

Высокомолекулярные соединения (серия Б), 2021, T. 63, № 2, стр. 83-88
АЦЕНАФТИЛЕН-БИС-(АРИЛАМИДНЫЕ) КОМПЛЕКСЫ АЛЮМИНИЯ И ГАЛЛИЯ В ПОЛИМЕРИЗАЦИИ ЛАКТИДА
О. В. Казарина a, А. Г. Морозов a, *, И. Л. Федюшкин a
a Институт металлоорганической химии им. Г.А. Разуваева Российской академии наук
603950 Нижний Новгород, бокс-445, ул. Тропинина, 49, Россия
* E-mail: morozov@iomc.ras.ru
Поступила в редакцию 02.07.2020
После доработки 05.11.2020
Принята к публикации 19.11.2020
Аннотация
Исследована каталитическая активность соединений алюминия и галлия с неинноцентным аценафтен-1,2-дииминовым лигандом dpp-bian(=1,2-бис-[(2,6-диизопропилфенил)имино]аценафтен) в полимеризации с раскрытием цикла лактидов молочной кислоты. Установлено, что в данном процессе гомобиметаллические соединения алюминия и галлия состава [(dpp-bian)Mt]2 проявляют низкую активность и не обеспечивают контроль молекулярно-массовых характеристик образующихся полимеров. Напротив, в присутствии металлокомплекса (dpp-bian)Ga(OCH2CH2NMe2) полимеризация лактида протекает в контролируемом режиме и реакция имеет первый порядок по мономеру. Алюминиевый аналог моноядерного алкоксидного комплекса галлия демонстрирует отсутствие каталитической активности в полимеризации лактида.
На протяжении последних двадцати лет сохраняется устойчивый интерес к синтетическим материалам, получаемым из возобновляемого растительного сырья, как альтернативе полимерам на основе нефтепродуктов. По данным немецких аналитических компаний “European Bioplastics e.V.” и “Nova-Institut GmbH” в 2019 году в Европейском Союзе было произведено 2.1 млн тонн биопластика, причем более половины – биоразлагаемые полимеры [1]. Лидером среди полимеров выступает полилактид (ПЛА), объем производства которого составил около 300 тыс. тонн. Это обусловлено, прежде всего, доступностью мономера ПЛА – молочной кислоты, синтезируемой в промышленных масштабах путем ферментативного гидролиза природных сахаров, добываемых из крахмалсодержащего сырья [2]. Несмотря на то что поликонденсацией молочной кислоты удается получать даже высокомолекулярный полимер (Mw > 105) [3], наиболее удобным методом синтеза ПЛА является катализируемая металлокомплексами полимеризация с раскрытием цикла (ПРЦ) димера молочной кислоты – лактида [4, 5]. Данный способ позволяет контролировать молекулярную массу ПЛА, а также его микроструктуру, зависящую от энантиомерного состава мономера и стереоселективности катализатора [6]. Указанные параметры критически влияют на физико-механические свойства и скорость разложения полимера, а также обусловливают его применение в тканевой инженерии в качестве материала для создания различных имплантов [7, 8] и систем целевой доставки лекарств [9–11]. Использование координационных соединений галлия и алюминия в ПРЦ лактида вызвано не только биосовместимостью металлов [12, 13], что играет ключевую роль в дальнейшем применении полимера в медицинских целях, но и высокой каталитической активностью металлокомплексов. Так, производные алюминия и галлия с тридентатными бисамидоэфирными N,O,N-лигандами конвертируют до 100 экв. rac-лактида в час в среде толуола при 80°С, с образованием изотактически-обогащенного ПЛА (Pm = 0.62–0.70) [14]. Демонстрируя более высокие скорость и изоселективность (Pm = 0.84), бисиминофеноляты галлия [15] в расплаве за час полимеризуют до 800 экв. лактида. В свою очередь, алкилалкоксидные производные алюминия, имеющие в своем составе хиральные (1,2)-дифенилэтилен-саленовые лиганды дают ПЛА высокой степени изотактичности (Pm = = 0.80–0.90) [16], в то время как алкоксид (S,S)-[Me2Ga(μ-OCH(Me)CO2Me)]2, полученный из (S)-метиллактата, дает гетеротактически-насыщенный полимер (Pr = 0.78) [17]. В ряде работ отмечается высокая активность алкоксидов металлов 13 группы, стабилизированных N-гетероциклическими карбеновыми лигандами [18–22]. В частности, диметилалкоксиды галлия с координированным 1,3-бис-(2,4,6-триметилфенил)имидазолин-2-илиденом активны в полимеризации rac-лактида в среде дихлорметана уже при –20°С, превращая за 16 ч 320 экв. мономера в изотактически-обогащенный ПЛА (Pm = 0.78) [18].
Ранне [23] нами было показано, что соединения алюминия и галлия состава (dpp-bian)Mt–Mt(dpp-bian) (Мt – атом металла), содержащие неинноцентный лиганд (dpp-bian2− = 1,2-бис-[(2,6-диизопропилфенил)амидо]аценафтилен), полимеризуют ε-капролактон в растворе при комнатной температуре, причем [(dpp-bian)Al]2 осуществляет данный процесс с высокой скоростью (TOF 5320 ч–1), превосходя известные катализаторы на основе комплексов Al(III). При этом лиганд, находящийся в бисамидной форме, выступает в качестве акцептора протонов, оставаясь бидентатно связанным с металлоцентром. В соответствии с предполагаемым механизмом, рассчитанным методом DFT, это обеспечивает образование соответствующего интермедиата в процессе полимеризации капролактона. Для стабилизированных dpp-bian алкоксихлоридов титана, активных в ПРЦ L-лактида, отмечалось значительное влияние степени восстановленности лиганда металлокомплекса на контроль молекулярно-массовых характеристик образующегося полимера [24]. Следует отметить, что ПЛА, полученный полимеризацией лактида на аценафтиленбисамидных комплексах магния и кальция [25, 26], в экспериментах in vitro наряду с высоким уровнем клеточной пролиферации продемонстрировал отсутствие цитотоксичности по отношению к дермальным фибробластам человека [27].
Таким образом, с целью расширения спектра биосовместимых катализаторов ПРЦ циклических эфиров, используемых для получения полиэфиров биомедицинского назначения, в настоящей работе представлены данные о каталитической активности моно- и биметаллических комплексов алюминия и галлия, содержащих бисамидный лиганд dpp-bian2-, в полимеризации L- и rac-лактида.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Соединения [(dpp-bian)Mt]2 (Mt = Al (1), Ga (2)) и (dpp-bian)Mt(OCH2CH2NMe2) (Mt = Al (3), Ga (4)) чувствительны к кислороду и влаге воздуха, поэтому все операции, связанные с их синтезом, выделением, а также полимеризацию лактида выполняли в вакууме или в атмосфере сухого азота с использованием техники Шленка. Толуол для синтеза металлокомплексов и полимеризационных экспериментов сушили над бензофенонкетилом натрия, перегоняли в атмосфере сухого азота, хранили над молекулярными ситами (4 Å) и отбирали непосредственно перед использованием. Соединения 1 [28], 2 [29], 3 [23] и 4 [30] синтезировали по опубликованным методикам; L- и rac-лактид (“Aldrich”) очищали перекристаллизацией из толуола однократной возгонкой в вакууме (Т = 95°С, 0.0375 мм рт.ст.). Конверсию мономера в процессе полимеризации определяли методом ЯМР на спектрометре “Bruker DPX-200”, химические сдвиги приводили в шкале δ (м.д.) и соотносили с химическими сдвигами остаточных протонов хлороформа-d1. ММР образцов полученного поли-L-лактида определяли с использованием хроматографа “Knauer Smartline” с колонками “Phenogel Phenomenex 5u” (300 × 7.8 мм) средний диаметр пор 104, 105 Å, детектор – рефрактометр. В качестве подвижной фазы применяли ТГФ, скорость потока – 2 мл/мин; Т = 40°C. Калибровку осуществляли с использованием полистирольных стандартов с ММ в диапазоне (2.7–2570) × 103. Значения среднечисловой ММ поли-L-лактида приводили с учетом поправочного коэффициента Марка–Хаувинка (0.58), применяемого вследствие разницы в гидродинамических объемах ПС и ПЛА. Масс-спектры MALDI-TOF регистрировали в положительном режиме на оборудовании Службы масс-спектрометрии Института химии университета Страсбурга, Франция. Образцы готовили с использованием растворов полимеров в CH2Cl2 с концентрацией 0.5 мг/100 мл. В качестве матрицы использовалась 2,5-дигидробензойная кислота.
Полимеризацию с раскрытием цикла лактида с использованием в качестве катализаторов комплексов 1, 2, 4 осуществляли следующим образом. В колбу Шленка объемом 25 мл, снабженную магнитной мешалкой, помещали 8 мл толуольного раствора лактида (10 ммоль). К содержимому колбы при интенсивном перемешивании добавляли 2 мл раствора катализатора в толуоле. Реакционную смесь перемешивали в течение 24 ч при 80°С. Конверсию мономера определяли методом спектроскопии ЯМР 1H, для чего аликвоту раствора (0.1 мл) помещали на воздухе в ампулу ЯМР, что обеспечивало разрушение катализатора, о чем свидетельствовало изменение цвета раствора с фиолетово-синего на желтый. Растворитель удаляли в вакууме, а сухой остаток растворяли в хлороформе-d1. ПЛА выделяли из реакционной смеси, обрабатывая ее трехкратным объемом петролейного эфира. Выделенный полимер промывали на фильтре петролейным эфиром и сушили в вакууме до постоянной массы.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В качестве объектов исследования – катализаторов ПРЦ лактида были выбраны биядерные (dpp-bian)Mt–Mt(dpp-bian) (соединения 1, 2) и моноядерные комплексы (dpp-bian)Mt(OCH2CH2NMe2) (соединения 3, 4) алюминия и галлия:
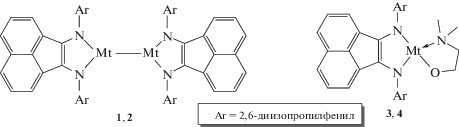
Соединения 1 и 2 могут быть получены прямым восстановлением нейтрального dpp-bian избытком соответствующего металла в среде толуола [28, 29], в то время как 3 и 4 образуются по обменной реакции галогенпроизводных (dpp-bian)AlCl(Et2O) и [(dpp-bian)GaI]2 с натриевой солью 2-(диметиламино)этанола [23, 30]. Во всех рассматриваемых соединениях лиганд dpp-bian является дианионом, бидентатно координированным атомами металлов. Атомы алюминия и галлия, входящие в состав комплексов 1 и 2, формально имеют степень окисления +2, в то время как алкоксидные производные 3 и 4 образованы ионами Al3+ и Ga3+ соответственно.
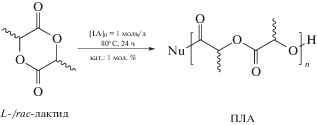
Полимеризация L- и rac-лактида проведена в среде толуола при повышенной температуре (80°С):
Оказалось, что из рассматриваемых соединений моноалюминиевое производное 3 не проявляет каталитической активности в описываемых условиях. Предположительно, четырехкоординационный центральный ион Al3+ комплекса 3, имея меньший, чем у галлия ионный радиус (53 против 61 пм [31]), в значительно бóльшей степени экранирован лигандным окружением. В частности, объемные изопропильные группы фенильных заместителей при атомах азота dpp-bian соединения 3 препятствуют координации мономера на металлоцентр, чего не происходит в случае галлиевого аналога 4. В свою очередь, ионы металла диалюминиевого комплекса 1, вследствие своей координационной ненасыщенности, способны координировать молекулы циклического эфира, инициируя тем самым ПРЦ, как это было показано ранее [23].
Соединения 1 и 2 активны в отношении как L-, так и rac-лактида. Однако во всех случаях (табл. 1, эксп. 1–4) можно видеть образование полимера, ММ которого значительно отличается от ожидаемой, а конверсия оказывается весьма низкой. Поскольку в составе катализаторов отсутствует уходящая группа, а биядерные комплексы 1 и 2 эффективно полимеризуют ε-капролактон в присутствии инициатора (бензиловый спирт) [23], можно полагать, что в данном случае в качестве инициатора выступают примеси (вода, спирты и т.д.) типа HNu (Nu – нуклеофильный остаток), содержащиеся в лактиде в следовых количествах, что обусловливает неконтролируемое протекание процесса.
Таблица 1.
ПРЦ лактида на комплексах 1, 2, 4 ([LA]0 = 1 моль/л, толуол, Т = 80°C, 24 ч)
| Эксперимент, № | Катализатор | Мономер (100 экв.) | Конверсия, % | Mnтеор* × 10–3 | Mnкорр × 10–3 | Đ |
|---|---|---|---|---|---|---|
| 1 | 1 | rac-лактид | 53 | 7.6 | 26.8 | 1.53 |
| 2 | 1 | L-лактид | 36 | 5.2 | 11.4 | 1.50 |
| 3 | 2 | rac-лактид | 26 | 3.8 | 9.4 | 1.45 |
| 4 | 2 | L-лактид | 24 | 3.4 | 8.6 | 1.48 |
| 5 | 4 | rac-лактид | 94 | 13.5 | 10.0 | 1.29 |
| 6 | 4 | L-лактид | 82 | 11.8 | 12.2 | 1.18 |
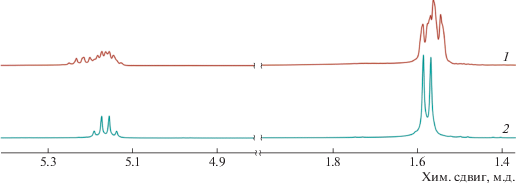
Алкоксидный комплекс галлия 4 в отличие от его алюминиевого аналога, активен в ПРЦ лактида. В присутствии соединения 4 процесс протекает с образованием полимеров, имеющих ММ, близкую к расчетной, и узкое ММР (табл. 1, эксп. 5 и 6), что свидетельствует в пользу контролируемой полимеризации. По данным ЯМР 1Н-спектроскопии ПРЦ L-лактида приводит к образованию изотактического полимера (рис. 1, спектр 2).
Рис. 1.
Спектры ЯМР 1Н (400 МГц, хлороформ-d1, 298 К) образцов полимера, полученных из рацемического (1) и L-лактида (2).
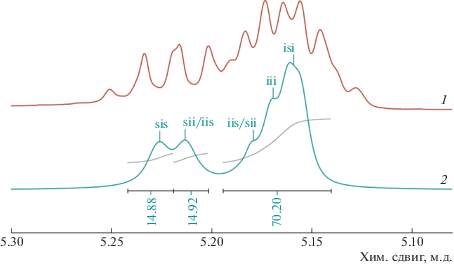
Интегрирование сигналов спектра ЯМР 1Н поли-D,L-лактида, записанного с подавлением спин-спинового взаимодействия между протонами (рис. 2, спектр 2) дает информацию о содержании последовательностей стереоцентров (тетрад) в полимере. В соответствии с распределением Бернулли [32] для [sii] = $\frac{{{{P}_{r}}{{P}_{m}}}}{2}$ и [sis] = $\frac{{P_{r}^{2}}}{2}$, полимер является атактическим (Pr ≈ Pm ≈ 0.55), что свидетельствует об отсутствии стереоселективности катализатора.
Рис. 2.
Область метиновых протонов спектра ЯМР 1Н (400 МГц, хлороформ-d1, 298 К) поли-D,L-лактида, полученного на комплексе 4 (1), та же область спектра ЯМР 1Н-{1Н} (2).
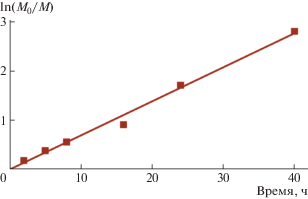
Используя в качестве модельной систему 4/L-лактид в соотношении 1/100 (табл. 2), методом избыточных концентраций были определены кинетические параметры полимеризации. Зависимость ln(M0/M) от времени линейна, что указывает на первый порядок реакции по мономеру (рис. 3), константа скорости равна 0.0694 ч–1.
Таблица 2.
ПРЦ лактида ([LA]0 = 1 моль/л, толуол, Т = 80°C) в системе 4/L-лактид 1/100
| Эксперимент, № | Время реакции, ч | Конверсия, % | Mnтеор* × 10–3 | Mnкорр × 10–3 | Đ | ln ([M]0/[M]) |
|---|---|---|---|---|---|---|
| 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 | 2 | 17 | 2.5 | 3.3 | 1.24 | 0.186 |
| 3 | 5 | 32 | 4.6 | 6.2 | 1.24 | 0.386 |
| 4 | 8 | 43 | 6.2 | 6.0 | 1.28 | 0.562 |
| 5 | 16 | 60 | 8.6 | 7.9 | 1.20 | 0.916 |
| 6 | 24 | 82 | 11.8 | 11.8 | 1.18 | 1.715 |
| 7 | 40 | 94 | 13.5 | 11.9 | 1.31 | 2.813 |
Рис. 3.
Зависимость ln(M0/M) от времени ПРЦ L-лактида на комплексе 4 (100 экв. L-лактида, 4, [LA]0 = 1 моль/л, толуол, Т = 80°C); y = 0.0694x, R2 = 0.9955.
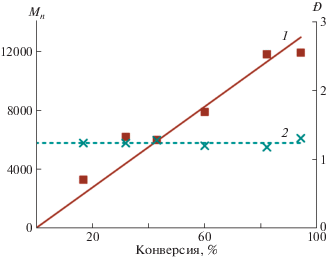
Следует отметить, что во всех экспериментах (табл. 2) регистрируется низкое и относительно постоянное значение полидисперсности образующегося ПЛА (рис. 4), что позволяет предполагать отсутствие побочных реакций обрыва/передачи цепи. Это подтверждается результатами MALDI-TOF масс-спектрометрии поли-L-лактида, согласно которым разница между соседними пиками спектра составляет 144 единицы, что соответствует массе звена лактида (рис. 4). Полученные данные в совокупности с линейной зависимостью ММ полимера от конверсии (рис. 5) подтверждают контролируемый характер полимеризации. В целом, анализ представленных результатов свидетельствует о протекании ПРЦ L-лактида на комплексе 4 по механизму координации–внедрения [33].
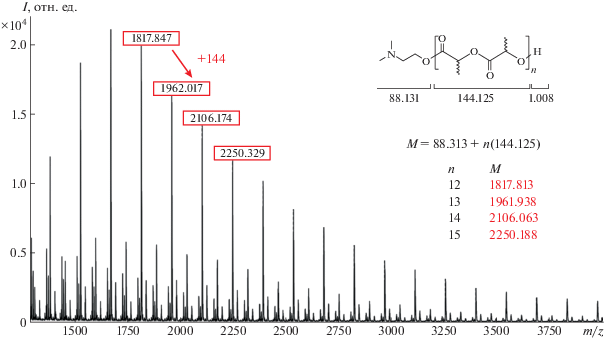
Рис. 5.
Зависимость Mn (1) и Đ (2) поли-L-лактида, полученного на комплексе 4, от конверсии мономера; y = 137.65x, R2 = 0.98.
Авторы выражают особую благодарность Самуэлю Дагорну (Университет Страсбурга) за плодотворное сотрудничество и неоценимую помощь в проведении исследований.
Работа выполнена при финансовой поддержке Российского научного фонда (код проекта 17-73-20356). В работе использовано научное оборудование Центра коллективного пользования “Аналитический центр ИМХ РАН” в Институте металлоорганической химии им. Г.А. Разуваева Российской академии наук (уникальный идентификатор проекта RFMEFI62120X0040).
Список литературы
https://www.european-bioplastics.org/news/publications/
Starr J.N., Westhoff G. Ullmann’s Encyclopedia of Industrial Chemistry. New York: Wiley, 2014.
Auras R. Encyclopedia of Polymer Science and Technology. New York: Wiley, 2010.
Kremer A.B., Mehrkhodavandi P. // Coord. Chem. Rev. 2019. V. 380. P. 35.
Lyubov D.M., Tolpygin A.O., Trifonov A.A. // Coord. Chem. Rev. 2019. V. 392. P. 83.
Sangeetha V.H., Deka H., Varghese T.O., Nayak S.K. // Polym. Compos. 2018. V. 39. P. 81.
Martina M., Hutmacher D.W. // Polym. Int. 2007. V. 56. P. 145.
da Silva D., Kaduri M., Poley M., Adir O., Krinsky N., Shainsky-Roitman J., Schroeder A. // Chem. Eng. J. 2018. V. 340. P. 9.
Mohamed F., van der Walle C.F. // J. Pharm. Sci. 2007. V. 97. P. 71.
Abbas A.O., Donovan M.D., Salem A.K. // J. Pharm. Sci. 2008. V. 97. P. 2448.
Hu X., Jing X. // Expert. Opin. Drug Delivery. 2009. V. 6. P. 1079.
Gray F., Kramer D.A., Bliss J.D. Kirk-Othmer Encyclopedia of Chemical Technology. New York: Wiley, 2010.
Klotz K., Weistenhöfer W., Neff F., Hartwig A., van Thriel C., Drexler H. // Dtsch. Arztebl. Int. 2017. V. 114. P. 653.
Hild F., Neehaul N., Bier F., Wirsum M., Gourlaouen C., Dagorne S. // Organometallics. 2013. V. 32. P. 587.
Ghosh S., Gowda R.R., Jagan R., Chakraborty D. // Dalton Trans. 2015. V. 44. P. 10410.
Maudoux N., Roisnel T., Dorcet V., Carpentier J.-F., Sarazin Y. // Chemistry. 2014. V. 20. P. 6131.
Horeglad P., Kruk P., Pecaut J. // Organometallics. 2010. V. 29. P. 3729.
Horeglad P., Szczepaniak G., Dranka M., Zachara J. // Chem. Commun. (Camb). 2012. V. 48. P. 1171.
Cybularczyk M., Dranka M., Zachara J., Horeglad P. // Organometallics. 2016. V. 35. P. 3311.
Schnee G., Bolley A., Gourlaouen C., Welter R., Dagorne S. // J. Organomet. Chem. 2016. V. 820. P. 8.
Schnee G., Bolley A., Hild F., Specklin D., Dagorne S. // Catal. Today. 2017. V. 289. P. 204.
Zaremba R., Dranka M., Trzaskowski B., Checinska L., Horeglad P. // Organometallics. 2018. V. 37. P. 4585.
Kazarina O.V., Gourlaouen C., Karmazin L., Morozov A.G., Fedushkin I.L., Dagorne S. // Dalton. Trans. 2018. V. 47. P. 13800.
Morozov A.G., Martemyanova T.V., Dodonov V.A., Kazarina O.V., Fedushkin I.L. // Eur. J. Inorg. Chem. 2019. V. 2019. P. 4198.
Fedushkin I.L., Morozov A.G., Chudakova V.A., Fukin G.K., Cherkasov V.K. // Eur. J. Inorg. Chem. 2009. V. P. 4995.
Morozov A.G., Markelova E.S., Fedyushkin I.L. // Russ. J. Appl. Chem. 2018. V. 91. P. 1044.
Morozov A.G., Razborov D.A., Egiazaryan T.A., Baten’kin M.A., Aleynik D.Y., Egorikhina M.N., Rubtsova Y.P., Charikova I.N., Chesnokov S.A., Fedushkin I.L. // J. Polym. Environ. 2020. https://doi.org/10.1007/s10924-020-01803-x
Fedushkin I.L., Lukoyanov A.N., Ketkov S.Y., Hummert M., Schumann H. // Chem. Eur. J. 2007. V. 13. P. 7050.
Fedushkin I.L., Moskalev M.V., Lukoyanov A.N., Tishkina A.N., Baranov E.V., Abakumov G.A. // Chem. Eur. J. 2012. V. 18. P. 11264.
Fedushkin I.L., Kazarina O.V., Lukoyanov A.N., Skatova A.A., Bazyakina N.L., Cherkasov A.V., Palamidis E. // Organometallics. 2015. V. 34. P. 1498.
Shannon R.D. // Acta Crystallogr. Sect. A. 1976. V. 32. P. 751.
Chamberlain B.M., Cheng M., Moore D.R., Ovitt T.M., Lobkovsky E.B., Coates G.W. // J. Am. Chem. Soc. 2001. V. 123. P. 3229.
Dechy-Cabaret O., Martin-Vaca B., Bourissou D. // Chem. Rev. 2004. V. 104. P. 6147.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)