

Высокомолекулярные соединения (серия Б), 2020, T. 62, № 6, стр. 403-415
НЕКОТОРЫЕ АСПЕКТЫ РАДИКАЛЬНОЙ ПОЛИМЕРИЗАЦИИ ПОД ДЕЙСТВИЕМ Cu(0)
М. Ю. Заремский a, *, Е. А. Букин a, К. О. Минеева a, С. Б. Зезин a
a Московский государственный университет имени М.В. Ломоносова. Химический факультет
119992 Москва, Ленинские горы, Россия
* E-mail: zaremski@mail.ru
Поступила в редакцию 17.04.2020
После доработки 26.06.2020
Принята к публикации 10.07.2020
Аннотация
Установлены некоторые количественные параметры, характеризующие SET-полимеризацию метил-акрилата с участием Сu(0): кинетические порядки скорости полимеризации по концентрации мономера и инициатора, а также значения константы скорости инициирования в различных условиях. Разработана методика раздельного определения ионов Cu(I) и Cu(II) по ходу полимеризации. Предложена новая эффективная форма катализатора – медное зеркало, позволяющая многократно повысить удельную скорость полимеризации и понизить остаточное содержание меди в полимере.
ВВЕДЕНИЕ
Одним из основных методов контролируемого синтеза полимеров является радикальная полимеризация с переносом атома галогена (ATRP) [1]. Среди различных способов ее воплощения наиболее интересным и наименее изученным представляется SET-полимеризация (Single Еlectron Тransfer – полимеризация с переносом одного электрона), также называемая Cu(0)-mediated RDRP (Reverse Deactivation Radical Polymerization) с участием нуль-валентной меди в качестве катализатора [2]. Реакция по этому механизму протекает достаточно быстро при низких значениях температуры с высоким выходом, она устойчива к наличию кислорода в системе и может осуществляться в воде, т.е. удобна в технологическом отношении. Кроме того, процесс отличается высокой степенью чистоты продукта.
SET-полимеризация проводится в присутствии гетерогенных катализаторов. К настоящему моменту наиболее устоявшимися формами медного катализатора, использующимися в этом процессе, считаются порошок и проволока. Медный порошок, хотя и обладает бόльшей удельной поверхностью, и, соответственно, выше скоростью инициирования, тем не менее демонстрирует, что полимеризация в его присутствии труднее контролируется и осложняется невоспроизводимостью [3]. Напротив, медная проволока отличается возможностью более простого контроля молекулярной массы полимера. Проволоку легче подготовить к реакции, она меньше загрязняет полимер ионами меди. Также описано использование наночастиц Cu(0) в качестве катализатора полимеризации. Эта форма отличается самой высокой скоростью полимеризации, например для метилакрилата в ДМСО реакция может пройти с конверсией 80% всего за 5 мин. Однако такой “сверхбыстрый” процесс нестационарен, характеризуется разогревом системы (вплоть до вскипания), а следовательно, нетехнологичен [4]. Поэтому медная проволока стала основной формой катализатора в SET-процессе [5].
Несмотря на то что SET-полимеризация была открыта в 2006 году [6], механизм реакции до сих пор выступает объектом дискуссии. Многие авторы, как правило, ограничиваются определением удельной скорости процесса, и основные кинетические параметры остаются неизвестными.
В настоящей работе определены некоторые количественные параметры, характеризующие полимеризацию метилакрилата с участием Сu(0), и предложена новая эффективная форма катализатора – медное зеркало.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Медную проволоку диаметром 0.15 мм механически дополнительно зачищали непосредственно перед полимеризацией. Мономер метилакрилата (МА, 99%) фирмы “Acros” и CuBr (“Sigma-Aldrich”) очищали по стандартной методике. Метил-2-бромпропионат (МБП, 9%) производства “Sigma-Aldrich” перегоняли в вакууме при 51 град/19 мм рт. ст., гексаметил-трис-(2-аминоэтил)амин (Mе6ТРЕН) синтезировали из ТРЕН – трис-(2-аминоэтил)амин (“Sigma-Aldrich”, 99%) по методике [7]. ДМСО, CuBr2, Ag2O, NaOH, HNO3 (конц.), глюкоза, CuSO4 ⋅ 5H2O, N2H4, ионообменная смола “Dowex”, тетрагидрат тартрата калия–натрия квалификации ч.д.а. и H2SO4 (конц., х.ч.) дополнительной очистке не подвергали.
Получение Сu-зеркала в две стадии
Cтеклянную ампулу тщательно промывали раствором хромовой смеси, дистиллированной водой и спиртом. Реакцию проводили по методике [8]. К 0.17 г Ag2O по каплям прибавляли раствор аммиака 25% при постоянном перемешивании до исчезновения крупных частиц вещества11. Раствор разбавляли водой до 20 мл. К 8 мл раствора глюкозы 4.7% в дистиллированной воде вводили одну каплю концентрированной азотной кислоты и кипятили 2 мин, после чего охлаждали и добавляли равный объем этилового спирта. Приготовленные растворы смешивали в соотношении 10 : 1. В ампулу вливали 0.5–1.0 мл реакционной смеси, нагревали на водяной бане при 50–60°С до плотного зеркального покрытия на внутренней поверхности ампулы. Образованное серебряное зеркало промывали дистиллированной водой, затем этиловым спиртом.
На второй стадии смешивали 6 мл раствора CuSO4 7% с 6 мл раствора, полученного растворением 8 г тетрагидрата тартрата калия–натрия и 2.6 г гидроксида натрия в 20 мл воды, и с 6 мл раствора формалина 37% в указанном порядке – все тщательно перемешивали. Смесь переносили в ампулу с нанесенным серебряным зеркалом и выдерживали 2–3 мин. Полученное зеркало промывали дистиллированной водой, а после – этиловым спиртом [9].
Получение Cu-зеркала в одну стадию с использованием гидразина
Реакцию осуществляли по методике [10]. Тщательно очищенные в хромовой смеси ампулы промывали дистиллированной водой, спиртом и хлористым метиленом, после чего высушивали. Далее опускали на 1–2 мин в раствор хлорида олова(II) 10%, подкисленного соляной кислотой, и тщательно промывали. Затем ампулы помещали в раствор, полученный растворением оксида серебра(I) 0.05 г в двух каплях концентрированной азотной кислоты с разбавлением водой до 100 мл, и, не промывая, высушивали при 40–100°C.
Смешивали 1 мл аммиачного раствора сульфата меди 4.3%, 2 мл раствора N2H4 0.15% и 1 мл раствора гидроксида натрия 2%. Полученную смесь заливали в подготовленные ампулы, нагревали их на водяной бане при 70–80°C в течение 15–20 мин до образования тонкой медной зеркальной пленки.
Получение Cu-зеркала в одну стадию с использованием формалина
За основу взяли методику, описанную в работе [11], и модифицировали ее. Стеклянные ампулы подвергали ультразвуковой обработке в водной среде в течение 5 мин с частотой 5 кГц. Затем обезжиривали под действием 2–3% раствора NaOH также в течение 5 минут при 60°С. Ампулы помещали в раствор азотной кислоты 30% на одну минуту при комнатной температуре. Поверхность стекла царапали стеклянной палочкой. После чего ампулу при комнатной температуре опускали на 1–2 мин в раствор хлорида олова(II) 10%, подкисленный соляной кислотой, и тщательно промывали. Далее поверхность обрабатывали раствором нитрата серебра(I) 1% в течение 1–2 мин при комнатной температуре. Затем ампулу, не промывая, помещали в метиловый спирт на одни сутки и высушивали в вакууме.
К 9 мл раствора, полученного растворением 0.075 г CuSO4 ⋅ 5H2O, 0.112 г динатриевой соли этилендиаминтетраацетата (ЭДТА), 0.04 г полиэтиленгликоля (Mn = 1 × 103) и 0.001 г 2,2-дипиридила в 20 мл дистиллированной воды, приливали 1 мл раствора формалина 40%, затем 9 мл раствора NaOH 2.3%. Смесь переливали в ампулу и помещали на водяную баню с температурой 70°С на 1–2 мин. После завершения процесса меднения ампулу промывали 3 раза дистиллированной водой, после чего погружали в метиловый спирт.
SET-полимеризация
В ампулу помещали медную проволоку длиной 15 см и диаметром 0.15 мм, тщательно зачищенную и скрученную в спираль длиной, равной высоте занимаемой жидкости. В типичном опыте в ампулу заливали 0.4 мл МА и 0.2 мл ДМСО, добавляли инициатор МБП и лиганд Mе6ТРЕН так, чтобы их концентрация составляла 0.03 и 0.003 моль/л соответственно. Полученную смесь вакуумировали путем трехкратного цикла замораживание–дегазация–размораживание. Затем ампулу запаивали. Полимеризацию проводили при 40°C. Продукт растворяли в бензоле и лиофильно высушивали в вакууме.
Аналогично осуществляли полимеризацию под действием медного зеркала. Для этого в ампулу диаметром 7 мм с нанесенным медным покрытием приливали 0.6 мл указанной выше смеси.
Очистка полимеров
Полимер в количестве 250–350 мг растворяли в 4 мл диоксана и добавляли 0.16 г смолы “Dowex”. Смесь перемешивали в течение 4 ч, после чего декантировали от смолы. Полученный раствор пропускали через хроматографическую колонку с основной окисью алюминия (Brockmann I, “Sigma-Aldrich”).
Инструментальные методы исследования
Рентгенофазовый анализ (РФА) полученных покрытий проводили с помощью дифрактометра “URD-6” (Германия) с использованием монохроматического излучения CuKα с λ = 1.54 Å.
Спектрофотометрические измерения выполняли на спектрофотометре “Ultra Spec 1100 Pro” (“General Electric Housecare”, Англия) с использованием кварцевых кювет с длиной оптического пути 1 см. Для расчета коэффициентов экстинкции готовили растворы CuBr2 (в интервале концентрации 6.8–0.7 мМ) в смеси МА : ДМСО = 2 : 1 по объему в присутствии 3 мМ Me6ТРЕН, а также раствор CuBr (6.8–1.7 мМ) в тех же условиях. Для измерения оптической плотности полимеризата готовили 2 мл рабочего раствора с концентрацией МБП и Mе6ТРЕН 30 и 3 мМ соответственно. Смесь дегазировали продуванием аргона, после чего с помощью шприца в инертной атмосфере переносили в герметичную кювету, содержащую спираль высотой 2.8 см и диаметром 0.5 см, свитую из медной проволоки длиной 30 см и диаметром 0.15 мм. Скорость записи спектра составляла 500 нм/мин.
Атомно-эмиссионную спектроскопию (АЭС) с индукционно связанной плазмой проводили на приборе “Agilent 5100 ICP-OES” (США) при рабочей длине волны 324.754 и 327.395 нм. Образцы полимеров помещали в 100 мл стеклянные стаканы. Добавляли 40 мл конц. H2SO4. Смесь нагревали при 25°C до образования устойчивой насыщенной черной окраски. Затем в стакан по каплям осторожно добавляли 3 мл конц. HNO3. После полного обесцвечивания черной окраски (допускалась светло-желтая окраска) раствор упаривали до 1–2 мл. Смесь охлаждали, добавляли 1–2 мл деионизированной воды (18.2 МОм/см) и продолжали упаривать до прекращения выделения бурых паров. Раствор переносили в пикнометры объемом 5.0 мл.
Градуировочную зависимость строили для серии растворов 10.0, 5.0, 3.0, 1.0, 0.5, 0.1 мкг/мл Cu.
Исследование кинетики радикальной полимеризации осуществляли калориметрическим методом на дифференциальном автоматическом микрокалориметре ДАК-1-1а в режиме прямой регистрации скорости тепловыделения dQ/dt в изотермических условиях при 40°С. Так, в рабочую ячейку помещали ампулу с исследуемой смесью, в компенсационную ячейку сравнения – ампулу с приблизительно равным количеством уже заполимеризовавшегося мономера. Количественную обработку данных проводили с помощью ЭВМ по программе, основанной на методе Симпсона; в расчетах использовали значение ΔH полимеризации метилакрилата – 17600 кал/моль [12].
Для определения скорости инициирования готовили раствор МБП (10–5–10–4 моль/л), Mе6ТРЕН (10 : 1) и фенил-N-трет-бутилнитрона (0.5–1.5 моль/л) в смеси МА : ДМСО = 2 : 1 по объему. Смесь в количестве 30 мкл помещали в ЭПР-ампулу и вакуумировали, как описано выше. К смеси добавляли отрезок медной проволоки 15.0 × 0.15 мм в вакууме непосредственно в ячейке прибора. Спектры записывали при комнатной температуре на радиоспектрометре РЕ-1307. В качестве эталона использовали спектр 0.1 мМ раствора ТЕМПО в циклогексане, записанного в тех же условиях.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Особенности кинетики полимеризации МА под действием Cu(0)
Радикальная полимеризация по SET-механизму под действием Cu(0) – это цепной процесс, включающий перечисленные ниже основные элементарные акты.
Инициирование – окислительно-восстановительная реакция между алкилгалогенидом и медью:
где RBr – инициатор, L – лиганд.Диспропорционирование и сопропорционирование между ионами меди:
Рост цепи, который происходит, как в обычной радикальной полимеризации:
Здесь $P_{n}^{\centerdot }$ – полимерный радикал, M – мономер.
Обратимый обрыв цепи – окислительно-восстановительная реакция между радикалом роста и CuBr2:
(4)
${\text{P}}_{n}^{\centerdot } + {\text{CuB}}{{{\text{r}}}_{2}}{\text{L}} \rightleftarrows {{{\text{P}}}_{n}}{\text{Br}} + {\text{CuBrL}}$Необратимый обрыв радикалов роста
(5)
${\text{P}}_{n}^{\centerdot } + {\text{P}}_{m}^{\centerdot } \to {{{\text{P}}}_{n}}{\kern 1pt} - {\kern 1pt} {{{\text{P}}}_{m}}$Основные исследования кинетики полимеризации МА в условиях SET-процесса были проведены с помощью гравиметрического определения выхода полимера. В большинстве случаев выполняется закон первого порядка по мономеру М, что графически отображается прямой линией в координатах ln([M]0/[M]) = f(t) [5].
Калориметрический эксперимент с непрерывной записью скорости полимеризации показал, что это не совсем так. Действительно, в основной области превращения полимеризация протекает с практически постоянной приведенной (деленной на текущую концентрацию мономера) скоростью (рис. 1), что характерно для процесса первого порядка. Более того, при концентрации мономера 4.4–8.8 моль/л приведенные значения скорости полимеризации, практически совпадают, что и должно наблюдаться, если скорость полимеризации прямо пропорциональна концентрации мономера. Однако при понижении концентрации мономера <2.2 моль/л уменьшение скорости полимеризации оказывается более сильным, чем по реакции первого порядка. Такое отклонение отмечается как при “естественном” расходе мономера по ходу полимеризации, так и при “искусственном” разбавлении мономера растворителем. Иными словами, полимеризация протекает в затухающем режиме.
Рис. 1.
Дифференциальные кинетические кривые полимеризации МА при [MA] = 8.8 (1), 6.6 (2), 4.4 (3) и 2.2 моль/л (4). [MБП] = 18 мМ, [Mе6ТРЕН] = 1.8 мМ, Cu-проволока 150 × 0.15 мм.
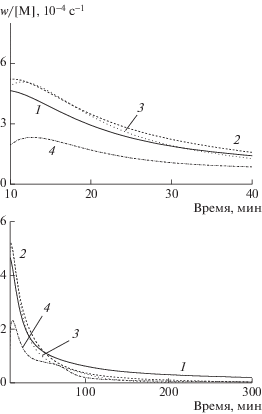
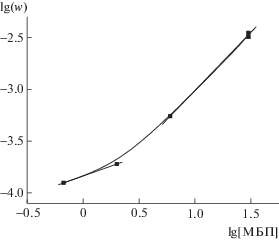
Хорошо известно, что порядок скорости полимеризации по площади поверхности меди близок к 0.5 (усреднение многочисленных данных дало значение 0.44 для Mе6ТРЕН [5] и 0.47 для трис-(2-пиридилметил)амина [13]). Только данных о влиянии концентрации инициатора на скорость полимеризации очень мало. Попытка их обобщить, предпринятая в работе [14], показывает, что зависимость скорости полимеризации от концентрации инициатора удовлетворяет как величине порядка 1/3 (такая величина характерна для “традиционной” cхемы Фишера–Фукуды [15]), так и 1/2. Если проанализировать данные работы [16], то оказывается, что n – величина переменная: она меняется от значения близкого к 1/3 при низкой концентрации инициатора до близкого к единице при высокой. Исследования настоящей работы подтверждают такую зависимость. В интервале концентрации инициатора МБП 0.67–30 мМ величина n меняется от 0.38 до 1.10 (рис. 2). Очевидно, что при этом меняется также механизм полимеризации.
Рис. 2.
Зависимость скорости полимеризации от концентрации инициатора в логарифмических координатах.
Ранее было показано [15], что порядок по инициатору 1/3 характерен для кинетики псевдоживой радикальной полимеризации в условиях, пока система не вышла в стационарное состояние. В этих условиях растущие цепи обрываются как обратимо по реакции с Cu(II) (4), так и необратимо вследствие квадратичного обрыва (5).
Если полимеризация протекает в указанном режиме, то следует ожидать накопление Cu(II) по закону
(6)
$[{\text{CuB}}{{{\text{r}}}_{2}}] = {{(3{{K}^{2}}{{k}_{t}}[{\text{I}}]_{0}^{2})}^{{1/3}}}{{t}^{{1/3}}},$Чтобы проверить эту закономерность, изучено накопление ионов меди в полимеризате на стадии выхода полимеризации на стационарность. Была разработана спектрофотометрическая лабораторная методика раздельного определения ионов одно- и двухвалентной меди непосредственно в полимеризационной системе.
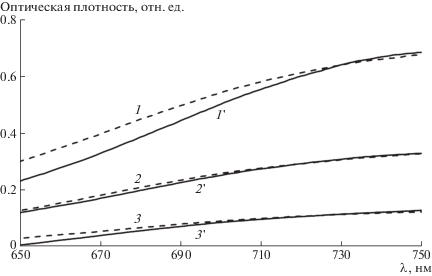
Для количественного определения использованы два известных из литературы факта: во-первых, в области ~900 нм свет поглощают только ионы Cu(II), а поглощение ионами Cu(I) пренебрежимо мало; во-вторых, существует изобестическая точка для ионов меди(I) и (II), в которой значения оптической плотности их растворов равны [6]. Зная коэффициенты экстинкции в этих двух точках, несложно рассчитать концентрацию обоих компонентов. Экспериментально (рис. 3) найдено, что изобестическая точка находится при λ = 730 нм. Значения мольных коэффициентов экстинкции в указанных точках составляют ε900 = (336 ± 5) и ε730= (176 ± 7) л/моль см.
Рис. 3.
Спектры поглощения растворов комплексов CuBr ⋅ Mе6ТРЕН (1–3) и CuBr2 ⋅ Mе6ТРЕН (1'–3') в ДМСО при их концентрации 6.9 (1, 1') 3.4 (2, 2') и 1.7 мМ (3, 3').
Изменение спектров полимеризата представлено на рис. 4. Видно, что оптическая плотность в интервале 720–900 нм повышается по ходу полимеризации, это связано с накоплением в системе ионов меди. Результаты обработки спектров даны на рис. 5, из которого следует, что практически с самого начала полимеризации в системе присутствуют как ионы меди(I), так и меди(II), причем последних всегда больше, чем первых. На всех полученных кривых можно выделить две кинетические области – короткую начальную (~8 мин) и основную. В начальной области концентрация Cu(I) растет (рис. 5б), а концентрация Cu(II) меняется сложным образом. В основной части кинетической кривой концентрация Cu(I) практически не меняется, а Cu(II) растет.
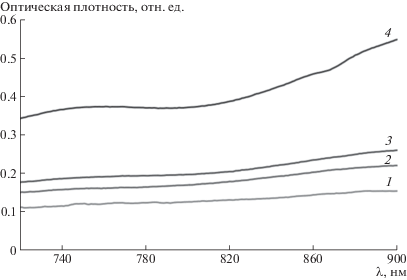
Рис. 5.
а, б – Изменение [CuBr ⋅ Mе6ТРЕН] (1–3) и [CuBr2 ⋅ Mе6ТРЕН] (4–6) по ходу полимеризации в смесях, подвергнутых предварительной дегазации в течение 5 (1, 4), 10 (2, 5) и 15 мин (3, 6); б – результаты обработки спектров (1–3) в увеличенном масштабе.
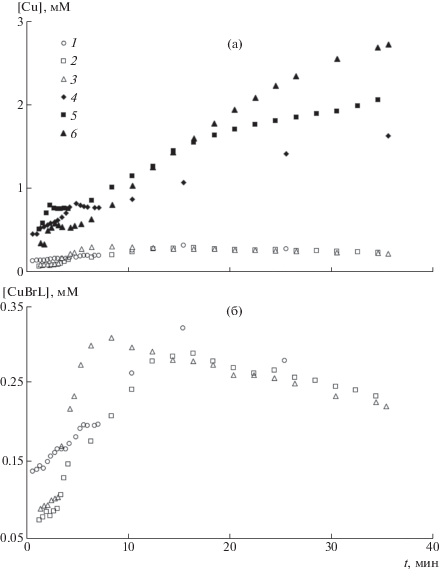
Изменение концентрации ионов меди в начальной кинетической области можно связать с периодом распада инициатора (см. ниже). Интенсивное окисление меди Cu(0) приводит к тому, что накопление Cu(I) в этой области превалирует над расходом по реакции диспропорционирования (2). Сложное изменение концентрации Cu(II), возможно, связано с нестационарной кинетикой быстрого зарождения и гибели первичных радикалов в начальный период процесса.
Во второй, основной, кинетической области концентрация Cu(I) в системе переходит в квазистационарное состояние, и постепенно убывает (рис. 5). Это означает, что скорость образования Cu(I) (1) и его расходования по реакции диспропорционирования (2) становятся примерно равными. Как и ожидалось, концентрация Cu(II) в этой области хорошо описывается законом (6), т.е. возрастает пропорционально кубическому корню из времени полимеризации (рис. 6) [17]. Причина такого роста обусловлена квадратичным обрывом растущих цепей (5). Два макрорадикала, взаимодействуя друг с другом, образуют “мертвый” полимер, при этом два иона Cu(II) остаются в растворе (4). Чем больше накапливается ионов Cu(II), тем меньше вероятность квадратичного обрыва и ниже скорость их последующего накопления.
Рис. 6.
Зависимость [CuBr2 · Mе6ТРЕН] от времени полимеризации в координатах уравнения (6) в смесях, подвергнутых предварительной дегазации в течение 5 (1), 10 (2) и 15 мин (3). R2 = 0.993 (1), 0.9842 (2), 0.99711 (3).
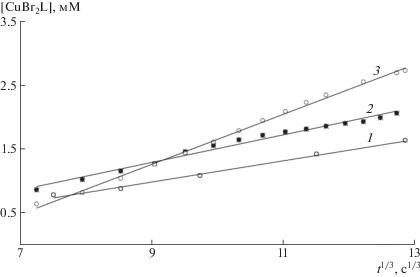
Полученные результаты позволяют сделать два важных вывода. Во-первых, не только в самом начале полимеризации, но, по крайне мере, в первые 40 мин роль необратимого квадратичного обрыва весьма ощутима. Во-вторых, в продолжающейся дискуссии о механизме активации в SET-полимеризации [5, 18, 19] данные настоящей работы подтверждают правоту мнения [18, 19] о заметном вкладе ионов Cu(I) в активацию полимеризации, а также опровергают предположение о мгновенном и количественном диспропорционировании Cu(I) с образованием Cu(0) и Cu(II) [6].
Исследование зарождения радикалов МА под действием Cu(0)
Скорость инициирования (1) полимеризации под действием медной проволоки позволяет определить, насколько быстро зарождаются макромолекулы в SET-процессе. Она была измерена по ранее разработанной методике конкурентного ингибирования [20]. Для чего предварительно были проведены исследования зарождения радикалов непосредственно в полимеризате в присутствии многократного избытка спиновой ловушки фенил-N-трет-бутилнитрона (ФБН):
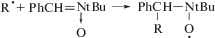
Методом ЭПР измерялось накопление нитроксилов – аддуктов нитрона с первичными радикалами на начальной стадии, когда расходом компонентов инициирующей системы можно пренебречь. При выборе условий необходимо было учитывать, что реакция протекает с высокой скоростью, причем ее продукты – ионы Cu(II) сами являются сильными ингибиторами, т.е. конкурентами ФБН. Ингибиторами представляются и нитроксильные аддукты нитрона, получающиеся по реакции (7). Кроме того, активность нитроксилов и ионов Cu(II) в реакции с первичными радикалами R на два порядка выше активности нитрона. Нельзя исключить и побочные реакции нитрона с комплексами одно- и двухвалентной меди, образующимися в системе. Поэтому измерения скорости инициирования данным методом достоверны только в условиях избытка ФБН на несколько порядков по отношению к нитроксилам и Cu(II). Графическим индикатором достоверности измерений служит линейный характер возрастания концентрации нитроксильных аддуктов нитрона во времени, с тангенсом угла наклона, не зависящим от концентрации нитрона. Опытным путем установлено, что этим требованиям удовлетворяет концентрация МБП 10–5–10–4 моль/л и ФБН 0.5–1.5 моль/л.
Кинетические кривые накопления аддуктов нитрона продемонстрированы на рис. 7, а результаты расчета константы инициирования приведены в табл. 1. Оказалось, что реакция инициирования – это реакция первого порядка по концентрации инициатора, о чем свидетельствует значение удельной (отнесенной к концентрации инициатора) скорости, а также линеаризация скорости в координатах уравнения первого порядка.
Рис. 7.
Накопление аддуктов нитрона в системе МА–МБП–Ме6ТРЕН–Сu(0)–ФБН. [МБП] = 0.140 (1), 0.090 (2), 0.070 (3), 0.063 (4), 0.043 (5), 0.039 (6) и 0.023 мМ (7).
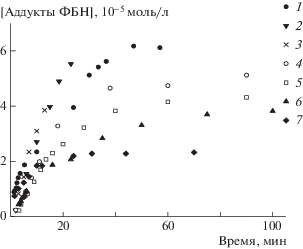
Таблица 1.
Кинетические параметры инициирования в системе МА–ДМСО–МБП–Mе6ТРЕН–Cu(0)–ФБН
| [МБП], 10–5 моль/л | Скорость инициирования, (10–6 моль/л)/мин | Удельная скорость w/[МБП], 10–2 мин–1 | Константа инициирования, 10–4 см/с |
|---|---|---|---|
| 7.5 | 3.8 | 5.0 | 3.7 |
| 6.0 | 2.1 | 3.5 | 2.5 |
| 4.3 | 1.7 | 4.6 | 2.9 |
| 3.9 | 1.6 | 4.1 | 3.0 |
| 2.3 | 1.2 | 5.2 | 3.8 |
| 1.4 | 0.7 | 5.0 | 3.6 |
| Средняя величина | 4.6 ± 0.6 | 3.2 ± 0.5 | |
Параллельно был проведен другой эксперимент, в котором в качестве сильного ингибитора–конкурента брали нитроксил ТЕМПО (8). В этом случае концентрацию инициатора можно повысить до величин, используемых при полимеризации. Скорость инициирования в присутствии ТЕМПО определяется по линейному участку кривой его расхода (рис. 8):
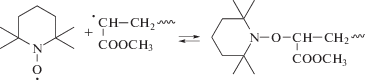
Рис. 8.
Кинетические кривые расхода [ТЕМПО]0 = = 3.0 (1) и 1.5 мМ (2) в системе МА–ДМСО–МБП – 30 мМ, Mе6ТРЕН – 3 мМ, Cu(0) – ТЕМПО.
В системе 30 мМ MБП–3 мМ Mе6ТРЕН–8.2 моль/л МА–ДМСО–Cu(0) скорость инициирования равна 2.9 × 10–3 и 2.4 × 10–3 моль/л мин при концентрации ТЕМПО 3.0 и 1.5 мМ соответственно. Это отвечает величине константы инициирования 3.1 × 10–4 и 2.6 × 10–4 см/с. Следовательно, оба способа измерения дают совпадающие значения. Период полупревращения инициатора, отвечающий данным константам, составляет 13 мин.
Полученные в данной работе величины лежат в интервале литературных оценок от 1 × 10–4 [21] до 1 × 10–3 см/с [18], сделанных путем кинетического моделирования.
Для исследования влияния среды на скорость инициирования был выбран ряд растворителей, различающихся полярностью. Результаты измерений приведены в табл. 2.
Таблица 2.
Константа инициирования в системе растворитель–МБП–Mе6ТРЕН–Cu(0)–ФБН
| Растворитель | Диэлектрическая проницаемость | Удельная скорость w/[МБП], 10–2 мин–1 | Константа инициирования, 10–4 см/с |
|---|---|---|---|
| Диоксан (абсолютный) | 2.2 | 0.6 | 0.44 |
| Бензол | 2.3 | 1.6 | 1.15 |
| Толуол | 2.4 | 0.5 | 3.60 |
| Гептан : бутанон (8 : 7) | 5.4 | 0.3 | 2.20 |
| Спирт (абсолютный) | 25.0 | 2.0 | 1.40 |
| Ацетонитрил | 37.0 | 3.0 | 2.20 |
| ДМСО | 49.0 | 4.5 | 3.30 |
| Спирт + 8% Н2О | – | 1.3 | 0.94 |
| ДМСО + 10% Н2О | – | 1.9 | 1.40 |
| Н2О | – | 0 | – |
| НСООН | – | 0 | – |
| НСООNH2 | – | 0 | – |
| ДМСО | – | 4.5 | 3.3 |
| ДМСО : МА (6 : 1) | – | 3.5 | 2.5 |
| ДМСО : МА (3 : 1) | – | 4.2 | 3.0 |
| ДМСО : МА (3 : 2) | – | 4.5 | 3.3 |
Как и следовало ожидать, константа инициирования возрастает с увеличением полярности или поляризуемости (бензол, толуол) растворителя, поскольку отрываемый ион брома – легко поляризуемое вещество. Те же тенденции изменения константы инициирования, даже более выраженные, были получены путем кинетического моделирования [18]. Скорость инициирования полимеризации МА практически не зависит от доли ДМСО в полимеризате в смеси с МА.
Неожиданным результатом стал факт полного подавления инициирования в протонных средах. Даже при добавлении 8–10% воды в растворитель скорость инициирования снижается почти в 2 раза. Известно [5], что в такой системе наблюдается прямо противоположный эффект: добавление нескольких процентов воды увеличивает скорость полимеризации. Причина этого пока не ясна и требует дальнейшего изучения.
Новая форма катализатора – медное зеркало
Как уже описано ранее, форма катализатора (порошок, проволока, наночастицы), а точнее – его удельная поверхность, существенно влияет на скорость полимеризации. В настоящей работе предложена новая форма катализатора – медное зеркало с высокой удельной поверхностью.
В основе метода химического нанесения зеркальных покрытий на стеклянную поверхность лежит реакция взаимодействия ионов металла с сорбированным на поверхности восстановителем. Окисление восстановителя и восстановление ионов металла протекают с заметной скоростью только для металлов, проявляющих автокаталитические свойства [9]. Реакция становится автокаталитической, если металлическая пленка, образовавшаяся в начальный период, катализирует дальнейшую реакцию восстановления этого же металла. Если осаждаемый металл не проявляет автокаталитические свойства, то реакция восстановления ионов металла протекает во всем объеме раствора и приводит к образованию металлического порошка.
Автокаталитическими свойствами обладают, главным образом, металлы d-элементов VIII группы, серебро и некоторые другие. Медь, как и подавляющее большинство остальных металлов, а также неметаллические материалы не являются катализаторами реакции окисления восстановителя. Поэтому напрямую – аналогично серебряному зеркалу – получить медное зеркало нельзя. Чтобы решить эту проблему, чаще всего используют предварительное покрытие стекла тонким слоем серебряного зеркала, поверх которого наносят медное [8]. Второй подход для придания каталитических свойств поверхности – ее специальная обработка – сенсибилизация и активация [9]. Сенсибилизация (повышение чувствительности стекла к ионам благородных металлов) заключается в обработке стеклянной поверхности растворами солей Sn2+, Fe2+, Ti3+ или Ge2+. Самым эффективным способом является использование SnCl2. Активация состоит в последующей обработке сенсибилизированной поверхности растворами соединений каталитически активных металлов: Ag, Pd, Pt, Au, Rh, Ru, Os или Ir [9].
В данной работе исследованы два приема нанесения медных покрытий на стеклянную подложку. Первый включал промежуточную стадию нанесения Ag-пленки, во втором – наносили медное покрытие непосредственно на поверхность сенсибилизированного и активированного стекла.
Согласно известной двухстадийной методике [8], на первом этапе была проведена реакция серебряного зеркала, стенки сосуда покрывались равномерным слоем серебра. Затем осуществлено осаждение меди из раствора медного купороса формальдегидом в щелочной среде:
При этом медь осаждается ровным слоем на поверхности серебряного зеркала. Через 20–25 с начинается бурная реакция с выделением газа. Спустя 1–2 мин поверхность серебряного зеркала приобретает красноватый оттенок, что объясняется проникновением частиц меди к границе серебро/стекло (рис. 9в). Такой характер процесса можно объяснить тем, что серебро играет роль центров кристаллизации и осаждения меди. При образовании на поверхности серебра первичного слоя меди, она сама начинает катализировать восстановление двухвалентной меди из раствора. Однако через 3–5 мин происходит отслоение полученного зеркала, поэтому далее продолжать процесс не следует.
Рис. 9.
Внешний вид медных зеркал, полученных с помощью гидразина (а), формалина (б) и Ag-пленки (в).

Поскольку биметаллическое покрытие может обладать иными окислительно-восстановительными свойствами, отличными от свойств меди в SET-полимеризации, были изучены возможности непосредственного нанесения медной пленки на стеклянную подложку. Для этого применялись две методики: с использованием в качестве восстановителя гидразина [10]:
Несмотря на то что первая методика достаточно проста, она плохо воспроизводима, что выражается в неоднородности покрытия (рис. 9а). В ряде случаев покрытие не образовывалось вообще, и начиналось осаждение бурого порошка меди.
При восстановлении меди формалином процесс идет существенно быстрее. Через 5–10 с раствор желтеет, а потом обесцвечивается. На поверхности стекла, которая подвергалась нагреванию, выделяется тонкая медная зеркальная пленка (рис. 9б). В ходе нескольких опытов было замечено, что на “молибденовое” стекло медь осаждается хуже, чем на обычное.
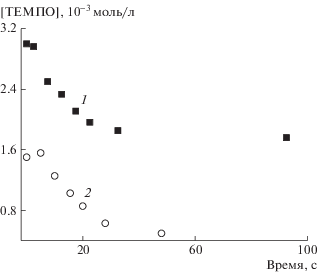
Полученные покрытия были исследованы методом РФА. Для идентификации данных рассчитаны межплоскостные расстояния D по уравнению Брэгга–Вульфа:
Cредний размер кристаллитов определен по формуле Шерера:
(θ – угол рассеяния, K – постоянная Шерера, β – ширина рефлекса на полувысоте).Рефрактограммы представлены на рис. 10; результаты их обработки сведены в табл. 3. Для сравнения приведена рефрактограмма серебряного зеркала (рис. 10, кривая 1). В образцах прямого осаждения видны основной 43.3° и дополнительный 50.8° рефлексы, отвечающие металлической меди. В образце, полученном на серебряной подложке, как и следовало ожидать, наблюдаются рефлексы, соответствующие меди (0) и серебру (0) (рис. 10, кривая 2).
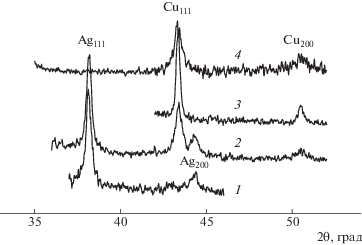
Таблица 3.
Литературные [22] и экспериментальные значения межплоскостных расстояний в полученных покрытиях и “полуширине на полувысоте” (ПШПВ)
| №, п/п | Образец | 2θ, град | Dhkl, Å | ПШПВ, град | Размер кристаллитов, нм |
|---|---|---|---|---|---|
| экспериментальные/ литературные данные | |||||
| 1 | Серебряное зеркало | 38.18 | 2.36/2.36 (Ag111) | 0.48 | 20.8 |
| 44.40 | 2.04/2.04 (Ag200) | ||||
| 2 | Медно-серебряное зеркало | 43.38 | 2.09/2.08 (Сu111) | 0.46 | 23.4 |
| 50.60 | 1.80/1.81 (Сu200) | ||||
| 3 | Медное зеркало (N2H4) | 43.36 | 2.09/2.08 (Сu111) | 0.34 | 35.3 |
| 51.00 | 1.79/1.81 (Сu200) | ||||
| 4 | Медное зеркало (HCHO) | 43.31 | 2.09/2.08 (Сu111) | 0.51 | 20.2 |
| 50.60 | 1.80/1.81 (Сu200) |
Как видно из табл. 3, все зеркала представляют собой поликристаллические покрытия с размером кристаллитов 20–25 нм. Исключение составляет медное покрытие, нанесенное под действием гидразина, с размером кристаллитов 35 нм.
Все полученные покрытия использовны для инициирования полимеризации в стандартной системе МА–ДМСО–МБП–Me6ТРЕН. Полимеризация проведена при 40оС. В качестве метода регистрации скорости полимеризации использована дифференциальная изотермическая калориметрия, позволяющая измерить скорость реакции во всем интервале степеней превращения по скорости тепловыделения. Полученные результаты представлены на рис. 11 в виде дифференциальных кинетических кривых.
Рис. 11.
Дифференциальные кинетические кривые полимеризации МА для разных типов катализаторов: 1 – Cu-проволока; 2 – Ag-пленка; 3 – Cu/Ag-пленка; 4 – Cu-пленка, полученная с гидразином; 5 – Cu-пленка, полученная с формалином.
Легко заметить, что вид кинетических кривых в образцах с участием медной проволоки и зеркала принципиально отличается. В последнем случае наблюдалась высокая скорость прохождения реакции вначале, которая затем быстро падает. Напротив, для полимеризации с участием проволоки характерна меньшая скорость вначале, которая лишь немного снижается в течение долгого времени. Видно, что максимальная скорость полимеризации проявляется в случае с чистым медным зеркалом, чуть меньшую каталитическую активность проявляет система Cu/Ag, и в несколько раз менее эффективным оказалось серебряное зеркало.
Сам факт, что серебряное покрытие проявляет каталитическую активность в процессе радикальной полимеризации, оказался полной неожиданностью, поскольку ранее было известно, что в SET-полимеризации могут участвовать только металлы с переменной степенью окисления. Вероятно, это связано с высоким сродством серебра по отношению к атому брома инициатора МБП [23]:
Отметим, что во всех изученных системах удельный (отнесенный к единице веса катализатора) выход полимера был намного выше, чем в “стандартных” условиях с использованием медной проволоки (табл. 4). Максимальное – 500-кратное повышение удельного выхода наблюдалось при полимеризации с участием зеркала, полученного под действием формалина.
Таблица 4.
Выход полимеров на разных катализаторах в форме зеркала
| Тип катализатора | Масса, мг | Удельный вес мг полимера/мг катализатора | ||
|---|---|---|---|---|
| катализатора | мономера | полимера | ||
| Медь (N2H4) | 7.7 | 396 | 306 | 40.0 |
| Медь (HCHO) | 0.1 | 398 | 265 | 2650.0 |
| Серебро | 1.0 | 367 | 337 | 337.0 |
| Медь/серебро | 0.8/0.4 | 376 | 289 | 241.0 |
| Медь (проволока) | 50.0 | 382 | 334 | 6.7 |
Таким образом, предложенный в настоящей работе вариант катализатора позволяет осуществить полимеризацию с очень высокой начальной скоростью. Важно, что использованный катализатор в виде медного зеркала был получен в количествах, в ~500 раз по весу меньших, чем обычно используемая медная проволока (табл. 4). Тем не менее начальная скорость полимеризации была в 5 раз выше. Логично предположить, что данный эффект связан с большой поверхностью медного катализатора в виде поликристаллического зеркала. Однако недостатком данной формы катализатора является то, что она не всегда позволяет провести полимеризацию до конца. Процесс тормозится в области 50–80%. Вероятно, это связано с недостаточным количеством нанесенного катализатора.
Очистка продуктов полимеризации от медного катализатора и количественное определение его остаточного содержания
Известны два основных метода очистки растворов полимеров от остатков меди: обработка ионообменными смолами [24] и пропускание через колонку с основной окисью алюминия [25]. Одним из преимуществ SET-полимеризации, как уже указывалось выше, служит низкая загрязненность полимера остатками катализатора. Об этом убедительно свидетельствует внешний вид продуктов [3]. Хотя количественная информация о содержании меди в полимере, насколько известно, отсутствует. Поэтому целью работы было установить ее. Для этого получены два образца полиметилакрилата (табл. 5) под действием медной проволоки и зеркала.
Таблица 5.
Образцы полимера для определения содержания остатков катализатора
| Образец, № | Тип катализатора | Масса катализатора, мг | Выход полимерного продукта, % |
|---|---|---|---|
| 1 | Проволока | 150 | 83.5 |
| 2 | Зеркало (N2H4) | 2 | 56.7 |
Для количественного определения меди в неочищенных продуктах был использован спектрофотометрический метод, основанный на поглощении света диэтилдитиокарбаматным комплексом меди в интервале 435–440 нм. Значение мольного коэффициента поглощения 9300 ± ± 100 л/(моль см). Рассчитанное содержание меди в неочищенных образцах 1 и 2 (табл. 5) составило 35.5 ± 0.4 и 6.3 ± 0.1 м.д. соответственно, что на порядок ниже, чем в случае обычной ATRP [2]. Как и следовало ожидать, медное зеркало является значительно более “чистым” катализатором, чем проволока.
Для анализа очищенных полимеров был использован более чувствительный метод АЭС с индукционно связанной плазмой. Результаты анализа всех полученных образцов этим методом представлены в табл. 6.
Таблица 6.
Определение остатков меди с помощью АЭС с индукционно связанной плазмой в очищенных и неочищенных образцах
| Образец*, № | Метод очистки | Содержание меди, м.д. |
|---|---|---|
| 1 | – | 34.0 ± 4.0 |
| 2 | – | 7.0 ± 0.7 |
| 1 | смола “Dowex” | 7.1 ± 0.7 |
| 2 | смола “Dowex” | 1.8 ± 0.2 |
| 1 | смола “Dowex” + Al2O3 | 1.9 ± 0.2 |
| 2 | смола “Dowex” + Al2O3 | 1.5 ± 0.1 |
* Номера образцов соответствуют образцам, приведенным в табл. 5.
Видно, что оба способа – УФ-спектроскопия и АЭС с индукционно связанной плазмой дают одинаковые результаты с точностью до ошибки метода. Оказалось, что обработка продукта ионообменной смолой “Dowex” в среднем снижает содержание меди в 5 раз. Дополнительная очистка на колонке с основной окисью алюминия уменьшает содержание меди до ультраследовых количеств.
Таким образом, известные методы очистки полимеров достаточно эффективны и для продуктов SET-полимеризации.
ЗАКЛЮЧЕНИЕ
После открытия SET-полимеризации за последнее время вышло уже более 500 публикаций [2], посвященных этой тематике. Основные усилия ученых были направлены на расширение круга используемых мономеров, инициаторов, растворителей, металлов и лигандов. В настоящей работе изучены вопросы, которые обычно остаются вне поля зрения экспериментаторов. Так, рассмотрены количественные параметры кинетики и механизма контролируемой полимеризации, загрязненность полимеров остатками соединений меди, а также новые формы катализатора все это может быть весьма полезным для осуществления синтеза полимеров с использованием Cu(0).
Работа выполнена в рамках проекта “Современные проблемы химии и физикохимии высокомолекулярных соединений” (Госбюджет № АААА-А16-116031050014-6).
Список литературы
Matyjaszewski K., Tsarevsky N.V., Braunecker W.A., Dong H., Huang J., Jakubowski W., Kwak Y., Nicolay R., Tang W., Yoon J.A. // Macromolecules. 2007. V. 40. № 22. P. 7795.
Anastasaki A., Nikolaou V., Nurumbetov G., Wilson P., Kempe K., Quinn J.F., Davis T.P., Whittaker M.R., Haddleton D.M. // Chem. Rev. 2016. V. 116. № 3. P. 835.
Percec V., Nguyen N., Rosen B., Lligadas G. // Macromolecules. 2009. V. 42. № 7. P. 2379.
Jiang X., Rosen B., Percec V. // J. Polym. Sci., Polym. Chem. 2009. V. 48. № 2. P. 403.
Rosen B., Percec V. // Chem. Rev. 2009. V. 109. № 11. P. 5069.
Percec V., Guliashvili T., Ladislaw J.S., Wistrand A., Stjerndahl A., Sienkowska M.J., Monteiro M.J., Sahoo S. // J. Am. Chem. Soc. 2006. V. 128. № 43. P. 14156.
Singh A. Ph. D. Thes. Akron. USA. 2008. https://etd.ohiolink.edu/!etd.send_file?accession=akron1207619514&disposition=inline
Hill J.W., Foss D.L., Scott L.W. // J. Chem. Ed. 1979. V. 56. № 11. P. 752.
Петрова Т.П. // Соросовский образовательный журн. 2000. Т. 6. Вып. 11. С. 57.
Казначей Б.Я. Гальванопластика в промышленности. М.: Гос. изд-во местной пром-ти РСФСР, 1955.
Watanabe M., Seida H., Honma H. // Technological Reports. 2007. V. 58. № 10. P. 612.
Brandrup E., Immergut H., Grulke E.A. // Polymer Handbook. New York: Wiley, 1999.
Zhang Y., Wang Yu., Peng C.-H., Zhong M., Zhu W., Konkolewicz D., Matyjaszewski K. // Macromolecules. 2012. V. 45. № 1. P. 78.
Harrisson S., Couvreur P., Nicolas J. // Macromolecules. 2012. V. 45. № 18. P. 7388.
Goto A., Fukuda T. // Progr. Polym. Sci. 2004. V. 29. № 4. P. 32.
Ligadas G., Percec V. // J. Polym. Sci., Polym. Chem. 2008. V. 46. № 8. P. 2745.
Fischer H., Souaille M. // Macromolecules. 2001. V. 34. № 9. P. 2830.
Nicolas J., Perrier S., Harrisson S. // ACS Symp. Ser. 2015. V. 1187. P. 129.
Konkolewicz D., Wang Yu., Krys P., Zhong M., Isse A.A., Gennarob A., Matyjaszewski K. // Polym. Chem. 2014. V. 5. № 15. P. 4409.
Zaremski M.Yu., Odintsova V.V., Plutalova A.V., Gurskii M.E., Bubnov Yu.N. // Polymer Science B. V. 60. № 2. P. 162.
Peng C.-H., Zhong M., Wang Yu., Kwak Y., Zhang Y., Zhu W., Tonge M., Buback J., Park S., Krys P., Konkolewicz D., Gennaro A., Matyjaszewski K. // Macromolecules. 2013. V. 46. № 10. P. 3803.
Миркин Л.И. // Справочник по рентгеноструктурному анализу поликристаллов. М.: Гос. изд-во физ.-мат. лит-ры, 1961.
Заремский М.Ю., Загрибельный Б.А., Орлова А.П., Зезин С.Б., Оленин А.В. Пат. 2566302 Россия. 2014.
Matyjaszewski K., Pintauer T., Gaynor S. // Macromolecules. 2000. V. 33. № 4. P. 1476.
Griller D., Ingold K. // Acc. Chem. Res. 1976. V. 9. № 1. P. 13.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)