

Высокомолекулярные соединения (серия Б), 2020, T. 62, № 6, стр. 458-464
ИНТЕРПОЛИМЕРНЫЕ КОМПЛЕКСЫ АЛЬГИНАТА НАТРИЯ – рН-ЧУВСТВИТЕЛЬНЫЕ НОСИТЕЛИ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ
М. Ю. Горшкова a, *, И. Ф. Волкова a, Э. С. Григорян a, Л. И. Валуев a
a Институт нефтехимического синтеза им. А.В. Топчиева Российской академии наук
119991 Москва, Ленинский пр., 29, Россия
* E-mail: mgor@ips.ac.ru
Поступила в редакцию 25.04.2020
После доработки 24.06.2020
Принята к публикации 08.07.2020
Аннотация
Разработан способ получения гидрогелей на основе интерполимерных комплексов альгината натрия и сополимера метилвинилового эфира с малеиновым ангидридом. Показано, что формирование интерполимерной сетки обусловлено образованием водородных связей между гидроксильными группами альгината и карбоксильными группами сополимера. Структура сетки – тип связей и их плотность – определена преимущественно температурой обработки смеси полимеров. На примере модельного лекарства лидокаина продемонстрирована возможность применения таких комплексов в качестве платформы для рН-чувствительных систем доставки лекарственных веществ.
ВВЕДЕНИЕ
Одна из главных задач современной химии высокомолекулярных соединений заключается в использовании результатов фундаментальных исследований для решения конкретных проблем по обеспечению комфортных условий жизнедеятельности человека, связанных, в частности, с охраной его здоровья. Природные нетоксичные, биосовместимые, биодеградируемые полисахариды, способные демонстрировать мукоадгезионные свойства, являются весьма перспективными для использования их при создании продуктов биомедицинского назначения, в том числе систем доставки лекарственных веществ [1]. Альгинат натрия, выделяемый из бурых водорослей, представляет собой полисахарид, несущий в каждом звене карбоксильные группы, наличие которых определяет возможность получения на его основе гидрогелей, микро- и наночастиц.
Именно гидрогели – набухшие в водном растворе трехмерные сетки сшитых ковалентными или солевыми связями полимерных цепей:
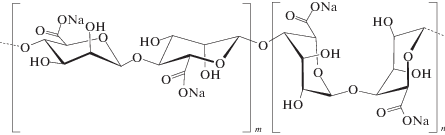
являются наиболее распространенной формой применения данного полисахарида. Для альгинатных гидрогелей чаще всего используют реакцию карбоксильных групп полимера с солями поливалентных металлов, например, с хлоридом кальция [2, 3]. Такие гидрогели обладают невысокой механической стабильностью, в связи с чем их применяют как мукоадгезивные носители лекарственных веществ, например для доставки лекарств непосредственно в мозг при интранозальном введении [4].
Более стабильные гидрогели образуются в результате ковалентной сшивки макромолекул альгината. Для синтеза таких гелей альгинат модифицируют введением в состав его макромолекул функциональных групп, способных образовывать ковалентные связи [5–8], либо используют полифункциональные сшиватели, реагирующие с кислотными или гидроксильными группами альгината [9, 10]. Альтернативным способом получения гидрогелей можно назвать формирование интерполимерной сетки реакцией двух различных полимерных компонентов с образованием водородных, солевых или ковалентных связей [11–17]. Наличие значительного количества карбоксильных и гидроксильных групп в альгинате натрия предполагает использование в качестве второго компонента полимера, содержащего ангидридные и(или) карбоксильные группы. Таким полимером в настоящей работе служил сополимер метилвинилового эфира с малеиновым ангидридом (МВЭМА).
Сополимер МВЭМА обладает собственной биологической активностью при низкой токсичности и широко применяется в фармацевтической промышленности в качестве вспомогательного компонента под коммерческим названием “Gantrezтм”. В гидролизованном виде МВЭМА представляет собой поликислоту с высокой плотностью зарядов:
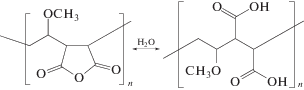
Цель данной работы – изучить особенности формирования сшитых интерполимерных комплексов (ИПК) альгината натрия и сополимера МВЭМА, исследовать зависимости свойств комплексов от условий получения, а также оценить потенциал их использования в качестве носителей лекарственных веществ. В качестве модельного лекарственного вещества выступал лидокаин – вещество с местным обезболивающим действием.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы
Альгинат натрия (“Foodchem International Corp.”, Китай), чередующийся сополимер метилвинилового эфира и малеинового ангидрида (“Polysciences Inc.”, США) и лидокаина гидрохлорид (“Spectrum”, США) использовали без дополнительной очистки. Молекулярную массу альгината натрия Mw = 17 × 105 определяли методом ГПХ. Образцы МВЭМА с молекулярными массами Mw = 41 × 103 и 8 × 103, найденными методом ГПХ, обозначали как МВЭМА-41 и МВЭМА-8 соответственно. Буферы MES с рН 5.5 и фосфатный с рН 7.2 готовили растворением концентратов (“Sigma-Aldrich”, США), тетраборатный буфер с рН 10 получали из борной кислоты и едкого натра. Хлористый натрий, едкий натр, соляную кислоту квалификации х.ч. (“ЛабТех”, Россия) применяли без очистки. Растворителем для приготовления всех растворов служила бидистиллированная вода.
Методы
Молекулярно-массовые характеристики полимеров определяли методом ГПХ на приборе “Gilson” (США–Франция), оснащенном рефрактометрическим детектором “RI-131 Gilson”, инжектором “Rheodyne 7125”, колонкой “TSK gel GM PWXL” (“Toson Bioscience”, Япония), а также пакетом сбора и обработки данных “MutiChrom” (“Ampersand”, Россия). Эксперименты проводили при комнатной температуре. В качестве растворителя и элюента использовали боратный буфер с рН 10.2, содержащий 0.2 моль/л NaCl; калибровку системы выполняли с помощью ПЭО-стандартов.
Интерполимерные комплексы получали смешиванием водных растворов полимеров. К раствору альгината натрия с концентрацией 20 мг/мл добавляли рассчитанное количество раствора МВЭМА (концентрация 40 мг/мл), смесь перемешивали 10–15 мин при комнатной температуре и выдерживали 1 ч. Далее смесь выливали в чашки Петри и высушивали при комнатной температуре не менее 48 ч до постоянной массы. Приготовленные пленки подвергали термической обработке, выдерживая их при заданной температуре в течение 24 ч. Состав и режим обработки гелей отражены в обозначении образцов. Так, например, А/МВЭМА-8/80 – композиция с соотношением альгината натрия и сополимера МВЭМА 1 : 1, молекулярной массой сополимера 8 × 103 и температурой обработки пленки 80°С.
Для приготовления ИПК, содержащих 5 мас.% лидокаина, к раствору альгината добавляли рассчитанное количество водного раствора лекарственного вещества с концентрацией 32 мг/мл, перемешивали раствор в течение 15 мин, после чего вводили раствор МВЭМА и далее проводили процесс по описанной выше методике.
Для нахождения степени набухания гелей в воде навеску ИПК (30 мг) помещали в стеклянную емкость и заливали 50 мл воды. Через установленные интервалы времени жидкость сливали, остатки ее удаляли фильтровальной бумагой, затем определяли массу влажного гидрогеля. Степень набухания вычисляли по уравнению
где m0 и mt – масса сухого и влажного геля соответственно.Для изучения кинетики набухания и скорости выделения лидокаина в растворах буферов навеску ИПК (200 мг) помещали в стеклянную емкость, добавляли 15 мл буферного раствора и оставляли без перемешивания при комнатной температуре. Через заданные промежутки времени отбирали пробы и измеряли их оптическую плотность; после измерения пробу возвращали обратно. Концентрацию лидокаина находили методом УФ-спектроскопии с помощью прибора “Specord M-40” (“Carl Zeiss Jena”, Германия) по разнице оптических плотностей проб, отобранных в опытах с лидокаин-содержащими ИПК и опытах с ИПК без лекарственного вещества. Концентрацию рассчитывали по полученным ранее калибровочным зависимостям оптической плотности на длине волны 271.4 нм от концентрации лидокаина: D = 1.47 С для буфера с рН 5.5 (MES) и D = 1.30 С для фосфатного буфера с рН 7.2.
Результаты представляли в виде зависимости величины (ЛВтек/ЛВисх) × 100% от времени, где ЛВтек – текущее количество лекарственного вещества лидокаина гидрохлорида в растворе (мг), ЛВисх – исходное количество лидокаина (мг).
ИК-спектры исходных полимеров и гелей регистрировали на приборе “Perkin Elmer Spectrum one” (“Perkin Elmer”, США), оснащенном универсальной приставкой ATR с однократным отражением от поверхности кристалла “Diamant Zn–Se”.
Для потенциометрических измерений использовали метр рН “Beckman Φ-70” с комбинированным электродом “Futura Plus” (США).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Возможность образования ИПК и его структура определяются, в первую очередь, строением исходных макромолекул – имеющимся количеством групп, способных к взаимодействию, длиной цепей полимеров, а также соотношением компонентов. Наличие значительного количества гидроксильных групп в альгинате натрия и ангидридных и карбоксильных групп в МВЭМА создает основу для формирования ИПК из этих полимеров. Необходимо отметить, что оба полимера представляют собой слабые полиэлектролиты, степень ионизации которых зависит от рН среды, что может обеспечить проявление рН чувствительности систем на их основе. Длина цепи у образцов МВЭМА, использованных в работе, существенно меньше, чем у макромолекул альгината, что позволяет рассматривать МВЭМА в качестве сшивающего агента. В табл. 1 показана зависимость степени набухания ИПК от условий его получения. Как и следовало ожидать, увеличение количества МВЭМА приводит к образованию более плотно сшитой сетки и уменьшению степени набухания ИПК. Аналогичный эффект наблюдается при уменьшении температуры термической обработки для гелей с одинаковым составом. Молекулярная масса сшивающего агента слабо влияет на степень набухания.
Таблица 1.
Степень набухания в воде гелей А/МВЭМА с различным соотношением компонентов и температурой обработки
| Соотношение А : МВЭМА | Температура обработки, °С | Степень набухания | |
|---|---|---|---|
| МВЭМА-8 | МВЭМА-41 | ||
| 1 : 1 | 23 | 35 ± 2.1 | 30 ± 1.8 |
| 1 : 1 | 37 | 41 ± 2.5 | 39 ± 2.3 |
| 1 : 1 | 80 | 51 ± 3.1 | 49 ± 2.9 |
| 1 : 2 | 80 | 23 ± 1.4 | 29 ± 1.7 |
| 1 : 5 | 80 | 11 ± 0.7 | 15 ± 0.9 |
Структуру ИПК изучали методом ИК-спектроскопии. Логично предположить, что формирование ИПК происходит за счет взаимодействия карбоксильных и гидроксильных групп, поэтому наиболее информативными являются области спектра, относящиеся к поглощению данных групп (рис. 1, 2).
Рис. 1.
ИК-спектры альгината натрия (1), МВЭМА (5) и ИПК А/МВЭМА-8 состава 1 : 1, выдержанного при 23 (2), 37 (3) и 80°С (4).
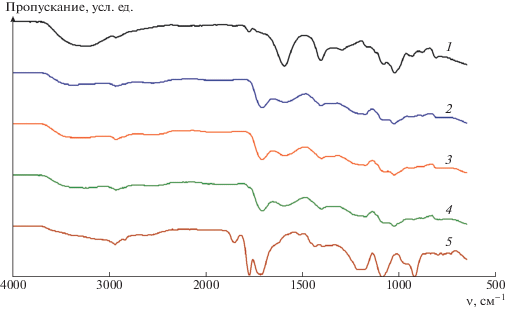
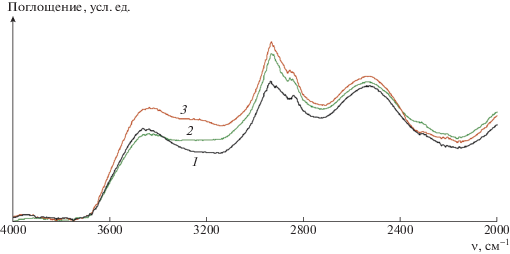
Рис. 2.
ИК-спектр ИПК А/МВЭМА-8 состава 1 : 1, выдержанного при 23 (1), 37 (2) и 80°С (3), в области поглощения гидроксильных групп.
Рассмотрим сначала область поглощения карбоксильных групп. В спектре исходного альгината натрия (рис. 1, спектр 1) присутствуют только полосы, относящиеся к карбоксильным группам в солевой форме (1593 и 1406 см–1). В спектре сополимера МВЭМА (спектр 5) наблюдаются полосы ангидридных групп 1851 и 1776 см–1, а также полоса карбоксильных групп 1717 см–1. Положение последней показывает, что карбоксильные группы находятся в связанном состоянии. В спектрах ИПК присутствуют полосы карбоксильных групп 1710–1707 см–1 и карбоксилат-ионов 1595 см–1. Сдвиг полосы карбоксильных групп в спектрах ИПК относительно их положения в спектре исходного МВЭМА на 7–10 см–1 указывает на их ассоциированное состояние.
В области поглощения гидроксильных групп (3000–3600 см–1), в спектрах альгината и ИПК наблюдается широкая полоса со сложным контуром, отвечающая гидроксильным группам как свободным, так и связанным [18, 19]. Поведение этой полосы сложнее проследить, поскольку в системе присутствует вода. Для количественной оценки спектры нормировали по внутреннему стандарту, за который принимали полосу валентных колебаний групп СН (2933 см–1), и рассчитывали интегральные интенсивности полос карбоксильных групп, гидроксильных групп и карбоксилат-ионов (табл. 2). Положение полосы 1596 см–1, относящейся к карбоксилат-ионам, и ее интегральная интенсивность практически не зависят от температуры обработки ИПК, это позволяет предположить, что данные группы не принимают участия в формировании интерполимерной сетки. Наличие в спектрах образцов ИПК полосы в области 2700–2500 см–1 (рис. 2) указывает на сильное водородное связывание [20] в исследуемых комплексах, что согласуется с поведением полосы в области 1710 см–1, относящейся в основном к МВЭМА. Как видно из табл. 2, интегральная интенсивность полосы 2700 см–1 (карбонил–гидроксил) для непрогретого ИПК (А/МВЭМА-8/23) выше, чем для образцов, подвергутых термообработке (А/МВЭМА-8/37 и А/МВЭМА-8/80), что, вероятно, определяется большим содержанием воды в этом образце.
Таблица 2.
Характеристика полос поглощения карбоксильных и гидроксильных групп в ИК-спектрах ИПК А/МВЭМА-8, температура обработки и содержание воды
| Температура обработки, °С | Интегральная интенсивность полосы | Содержание воды (данные ДСК), мас.% | |||
|---|---|---|---|---|---|
| 1596 см–1 [СОО–] | 1708–1710 см–1 [СООН…СООН] | 3250–3414 см–1 [–OH] | 2700 см–1 [C=O…OH] | ||
| 23 | 1.67 | 3.79 | 6.04 | 0.97 | 10.4 |
| 37 | 1.69 | 2.95 | 5.39 | 0.64 | 9.0 |
| 80 | 1.55 | 3.0 | 4.75 | 0.67 | 7.7 |
Действительно, согласно данным ДСК, содержание воды в образце уменьшается после прогревания, однако разница не превышает 1–2%. Вероятно, в образцах, не подвергнутых термообработке, формирование Н-связей происходит с участием воды. Это предположение подтверждается поведением полосы гидроксилов (3400 см–1). В спектрах прогретых ИПК она уширяется и здесь появляется второй максимум при 3200 см–1, причем интенсивности полос уменьшаются. Наблюдаемые изменения могут свидетельствовать об увеличении вклада Н-связей, что хорошо согласуется со смещением полосы карбонилов в коротковолновую область и данными ДСК о содержании воды в ИПК. Аналогичные закономерности наблюдаются и для гелей, полученных с сополимером более высокой молекулярной массы – МВЭМА-41.
Анализ спектральных данных позволяет заключить, что именно водородные связи обеспечивают формирование ИПК, причем увеличение температуры термической обработки влечет перестройку их структуры. Вероятно, прогревание при 37°С приводит к частичному разрушению Н-связей, а при температуре 80°С они формируются вновь, но с участием меньшего количества молекул воды. Заметим, ранее предполагалось, что именно выдержка смесей полисахаридов с МВЭМА при повышенных значениях температуры обеспечивает формирование интерполимерной сетки, причем за счет образования эфирных связей. Так, по мнению авторов работы [11], уширение полосы карбонилов и появление новой полосы 1780 см–1 при прогревании при 80°С пленок, полученных из смесей гиалуроновой кислоты и МВЭМА в гидролизованной форме, свидетельствует об образовании эфирных связей между карбоксильными группами МВЭМА и гидроксильными группами кислоты. Этот вывод, однако, представляется неубедительным, поскольку появление полосы 1780 см–1 в ИК-спектрах указывает на образование ангидридных циклов МВЭМА в результате его чаcтичной дегидратации при прогревании, а более детального рассмотрения областей спектра, отвечающих эфирным связям, в работе [11] не приводится. По-видимому, в данных системах формирование интерполимерной сетки происходит также за счет водородных связей.
Возможность использования полученных ИПК в качестве систем доставки лекарственных веществ изучали на примере лидокаина, находящего применение при лечении раневых поверхностей. Профили выделения лидокаина из ИПК, различающихся длиной цепи сшивающего агента и температурой термической обработки, в среды, моделирующие физиологические условия, приведены на рис. 3.
Рис. 3.
Профиль выделения лидокаина из геля А/МВЭМА-8/23 (1), А/МВЭМА-41/23 (2), А/МВЭМА-41/80 (3) и А/МВЭМА-8/80 (4) в фосфатном буфере с рН 7.2 при комнатной температуре.
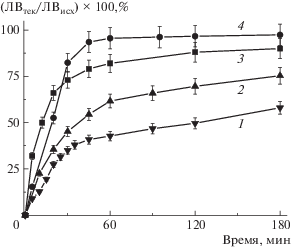
Для ИПК, не подвергшихся термообработке, характерны невысокие значения скорости выделения лекарственных веществ. Минимальные значения скорости выделения наблюдались для ИПК с низкомолекулярным образцом МВЭМА-8. Увеличение молекулярной массы МВЭМА приводило к некоторому ускорению выделения лидокаина. Так, количество выделившегося лидокаина для образцов А/МВЭМА-8 и А/МВЭМА-41 составило за 30 мин 30 и 40%, а за 60 мин 40 и 60% соответственно.
Прогревание образцов при температуре 80°С приводило к ускорению выделения лидокаина, особенно выраженному для образцов с МВЭМА-8. Количество лидокаина, выделившегося из композиции за 30 мин, для А/МВЭМА-8/80 было в ~2.7 раза выше, чем для непрогретых А/МВЭМА-8/23. Для образцов с более высомолекулярным МВЭМА-41 прослеживалась аналогичная тенденция, однако скорость выделения лидокаина для прогретых образцов была выше только в ~1.7 раза.
Как упомянуто выше, оба компонента, входящие в состав ИПК, являются слабыми полиэлектролитами, степень ионизации карбоксильных групп которых зависит от рН среды. Это обстоятельство может быть решающим в функционировании таких систем, обеспечивая, например, преимущественный выход лекарственного вещества в слабокислых средах. Возможность реализации рН-зависимого поведения гидрогелей была исследована на примере выделения лидокаина из композиций А/МВЭМА-41/23 в нейтральных и слабокислых средах. Из табл. 3 видно, что такое поведение действительно реализуется. На начальном этапе (до 30 мин) скорость выделения лидокаина в слабокислых средах в ~2 раза выше, чем в нейтральных. Затем выделение лекарственного вещества замедляется, но остается выше в слабокислых средах.
Таблица 3.
Кинетика высвобождения лидокаина гидрохлорида из ИПК А/МВЭМА-41/23 в нейтральных и слабокислых средах
| Время, мин | Количество лекарственного вещества, мас.% | |
|---|---|---|
| рН 7.2 | рН 5.5 | |
| 10 | 22.5 ± 1.3 | 39.9 ± 1.9 |
| 20 | 35.5 ± 1.8 | 71.7 ± 3.6 |
| 30 | 45.2 ± 2.3 | 81.2 ± 4.1 |
| 45 | 54.5 ± 2.7 | 88.2 ± 4.4 |
| 60 | 61.6 ± 3.7 | 91.6 ± 4.6 |
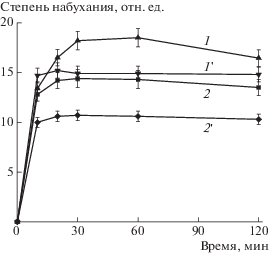
Данные экспериментов по кинетике выделения лидокаина хорошо коррелируют с результатами изучения кинетики набухания ИПК в средах с разными значениями рН. Набухание ИПК в нейтральных средах происходит постепенно, а в кислых средах ИПК практически сразу набухают до максимальных значений (рис. 4). При этом степени набухания гелей в кислых средах были ниже, чем в нейтральных для всех исследованных образцов независимо от температуры обработки и молекулярной массы МВЭМА. Наблюдаемое уменьшение степени набухания ИПК, вероятно, определяется изменением плотности сшивки. При понижении рН среды степень ионизации карбоксильных групп как в альгинате, так и в МВЭМА уменьшается, а количество незаряженных групп соответственно возрастает, в результате чего количество водородных связей увеличивается, и ИПК приобретают более плотную структуру. Изменение структуры ИПК, в свою очередь, определяет рост скорости выделения лидокаина в слабокислых средах, позволяя тем самым обеспечить достаточное выделение лекарства в области нарушения кожного покрова, которое всегда сопровождается подкислением поверхности.
ЗАКЛЮЧЕНИЕ
Получены гидрогели интерполимерных комплексов на основе альгината натрия и сополимера метилвинилового эфира с малеиновым ангидридом. Изучено влияние состава и режимов термической обработки ИПК, а также характеристик исходных полимеров на свойства гелей – степень набухания, стабильность и скорость выделения модельного лекарственного вещества. Исследование структуры гидрогелей методом ИК-спектроскопии показало, что ИПК образуются за счет формирования водородных связей между гидроксильными группами альгината и карбоксильными группами сополимера МВЭМА. Тип водородных связей и их количество определяют степень набухания ИПК и кинетику выделения лекарственного вещества. Результаты исследования могут быть основой для создания рН- чувствительных систем доставки лекарственных средств.
Работа выполнена в рамках Госзадания ИНХС РАН.
Авторы выражают благодарность сотрудникам Акционерного общества “Институт пластмасс” Н.Н. Молотковой и И.М. Шелониной за измерение и анализ ИК-спектров; а также сотрудникам Института нефтехимического синтеза им. Топчиева РАН Г.Н. Бондаренко за обсуждение результатов спектральных исследований и Г.А. Шандрюку за проведение ДСК-исследований.
Список литературы
Kuen Yong Lee, Mooney D.J. // Prog. Polym. Sci. 2012. V. 37. № 1. P. 106. https://doi.org/10.1016/j.progpolymsci.2011.06.003
Stokke B.T., Draget K.I., Smidsrød O., Yuguchi Y., Urakawa H., Kajiwara K. // Macromolecules. 2000. V. 33. № 5. P. 1853.
Draget K.I., Skjak-Braek G., Smidsrod O. // Carbohydr. Polym. 1994. V. 25. P. 31.
Gorshkova M.Yu., Volkova I.F., Vanchugova L.V., Va-luev L.I., Valueva T. // Appl. Biochem. Microbiol. 2019. V. 55. № 4. P. 371.
Xiaokun Wang, Tong Hao, Jing Qu, Changyong Wang, Haifeng Chen // J. Nanomaterials. V. 2015. Article ID 970619.
Ariel W., Chan R., Neufeld J. // Biomaterials. 2009. V. 30. № 30. P. 6119.
Himadri Sekhar Samanta Samit, Kumar Ray // Carbohydr. Polym. 2014. V. 99. P. 666.
Benettayeb A., Guibal E., Morsli A., Kessas R. // Chem. Eng. J. 2017. V. 316. P. 704.
Mollah M.Z.I., Mubarak A. Khan, Hoque M.A. // Carbohydr. Polym. 2008. V. 72. № 2. P. 349.
Wu Mingyue, Hu Xiangming, Zhang Qian, Zhao Yanyun, He Zhenglong // J. Cleaner Prod. 2020. V. 259. P. 120870.
Larraneta E., Megan Henry M., Irwin N.J., Trotter J., Perminova A.A. // Carbohydr. Polym. 2018. V. 181. P. 1194.
Dhubojyoti Mukherjee, Md. Azamthulla, S. Santhosh, Guru Dath, Arijit Ghosh, Rahul Natholia, J. Anbu, B. Venkatesh Tja, K. Mohammed Mazammil // J. Drug Delivery Sci. Tech. 2018. V. 46. P. 498.
Anqi Li, Tian Gong, Xi Yang, Yurong Guo // Int. J. Biol. Macromol. 2020. V. 151. P. 257.
Yuge Niu, Qi Xia, Na Li, Ziyuan Wang, Liangli Lucy Yu. // Food Chem. 2019. V. 270. P. 223.
Rapee Khlibsuwan, Watcharee Khunkitti, Thaned Pongjanyakul // Int. J. Biol. Macromol. 2020. V. 1481. P. 1061.
Xuhong Guo, Frank Deng, Li Li, Robert K. Prud’homme // Biomacromol. 2008. V. 9. № 6. P. 1637.
Calo E., Barros J., Ballamy L., Khutoryanskiy V.V. // RSC Adv. 2016. V. 6. P. 105487.18.
Lebedeva T.L., Feldstein M.M., Kuptsov S.A., Platé N.A. // Polymer Science A. 2000. V. 42. № 9. P. 989.
Фельдштейн М.М., Лебедева Т.Л., Шандрюк Г.А., Котомин С.В., Купцов С.А., Игонин В.Е., Гроховская Т.Е., Куличихин В.Г. // Высокомолек. соед. A. 1999. Т. 41. № 8. С. 1316.
Белами Л. Новые данные по ИК-спектроскопии сложных молекул / Пер. с англ. В.М. Акимова, Э.Г. Тетерина. М.: Мир, 1971.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)