метана фиксировали по газовой бюретке, а содержание спиртовых и карбоксильных групп принимали эквивалентным количеству метана.
Получение композитов полимеризацией метилметакрилата в присутствии ДНА
Реакционную смесь готовили растворением расчетного количества инициатора ДАК и, если необходимо, агента обратимой передачи цепи БТК в мономере ММА. Приготовленный раствор с расчетным количеством ДНА помещали в стеклянные ампулы объемом 10 см3 и создавали в них инертную атмосферу аргона путем трехкратного повторения цикла: замораживание–вакуумирование–размораживание–заполнение аргоном, затем ампулы отпаивали. Полимеризацию проводили при 70°С в водяном термостате, воздействуя периодически на смесь ультразвуком с частотой 44 кГц до загущения реакционной смеси. Продолжительность полимеризации 8 ч. Композит извлекали из ампул и под вакуумом удаляли остаточный мономер.
Методы исследования
Дифференциальную сканирующую калориметрию проводили на приборе “Shimadzu DSC-60 Plus”. Обработку данных осуществляли с помощью программного обеспечения “Shimadzu Corporation ©ta60 Version 2.21”. Условия анализа: материал тигля – Al, поток газа – отсутствовал, масса образца – 4.5 мг. В качестве эталона использовали навеску Al2O3, помещенную в алюминиевый тигель, массой в ∼2 раза больше массы образца. Температурная программа: нагревание вели со скоростью 30 град/мин от 25 до 250°С. Базовую линию снимали по той же температурной программе в отсутствие образца, корректируя по температуре. Испытания проводили трижды, повторяя после полного охлаждения образца. Данные первого испытания не учитывали.
Исследования методом малоуглового рентгеновского рассеяния(SAXS) осуществляли на дифрактометре “Rigaku SmartLab 3”. Измерения проводили на длине волны λKα = 0.154 c детектором“D/teX Ultra 250”. Все полимерные образцы представляли собой пластины толщиной 0.5 мм.
Гель-проникающую хроматографию осуществляли на приборе фирмы “Waters”, система “Breeze”, снабженным рефрактометрическим детектором, растворитель – тетрагидрофуран, использовали стандартный набор стирогелевых колонок 1HR, 2HR, 4HR, эффективных в области молекулярных масс 1 × 102–5 × 103, 1 × 102–1 × 104, 5 × 103–5 × 105 соответственно; калибровка по стандартным образцам полистирола. Для высокомолекулярных образцов использовали набор колонок 2HR, 4HR и 5HT, последняя из которых эффективна в области молекулярных масс 3 × 104–3 × 106.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Модификация поверхности ДНА
Поверхностная модификация ДНА призвана подавить склонность частиц ДНА к агрегации и обеспечить их эффективное распределение в органических средах, в частности в мономерах, что подтверждают результаты ранних исследований [71–74]. В данной работе поверхностная модификация была осуществлена тремя различными функциональными группами: компактными и объемными кремний органическими фрагментами и перфторалкильным радикалом.
Модификация гептаметилтрисилоксановыми группами (обозначена как тип модификации I, а модифицированные ДНА как ДНА-I) может быть описана следующими реакциями:
взаимодействие с тионилхлоридом
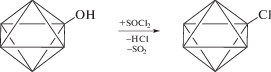
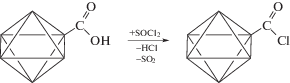
взаимодействие с аллилатом натрия
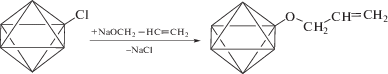
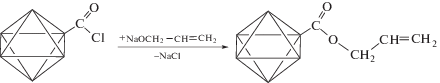
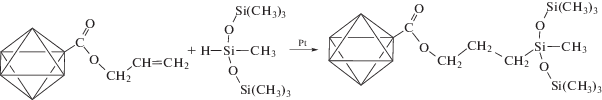
гидросилилирование по аллильным группам
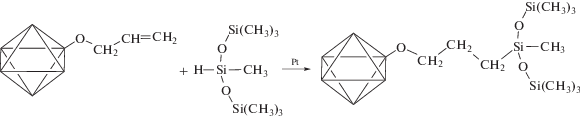
Модификация триметилсилильными группами (обозначена как тип модификации II, а модифицированные ДНА как ДНА-II) может быть описана следующими реакциями:
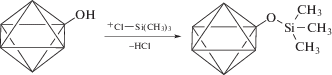
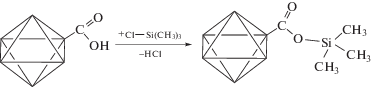
Недостатком этого способа модификации является относительная неустойчивость фрагмента Si–O–C к гидролизу.
Модификация 2,2′,3,3′,4,4′,5,5′,5′′-нонафтор-амилокси группами (обозначена как тип модификации III, а модифицированные ДНА как ДНА-III) может быть описана взаимодействием с тионилхлоридом, как это представлено выше, затем взаимодействием с алкоголятом:
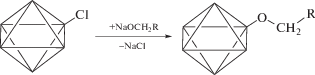
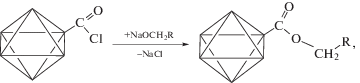
где R = –CF2–CF2–CF2–CF3.
Первой стадией для типов модификации I и III является реакция с тионилхлоридом поверхностных гидроксильных и карбоксильных групп. Это удобный прием повышения реакционной способности функциональных групп для дальнейших их превращений, использованный ранее для модификации карбоксильных групп на поверхности углеродных нанотрубок [75–77] и ДНА [78–81]. Надежным доказательством успешного протекания данной реакции можно полагать уменьшение концентрации функциональных групп, содержащих активный водород. Такими группами в случае ДНА считаются гидроксильные и карбоксильные группы. В данной работе удельное содержание этих групп до и после модификации определялось по методу Чугаева–Церевитинова. Количество групп, содержащих активный водород, снизилось с 18.0 до 1.5 ммоль/г. Это же подтверждают данные ИК-спектроскопии: при обработке тионилхлоридом уменьшается интенсивность широкого сигнала в области 3000–3500 см–1, отвечающего колебаниям гидроксильных групп, а самое главное, появляется интенсивное поглощение при 810 см–1, отсутствующее в спектре исходных ДНА и относящееся к валентным колебаниям ковалентной связи C–Cl.
Прививка на поверхность объемных кремнийсодержащих групп осуществлялась гидросилилированием. Доказано [82], что всякое вещество, в составе которого есть терминальная двойная связь в виде винильной или аллильной группы, независимо от строения другой части соединения, в присутствии катализатора Карстеда реагирует с любой группой Si–H почти количественно. Присоединение проходит строго по правилу Фармера. Протекание реакций при модификации типа I и II подтверждается появлением в ИК-спектрах характерных полос поглощения: группы Si–CH3 – 780 см–1 (сильная) и Si–O–Si 1000–1200 см–1 (очень сильная).
Присоединение алкоголята к хлоридным и ацилхлоридным группам доказано твердотельным спектром ЯМР 19F. Наличие фторсодержащих фрагментов на поверхности ДНА подтверждается сигналом фтора группы CF3 при –82 м.д. и групп СF2 от –123 до –127 м.д. Образование ковалентной связи перфторалкильного радикала с ДНА доказано сравнением соотношения сиг-нал : шум в спектрах ЯМР образца, полученного по описанной выше схеме, и образца исходных ДНА, обработанных нонафторамиловым спиртом, который может адсорбироваться за счет физических взаимодействий на поверхности ДНА. После отмывки обоих образцов отношение сигнал:шум модифицированного образца на два порядка превышало отношение для образца сравнения. Именно столь существенная разница интенсивностей в сигналах 19F перфторбутильного фрагмента позволяет сделать однозначный вывод о формировании поверхностного слоя перфторбутильных радикалов, химически связанных с кристаллом ДНА.
Модификация поверхности объемными силоксановыми группами (тип I) привела к появлению свойства самодиспергируемости таких ДНА в метилметакрилате за счет теплового движения и диффузии, кажется, впервые обнаруженного для таких систем (ДНА – органическая жидкость). ДНА, модифицированные по типу II и III, легко диспергировались в метилметакрилате при непродолжительном (1–3 мин) воздействии ультразвука и сохраняли устойчивость в течение времени, необходимого для проведения полимеризации. Лучшая совместимость ДНА-I с мономером сделала их основным объектом исследования влияния ДНА на свойства полученного композита. Поскольку главной задачей являлось посредством модификации поверхности и проведения полимеризации непосредственно в полученных дисперсиях избежать агрегирования и достичь максимально эффективного распределения ДНА в матрице, то первоначально были получены композиты, содержащие ДНА всех трех типов модификации, и определен размер их частиц в композите.
Диспергирование ДНА в полимерной матрице
Первый вопрос, на который должен быть дан ответ при решении подобных задач создания полимерных нанокомпозитов: каков размер частиц наполнителя, оказавшихся в итоге в полимерной матрице? С этой целью были изучены образцы ПММА, наполненные ДНА всех трех типов поверхностной модификации (табл. 1, образцы 2–5), полученные при концентрации инициатора 5 × × 10–2 моль/л. Методом SAXS, помимо композитных образцов, исследован ПММА, не содержащий ДНА, и порошки модифицированных ДНА. (Для всех типов модификации были получены идентичные данные по размеру частиц ДНА, поэтому здесь в качестве примера приведены результаты только для типа модификации III – образец “порошок ДНА”.)
Таблица 1.
Свойства композитов, содержащих 0.05 мас. % ДНА
| Тип ДНА | Mn × 10–3 | Ð | Tс, °C |
|---|---|---|---|
| [ДАК] = 5 × 10–2 моль/л | |||
| ПММА – образец 1 | 90 | 3.60 | 127 |
| I – образец 2 | 124 | 3.76 | 120 |
| I – образец 3* | 43.7 | 2.70 | 108 |
| II – образец 4 | 94 | 3.05 | 123 |
| III – образец 5 | 103 | 3.87 | 128 |
| [ДАК] = 1 × 10–3 моль/л | |||
| – | 740 | 5.20 | 128 |
| I | 650 | 5.65 | 131 |
| II | 730 | 4.90 | 127 |
| III | 564 | 2.90 | 128 |
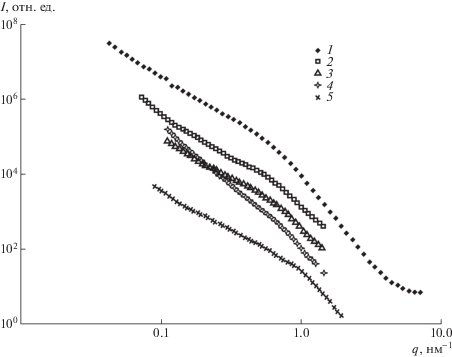
На рис. 2 представлена зависимость интенсивности рассеяния от модуля вектора рассеяния q для перечисленных образцов. Модуль вектора рассеяния определяется формулой q = 4 π sinθ/λ (2$\theta $ – угол рассеяния, λ – длина волны рентгеновских лучей).
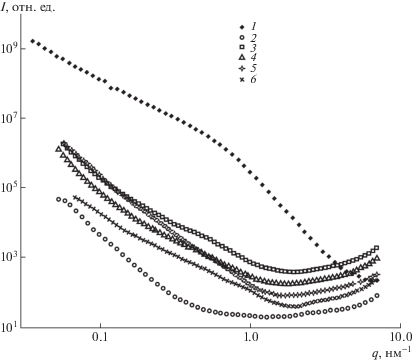
В композитных образцах рассеяние обусловлено в основном скачком плотности на границах наноалмазных частиц. Однако интенсивность рассеяния от наноалмазных частиц достаточно быстро спадает при значениях q > 1 нм–1. Это приводит к тому, что интенсивность рассеяния у композитных образцов при достаточно больших значениях вектора рассеяния определяется исключительно матрицей ПMMA. В частности, в диапазоне q = 1.8…3.7 нм–1 поведение интенсивности от образца чистого ПMMA и от образцов 2–5 на рис. 2 практически совпадает.
Предположим, что отсутствует интерференция между рассеянием от частиц и матрицы. В этом случае интенсивность рассеяния от наноалмазных частиц в смешанных образцах может быть найдена как разность рассеяния от композитного и чистого образца, приведенных к одинаковым значениям интенсивности первичного пучка и толщине образца. Получившиеся после такого вычитания интенсивности показаны на рис. 3. Также здесь для сравнения представлена интенсивность рассеяния от исходного порошка наноалмазных частиц.
Рис. 3.
Интенсивность рассеяния от наноалмазных частиц в образцах порошка детонационных наноалмазов ДНА-III и полимерных композитов, содержащих ДНА-I, ДНА-II и ДНА-III (см. табл. 1): 1 – порошок ДНА, 2–5 – образцы 2–5 соответственно.
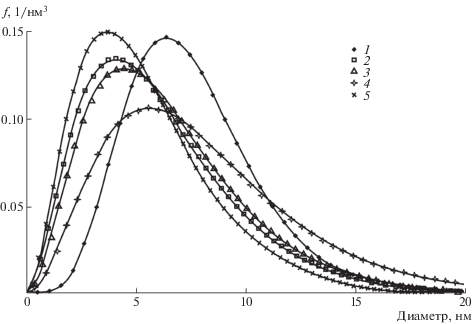
На основе интенсивностей, представленных на рис. 3, были найдены распределения по размерам частиц в каждом из образцов. Частицы рассматривались как шарообразные. В качестве базового использовано гамма-распределение для расположения объемной доли частиц по их диаметру. Для учета межчастичной интерференции был использован структурный фактор фрактального распределения массы. Найденные распределения представлены на рис. 4, а соответствующие каждому распределению наиболее вероятный размер (мода), средний размер и среднеквадратичный разброс приведены в табл. 2. В композитных образцах 2, 3, 5 максимум распределения приходится на размеры меньше 5 нм. Это означает, что все три использованных в работе типа поверхностной модификации ДНА обеспечивают эффективность их диспергирования в жидком ММА под действием ультразвука и получение на их основе композитов, размер частиц ДНА в которых составляет порядка единиц нм.
Влияние ДНА на полимеризацию ММА и свойства композитов на его основе
Данные табл. 1 свидетельствуют о неожиданном влиянии ДНА на молекулярно-массовые характеристики ПММА, полученного в их присутствии. Если уменьшение ММ при введении ДНА легко объясняется увеличением вероятности обрыва цепи в присутствии высокодисперсного наполнителя, что сопровождается также сужением ММР (как это ярко проявилось в случае ДНА-III при низкой концентрации инициатора 10–3 моль/л), то причина возрастания ММ не столь очевидна. Возможно, что это связано с определенным структурированием в полученных золях. Влияние ДНА на ММ больше видно при малом содержании ДАК, когда вероятность обрыва цепи диспропорционированием или рекомбинацией значительно меньше, чем в серии экспериментов с концентрацией ДАК в 50 раз выше (табл. 1).
Для более детального рассмотрения влияния ДНА на молекулярно-массовые характеристики и Tс, полученного в их присутствии ПММА, представлены три серии образцов (табл. 3). Поскольку наибольшую совместимость с мономером продемонстрировали ДНА, модифицированные гептаметилтрисилоксановыми группами (ДНА-I), то именно они были выбраны для поставленной цели. Область концентрации ДНА обусловлена известным диапазоном, ниже границ которого эффект от ДНА уже не проявляется, а выше – чаще отрицательный, нежели положительный из-за агрегирования ДНА. Поэтому композиты получены с содержанием ДНА 0.01–0.10 мас. %. Три серии композитов отличаются друг от друга величиной ММ и шириной ММР, которые задавались концентрацией инициатора и присутствием или отсутствием агента обратимой передачи цепи [83]).
Таблица 3.
Характеристики композитов, полученных в присутствии ДНА-I
| Содержание ДНА-I, мас. % | Mn × 10–3 | Ð | Tс, °C |
|---|---|---|---|
| [ДАК] = 1 × 10–3 моль/л | |||
| 0 | 740 | 5.2 | 128 |
| 0.01 | 620 | 6.0 | 128 |
| 0.05 | 650 | 5.6 | 131 |
| 0.10 | 610 | 5.9 | 128 |
| [ДАК] = 5 × 10–2 моль/л | |||
| 0 | 90 | 3.6 | 127 |
| 0.01 | 80 | 4.9 | 127 |
| 0.05 | 123 | 3.6 | 126 |
| 0.10 | 109 | 3.6 | 126 |
| [ДАК] = 5 × 10–2 моль/л; [БТК] = 5 × 10–2 моль/л | |||
| 0 | 40.0 | 2.9 | 117 |
| 0.01 | 41.2 | 2.8 | 109 |
| 0.05 | 43.7 | 2.7 | 108 |
| 0.10 | 43.4 | 2.8 | 108 |
Особенностью полимеризации ММА в присутствии агента обратимой передачи цепи, которым в данном случае был БТК, является наличие реакций передачи цепи по двойной связи C=S, которые обеспечивают попеременное существование растущего радикала в активном и “спящем” состоянии, что является условием осуществления реакции в контролируемом или “псевдоживом” режиме. Схема полимеризации включает как стадии типичные для классической радикальной полимеризации (1), так и специфические для ОПЦ-полимеризации в присутствии симметричных тритиокарбонатов (2)–(4) [84]:
(1)
$\begin{gathered} {\text{I}} \to 2{\dot {I}} \\ {\dot {I}} + n{\text{M}} \to {{{{\dot {P}}}}_{n}} \\ \end{gathered} $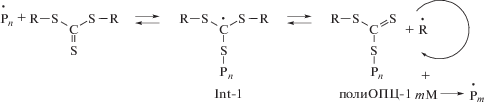
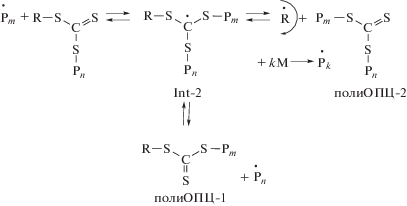
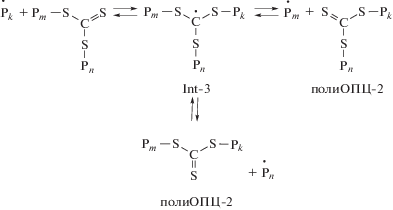
Реакционный центр формируется, как и в классической радикальной полимеризации, в результате термического распада инициатора и присоединения образовавшихся радикалов к первой молекуле мономера. Однако далее в конкуренции за взаимодействие с растущим радикалом участвует ОПЦ-агент. Он выбирается так, чтобы вероятность присоединения радикала к двойной связи “углерод–сера” превышала вероятность роста цепи и тем более обрыва. В результате реакции (2) происходит образование радикального интермедиата Int-1, который может фрагментировать как в сторону образования исходных продуктов, так и с отщеплением хорошо уходящей группы R. В данном случае R представляет собой относительно устойчивый бензильный радикал, поэтому равновесие (2) сдвинуто вправо. Продуктом, образующимся на стадии (2), является монозамещенный тритиокарбонат полиОПЦ-1, который также может работать в качестве агента обратимой передачи цепи. При этом реализуется равновесие (3), в котором фрагментация радикального интермедиата Int-2 обычно происходит с отщеплением второй группы R, и образуется дизамещенный тритиокарбонат полиОПЦ-2. Последняя стадия представляет собой лишь обмен радикалами, происходящий через присоединение к полиОПЦ-2 с образованием радикального интермедиата Int-3 и его равновероятной фрагментацией с отщеплением любого из заместителей. Пока радикал находится присоединенным к тритиокарбонатному фрагменту, реализуется его “спящее” состояние. При отщеплении от полиОПЦ-1 или полиОПЦ-2 он переходит в активное состояние и становится способен участвовать в росте цепи. На схеме показано также, что радикал R может реинициировать полимеризацию.
На примере полимеризации ММА в присутствии S,S'-бис-(метил-2-изобутират)тритиокарбоната, однако, показано, что стадия (4) не реализуется [85], т.е. ОПЦ-полимеризация ММА в присутствии симметричных тритиокарбонатов является двухстадийной, а реакция (3) никогда не происходит с отщеплением радикала R:
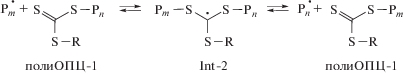 (3*)
(3*)
В результате представленного обмена радикалами макромолекулы ПММА усредняются по длине, ММР сужается, а максимальная молекулярная масса определяется концентрацией БТК, что и видно из табл. 3. Проведение полимеризации в режиме обратимой передачи цепи имеет также преимущества чисто практического плана. Благодаря реализации описанной схемы, кинетика полимеризации становится более сглаженной, характеризуется уменьшением или полным отсутствием гель-эффекта, устраняются перегревы реакционной массы, характерные для блочной полимеризации. Таким способом удалось получить качественные однородные образцы композитов, лишенные включения пузырей, трещин и подобных дефектов.
Очевидно наличие зависимости молекулярно-массовых характеристик ПММА от типа поверхностной модификации ДНА, в присутствии которых был получен полимер (см. табл. 1). Помимо типа поверхностной модификации на ММ оказывает влияние содержание ДНА (как это показано на примере ДНА-I), особенно для серии, полученной при концентрации ДАК 1 × 10–3 моль/л. При несомненном влиянии ДНА-I на молекулярно-массовые характеристики вывести закономерности, однако, затруднительно. Интересно, что введение минимального количества ДНА-I в количестве 0.01% влечет уширение ММР, которое при дальнейшем увеличении концентрации ДНА-I исчезает. Обращает на себя внимание, что при низкой концентрации ДАК введение ДНА приводит только к уменьшению ММ, вероятно, за счет обрыва цепей на развитой поверхности мелкодисперсного наполнителя. Хотя при высокой концентрации ДАК введение ДНА четко вызывает рост ММ, что можно связать только с уменьшением скорости обрыва цепей. Причиной может быть снижение молекулярной подвижности при определенной концентрации ДНА за счет появления структуры в золях ДНА. Как и следовало ожидать, в случае полимеризации с обратимой передачей цепи, влияние ДНА на молекулярно-массовые характеристики практически отсутствует, поскольку определяющим является наличие БТК и взаимодействия, представленные выше равновесиями (2) и (3*).
Для полученных композитов было изучено их поведение при нагревании и найдены значения Tс, представленные в табл. 1 и 3. Неоднозначность зависимости свойств композитов от содержания наполнителя – общее для подобных работ. Например, в работе [86] приводятся данные о зависимости Tс от концентрации наполнителя для ПММА, наполненного органомодифицированным неорганическим гидроксидом (это очень роднит с объектами исследования в нашей работе: неорганическими гидрофильными ДНА, покрытыми слоем органических радикалов). Авторы утверждают, что взаимодействие гидроксильных групп наполнителя со сложноэфирными ПММА ведет к увеличению Tс, не давая больше комментариев. Только все представляется намного сложнее: Tс повышается на 6° при увеличении концентрации наполнителя до 4% и возвращается к первоначальному значению для чистого ПММА при концентрации 12.5%. Совершенно противоположный эффект можно видеть на образцах ПММА, наполненного титанатом бария [87]: наблюдается монотонное снижение Tс с увеличением содержания наполнителя при максимальном эффекте 5°.
В нашем случае нет предпосылок для активного взаимодействия ДНА с полимером. Поэтому на большинстве серий образцов влияние ДНА на температуру стеклования практически отсутствует (что чаще всего и наблюдается при наполнении ПММА неорганическими наполнителями в таких количествах, как в настоящей работе), за исключением серии образцов, полученных в присутствии БТК. Кривые ДСК для них представлены на рис. 5. Данные табл. 1 свидетельствуют о том, что Tс определяется не столько молекулярно-массовыми характеристиками, сколько типом ДНА, влияние которого несомненно. Для ДНА-I Tс предсказуемо несколько выше для серии с бόльшей ММ, при этом зависимость от концентрации ДНА-I практически отсутствует, как и для серии с меньшей молекулярной массой, полученной при концентрации ДАК в 50 раз больше (табл. 3).
Рис. 5.
Кривые ДСК композитов, полученных в условиях обратимой передачи цепи. Содержание ДНА-I 0 (1), 0.01 (2), 0.05 (3) и 0.10 мас. % (4).
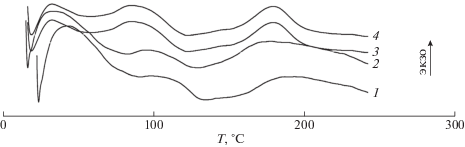
Наиболее интересные результаты получены для серии композитов, синтезированных в присутствии БТК. При практически одинаковых ММ характеристиках наблюдается яркая зависимость Tс от содержания ДНА-I: при введении ДНА-I температура стеклования снижается почти на десять градусов (табл. 3). Это можно объяснить повышением молекулярной подвижности в образцах за счет нарушения порядка, который, видимо, присущ ПММА, синтезированному в условиях обратимой передачи цепи и отличающемуся однородностью по ММ макромолекул. Ни для одной другой из исследованных серий не наблюдалось такого влияния ДНА на температуру стеклования.
ЗАКЛЮЧЕНИЕ
Используя традиционную радикальную полимеризацию и контролируемую полимеризацию с обратимой передачей цепи получены in situ полимерные нанокомпозиты, содержащие в полиметилметакрилатной матрице 0.01–0.10 мас. % частиц ДНА со средним диаметром 5–7 нм. Такое распределение ДНА достигнуто благодаря проведенной поверхностной функционализации ДНА гептаметилтрисилоксановыми, триметилсилановыми и 2,2′,3,3′,4,4′,5,5′,5′′-нонафторамилокси группами, обеспечившей им легкую диспергируемость в метилметакрилате с получением устойчивых на протяжении полимеризации золей.
Показано, что введение ДНА в мономер оказывает влияние на молекулярно-массовые характеристики образующегося полимера, приводя как к изменению молекулярной массы, так и ширины молекулярно-массового распределения. Для композитов, полученных методом полимеризации с обратимой передачей цепи, обнаружена зависимость температуры стеклования от содержания ДНА в композите при фактически одинаковых молекулярно-массовых характеристиках полимера: введение 0.05–0.10 мас. % ДНА снижают температуру стеклования на девять градусов. Уменьшение температуры стеклования с ростом концентрации ДНА связано с нарушением упорядоченности в упаковке узкодисперсных макромолекул полимеров, получающихся в условиях контролируемой радикальной полимеризации, а, следовательно, и с повышением молекулярной подвижности.
Авторы выражают благодарность В.Ю. Долматову (СКТБ “Технолог”) за любезно предоставленный образец ДНА.
Исследования методом малоуглового рентгеновского рассеяния и дифференциальной сканирующей калориметрии выполнены с использованием оборудования инжинирингового центра СПбГТИ(ТУ).
Список литературы
Shenderova O.A, Gruen D.M. Ultrananocrystalline Diamond: Synthesis, Properties, and Applications. New York: William Andrew Publ., 2012.
Долматов В.Ю. Детонационные наноалмазы. Получение, свойства, применение. СПб.: НПО “Профессионал”, 2011.
Whitlow J., Pacelli S., Paul A. // J. Controlled Release. 2017. V. 261. P. 62.
Shakun A., Vuorinen J., Hoikkanen M., Poikelispää, Das M., Hard A. // Composites. A. 2014. V. 64. P. 49.
Rifai A., Pirogova E., Fox K. // Encyclopedia of Biomedical Engineering. Amsterdam: Elsevier Inc., 2019. P. 97. V. 1.
Шугалей И.В., Судариков А.М., Возняковский А.П., Целинский И.В., Гарабаджиу А.В., Илюшин М.А. Химия поверхности детонационных наноалмазов как основа создания продукции биомедицинского назначения. СПб.: ЛГУ имени А.С. Пушкина, 2012.
Neitzel I., Mochalin V.N., Gogotsi Y. // Nanodiamonds. Advanced Material Analysis, Properties and Applications. Micro and Nano Technologies. Amsterdam: Elsevier Inc., 2017. P. 365.
Mochalin V.N., Gogotsi Y. // Diamond Relat. Mater. 2015. V. 58. P. 161.
Lee J.K.Y., Chen N., Peng S., Li L., Tian L., Thakor N., Ramakrishna S. // Prog. Polym. Sci. 2018. V. 86. P. 40.
Kausar A. // Hybrid Polymer Composite Materials. Structure and Chemistry. Amsterdam: Elsevier Inc., 2017.
Nunn N., Torelli M., McGuire G., Shenderova O. // Curr. Opinion Solid State Mater. Sci. 2017. V. 21. P. 1.
Detonation Nanodiamonds / Ed. by A.Y. Vul’, O.A. Shenderova. Boca Raton: CRC Press, Taylor & Francis Group, 2013.
Nanodiamonds. Advanced Material Analysis, Properties and Applications / Ed. by J.-C. Arnault. Saint Louis: Elsevier, 2017.
Schmidlin L., Pichot V., Comet M., Josset S., Rabu P., Spitzer D. // Diamond Relat. Mater. 2012. V.22. P. 113.
Neitzel I., Mochalin V., Knoke I., Palmese G.R., Gogotsi Y. // Comp. Sci. Techn. 2011. V. 71. P. 710.
Špitalský Z., Kromka A., Matějka L., Černoch P., Kovářová J., Kotek J., Šlouf M. // Adv. Comp. Lett. 2008. V. 17. № 1. P. 29.
Zhang Y., Rhee K.Y., Hui D., Park S.-J. // Composites. B. 2018. V. 143. P. 19.
Xu Q., Zhao X. // J. Mater. Chem. 2012. V. 22. P. 16416.
Zhu Y., Xu X., Wang B., Feng Z. // Chin. Particuology. 2004. V. 2. № 3. P. 132.
Khalilnezhad P., Sajjadi S.A., Zebarjad S.M. // Diamond Relat. Mater. 2014. V. 45. P. 7.
Kulvelis Yu.V., Shvidchenko A.V., Aleksenskii A.E., Yudina E.B., Lebedev V.T., Shestakov M.S., Dideikin A.T., Khozyaeva L.O., Kuklin A.I., Török Gy., Rulev M.I., Vul A.Ya. // Diamond Relat. Mater. 2018. V. 87. P. 78.
Chin R.-M., Chang S.-J., Li C.-C., Chang C.-W., Yu R.-H. // J. Colloid Interface Sci. 2018. V. 520. P. 119.
Zhao Y.-Q., Lau K.-T., Kim J.-K., Xu C.-L., Zhao D.-D., Li H.-L. // Composites. B. 2010. V. 41. P. 646.
Presti C., Ferry L., Alauzun J.G., Dumazert L., Gallard B., Quantin J.-C., Lopez Cuesta J.-M., Mutin P.H. // Diamond Relat. Mater. 2017. V. 76. P. 141.
Korobko A.P., Bessonova N.P., Krasheninnikov S.V., Konyukhova E.V., Drozd S.N., Chvalun S.N. // Diamond Relat. Mater. 2007. V. 16. P. 2141.
Zhaia Y.-J., Wanga Z.-C., Huanga W., Huanga J.-J., Wangb Y.-Y., Zhao Y.-Q. // Mater. Sci. Eng. A. 2011. V. 528. P. 7295.
Neitzel I., Mochalin V.N., Niu J., Cuadra J., Kontsos A., Palmese G.R., Gogotsi Y. // Polymer. 2012. V. 53. P. 5965.
Subhani T., Latif M., Ahmad I., Rakha S.A., Ali N., Khurram A.A. // Mater. Design. 2015. V. 87. P. 436.
Haleem Y.A., Liu D., Chen W., Wang C., Hong C., He Z., Liu J., Song P., Yu S., Song L. // Composites. B. 2015. V. 78. P. 480.
Vatanpour V., Salehi E., Sahebjamee N., Ashrafi M. // Arab. J. Chem. 2018. V. 13. № 1. P. 1731.
Maitra U., Prasad K.E., Ramamurty U., Rao C.N.R. // Solid State Commun. 2009. V. 149. P. 1693.
Passeri D., Biagioni A., Rossi M., Tamburri E., Terranova M.L. // Eur. Polym. J. 2013. V. 49. P. 991.
Krueger A., Lang D. // Adv. Funct. Mater. 2012. V. 22. P. 890.
Krueger A. // Comprehensive Hard Materials. Amsterdam: Elsevier Inc., 2014. V. 3. P. 379.
Lisichkin G.V., Korol’kov V.V., Tarasevich B.N., Kulakova I.I., Karpukhin A.V. // Russ. Chem. Bull. 2006. V. 55. P. 2212.
Lisichkin G.V., Kulakova I.I., Gerasimov Y.A., Karpuk-hin A.V., Yakovlev R.Y. // Mendeleev Commun. 2009. V. 19. P. 309.
Liu Y., Khabashesku V.N., Halas N.J. // J. Am. Chem. Soc. 2005. V. 127. P. 3712.
Krueger A., Boedeker T. // Diamond Relat. Mater. 2008. V. 17. P. 1367.
Krueger A., Liang Y.J., Jarre G., Stegk J. // J. Mater. Chem. 2006. V. 16. № 24. P. 2322.
Duffy E., Mitev D.P., Thickett S.C., Townsend A.T., Paull B., Nesterenko P.N. // Appl. Surf. Sci. 2015. V. 357. P. 397.
Wahab Z., Foley E.A., Pellechia P.J., Anneaux B.L., Ploehn H.J. // J. Colloid Interface Sci. 2015. V. 450. P. 301.
Zhang H.H., Liu Y.T., Wang R., Yu X.Y., Qu X.W., Zhang Q.X. // Chin. Chem. Lett. 2011. V. 22. P. 485.
Lau X.C., Desai C., Mitra S. // Solar Energy. 2013. V. 91. P. 204.
Krueger A., Boedeker T. // Diamond Relat. Mater. 2008. V. 17. P. 1367.
Cha I., Hashimoto K., Fujiki K., Yamauchi T., Tsubokawa N. // Mater. Chem. Phys. 2014. V. 143. P. 1131.
Basiuk E.V., Santamaria-Bonfil A., Meza-Laguna V., Gromovoy T.Yu., Alvares-Zauco E., Contreras-Torres F.F., Rizo J., Zavala G., Basiuk V.A. // Appl. Surf. Sci. 2013. V. 275. P. 324.
Spitsyn B.V., Davidson J.L., Gradoboev M.N., Galushko T.B., Serebryakova N.V., Karpukhina T.A. // Diamond Relat. Mater. 2006. V. 15. P. 296.
Lang D., Krueger A. // Diamond Relat. Mater. 2011. V. 20. P. 101.
Jee A.-Y., Lee M. // Current Appl. Phys. 2009. V. 9. P. 144.
Arnault J.C. // Novel Aspects of Diamond. Topics in Applied Physics / Ed. by N. Yang. Cham: Springer, 2015. P. 85. V. 121.
Yakovlev R.Yu., Kulakova I.I., Leonidov N.B., Lisichkin G.V. // Mendeleev Commun. 2012. V. 22. P. 213.
Voznyakovskii A.P., Klyubin V.V., Dolmatov V.Y., Agibalova L.V. // J. Superhard Mater. 2000. V. 22. № 2. P. 58.
Voznyakovsky A.P., Fujimura T., Dolmatov V.Y., Veretennikova M.V. // J. Superhard Mater. 2002. V. 24. № 6. P. 21.
Barras A., Szunerits S., Marcon L., Monfilliette-Dupont N., Boukherroub R. // Langmuir. 2010. V. 26. № 16. P. 13168.
Huang H., Liu M., Tuo X., Chen J., Mao L., Wen Y., Tian J., Zhou N., Zhang X., Wei Y. // Appl. Surf. Sci. 2018. V. 439. P. 1143.
Chang I.P., Hwang K.C., Ho J.-a.A., Lin C.-C., Hwu R.J.-R., Horng J.-C. // Langmuir. 2010. V. 26. № 5. P. 3685.
Huang H., Liu M., Jiang R., Chen J., Mao L., Wen Y., Tian J., Zhou N., Zhang X., Wei Y. // J. Colloid Interface Sci. 2018. V. 513. P. 198.
Xu D., Liu M., Zhang Q., Huang Q., Huang H., Tian J., Jiang R., Wen Y., Zhang X., Wei Y. // Mater. Sci. Eng. C. 2018. V. 91. P. 496.
Liu R., Zhao F., Yu X., Naito K., Ding H., Qu X., Zhang Q. // Diamond Related Mater. 2014. V. 50. P. 26.
Li L., Davidson J.L., Lukehart C.M. // Carbon. 2006. V. 44. P. 2308.
Dahoumane S.A., Nguyen M.N., Thorel A., Boudou J.-P., Chehimi M.M., Mangeney C. // Langmuir. 2009. V. 25. № 17. P. 9633.
Zeng G., Liu M., Shi K., Heng C., Mao L., Wan Q., Huang H., Deng F., Zhang X., Wei Y. // Appl. Surf. Sci. 2016. V. 390. P. 710.
Chen J., Liu M., Huang Q., Huang L., Huang H., Deng F., Wen Y., Tian J., Zhang X., Wei Y. // Chem. Eng. J. 2018. V. 337. P. 82.
Ayatollahi M.R., Alishahi E., Doagou R S., Shadlou S. // Composites. B. 2012. V. 43. P. 3425.
Avazkonandeh-Gharavol M.H., Sajjadi S.A., Zebarjad S.M., Mohammadtaheri M., Abbasi M., Alimardani M., Mossaddegh K. // Prog. Organic Coat. 2013. V. 76. P. 1258.
Jee A.-Y., Lee M. // Curr. Appl. Phys. 2011. V. 11. P. 1183e1187.
Lee J.-Y., Lim D.-S. // Surf. Coat. Technol. 2004. V. 188–189. P. 534.
Senyek M.L., Kulig J.J., Parker D.K. Pat. US6369158 B1 USA. 2002.
Sivtsov E.V., Gostev A.I., Parilova E.V., Dobrodumov A.V., Chernikova E.V. // Polymer Science C. 2015. V. 57. № 1. P. 110.
Несмеянов А.Н., Кочешков К.А. Методы элементоорганической химии (магний, бериллий, кальций, стронций, барий). М.: Наука, 1963.
Возняковский А.П., Калинин А.В., Фурсенко А.В. // Сверхтвердые материалы. 2011. Т. 192. № 4. С. 38.
Voznyakovskii A.P., Kalinin A.V., Mokeev M.V., Agibalova L.V., Vlasova E.N. // Russ. J. Appl. Chem. 2012. V. 85. № 7. P.1090.
Возняковский А.П., Калинин А.В., Агибалова Л.В., Шугалей И.В. // Породоразрушающий инструмент. 2014. № 17. С. 390.
Voznyakovskii A.P., Kalinin A.V., Agibalova L.V. // J. Superhard Mater. 2011. V. 33. P. 244.
Kong H., Gao C., Yan D. // Macromolecules. 2004. V. 37. № 11. P. 4022.
Philip B., Xie J., Abraham J.K., Varadan V.K. // Polymer Bull. 2005. V. 53. № 2. P. 127.
Sainsbury T., Erickson K., Okawa D., Zonte C.S., Fréchet J.M.J., Zettl A. // Chem. Mater. 2010. V. 22. № 6. P. 2164.
Zhang Q., Naito K., Tanaka Y., Kagawa Y. // Macromolecules. 2008. V. 41. P. 536.
Zhang X., Fu C., Feng L., Ji Y., Tao L., Huang Q., Li S., Wei Y. // Polymer. 2012. V. 53. P. 3178.
Maitra U., Gomathi A., Rao C.N.R. // J. Exp. Nanosci. 2008. V. 3. № 4. P. 271.
Kaur R., Chitanda J.M., Michel D., Maley J., Borondics F., Yang P., Verrall R.E., Badea I. // Int. J. Nanomed. 2012. V. 7. P. 3851.
Лукевиц Э.Я., Воронков М.Г. Гидросилилирование, гидрогермилирование, гидростаннилирование. Рига: Изд-во АН Латвийской ССР, 1964.
Handbook of RAFT Polymerization / Ed. by C. Barner-Kowollik. Weinheim: Wiley, 2008.
Chernikova E.V., Sivtsov E.V. // Polymer Science B. 2017. V. 59. № 2. P. 117.
Chernikova E.V., Terpugova P.S., Baskakov A.A., Plutalova A.V., Garina E.S., Sivtsov E.V. // Polymer Science B. 2010. V. 52. № 3–4. P. 119.
Li X., Chen L., Li Q., Zhang J., Su Z., Zhang X., Zheng K., Tian X. // RSC Advances. 2016. V. 6. P. 101941.
Elshereksi N.W., Mohamed S.H., Arifin A., Ishak Z.A.M. // J. Phys. Sci. 2014. V. 25. № 2. P. 15.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)