

Высокомолекулярные соединения (серия Б), 2021, T. 63, № 3, стр. 190-206
ЛИНЕЙНЫЕ И РАЗВЕТВЛЕННЫЕ ПОЛИМЕРЫ ЛАКТИДА ДЛЯ СИСТЕМ НАПРАВЛЕННОЙ ДОСТАВКИ ЛЕКАРСТВЕННЫХ СРЕДСТВ
В. И. Гомзяк a, *, Н. Г. Седуш a, А. А. Пучков a, Д. К. Поляков a, С. Н. Чвалун a
a Национальный исследовательский центр “Курчатовский институт”
123182 Москва, пл. Академика Курчатова, 1, Россия
* E-mail: vgomzyak@gmail.com
Поступила в редакцию 15.12.2020
После доработки 30.01.2021
Принята к публикации 12.02.2021
Аннотация
В обзоре представлены современные достижения в области синтеза биоразлагаемых полимеров на основе лактида различной топологии, а также проведен анализ основных методов получения наночастиц, перспективных для создания систем адресной доставки лекарственных препаратов.
ВВЕДЕНИЕ
Полимерные нано- и микрочастицы являются перспективной платформой для создания на их основе терапевтических систем адресного и пролонгированного действия. Наносомальные лекарственные формы характеризуются повышенной эффективностью и сниженной выраженностью побочных эффектов по сравнению с классическими лекарственными формами. Большинство таких систем основано на биоразлагаемых и биосовместимых материалах, в основном на алифатических сложных и простых полиэфирах, полиангидридах, полиамидах, полиортоэфирах и т.д. Наиболее востребованными из них являются сложные полиэфиры α-гидроксикислот: полилактид, полигликолид, поли(ε-капролактон), полидиоксанон, а также их сополимеры [1–6]. Регулирование молекулярной и надмолекулярной структуры, а также архитектуры биоразлагаемых полимеров позволяет управлять их физико-химическими свойствами и профилем высвобождения активного вещества, что имеет важное значение для создания фармацевтических систем пролонгированного действия. В настоящее время в литературе описано огромное количество полимеров лактида разной топологии: линейные, разветвленные, звездообразные, привитые и т.д. В настоящем обзоре обобщены литературные данные о методах синтеза биоразлагаемых полимеров на основе лактида различной топологии, а также об основных технологиях получения наноструктур для применения в системах адресной доставки лекарственных агентов.
СОПОЛИМЕРЫ ЛАКТИДА С ГЛИКОЛИДОМ
Полимеризация с раскрытием цикла (ring-opening polymerization – ROP) в последние десятилетия привлекает повышенное внимание в связи с возможностью синтеза широкого круга биоразлагаемых полимерных структур, совместимых с организмом человека. В организме протекает процесс гидролитической деструкции (разрыв сложноэфирных связей) этих полимеров, который на позднем этапе дополняется биодеградацией под действием ферментов. Продукты гидролиза выводятся, включаясь в метаболизм, а скорость гидролиза можно легко регулировать, изменяя химический состав и структуру сополимера [2, 4].
Ниже представлены некоторые циклические мономеры, полимеризуемые методом ROP.
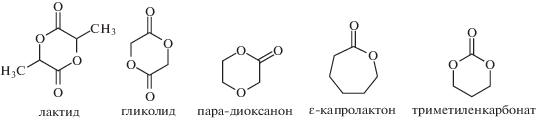
Среди них лактид и его сополимеры привлекают наибольшее внимание как материалы для биодеградируемых хирургических шовных нитей, систем доставки лекарственных веществ, сердечно-сосудистых, стоматологических и ортопедических временных крепежных элементов, матриксов для тканевой инженерии и биоискусственных органов и т.д. Основным сырьем для получения полимолочной кислоты, а также внутреннего циклического диэфира молочной кислоты – лактида, является молочная кислота. Молочная кислота (α-гидроксипропионовая) – простейшая α-гидроксикислота, имеющая асимметрический атом углерода. Она существует в трех формах: две оптически активные (энантиомеры) и оптически неактивная рацемическая смесь, из которых получают оптически активные L- и D-изомеры лактида и оптически неактивные мезо- и D,L-формы [7–11]:
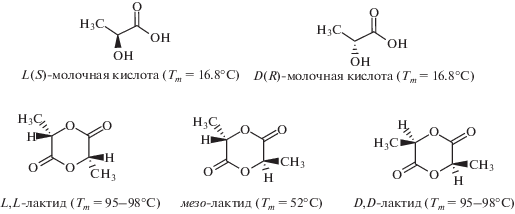
В промышленности молочную кислоту обычно получают из таких видов сырья, в которых содержатся глюкоза, сахароза и мальтоза, например рафинадная патока, меласса, крахмал (кукурузный, картофельный). Молочную кислоту можно получать также из нефтехимического сырья, однако ферментативный способ гораздо экологичнее. Более того, нефтехимическим синтезом получают, как правило, оптически неактивную смесь (примерно 50 : 50) L- и D-форм молочной кислоты, тогда как молочная кислота, производимая бактериальной ферментацией, образуется почти исключительно в L-форме [8, 9].
Существует два основных метода синтеза полилактида: поликонденсация молочной кислоты и полимеризация лактида с раскрытием цикла. Методом поликонденсации, ввиду необходимости отвода выделяющейся воды из реакционной среды, довольно сложно синтезировать полимер с высокой молекулярной массой и хорошими физико-механическими характеристиками, пригодный для изготовления изделий медицинского назначения. Для смещения равновесия и получения высокомолекулярного продукта, необходимо отгонять или химически связывать образующуюся в процессе поликонденсации воду. Молекулярная масса полимолочной кислоты, синтезированной методом поликонденсации без использования дополнительных сшивающих агентов, как правило, не превышает 10 × 103.
Полимеризация с раскрытием цикла алифатических циклических сложных эфиров – это постоянно и динамично развивающаяся область исследований. Преимуществами ROP перед поликонденсацией являются: более мягкие условия реакции, отсутствие побочных продуктов и возможность использования даже шестичленных или семичленных лактонов [4, 6]. Механизмы ионных и координационных процессов полимеризации алифатических циклических эфиров систематически рассматриваются с конца 1960-х годов. Эти механизмы обеспечивают достаточно хороший контроль характеристик полимера (т.е. предсказуемую молекулярную массу, узкое молекулярно-массовое распределение, высокую степень конверсии) и особенно хорошо подходят для макромолекулярной инженерии с получением гомо- и сополимеров различной архитектуры (например, двойных и тройных блок-сополимеров, привитых, звездообразных, сверхразветвленных и других) [4].
В зависимости от структуры мономера, каталитической/инициирующей системы и природы образующихся активных частиц ROP может протекать по свободнорадикальному механизму, механизму координации–внедрения, по анионному (или катионному) механизму [3]. Анионная и координационная ROP позволяют достичь самых высоких выходов полимеризации и молекулярных масс за короткое время реакции. В анионной полимеризации с раскрытием цикла (AROP) в качестве инициаторов используются нуклеофильные реагенты (металлоорганические соединения, амиды металлов, амины, алкоксиды, спирты, вода и т.д.). В литературе описано огромное количество каталитических систем для ROP [4, 12].
В зависимости от используемых металлоорганических производных металлов (таких как Al, Sn, Nd, Y, Yb, Sm, La, Fe, Zn, Zr, Ca, Ti и Mg) ROP протекает главным образом через два основных механизма. Некоторые из них действуют как катализаторы и активируют мономер за счет образования комплекса с карбонильной группой. Затем полимеризация инициируется любым нуклеофилом, например водой или спиртом, которые присутствуют в полимеризационной среде в виде примесей, или добавляются в контролируемом количестве. Металлоорганическое соединение играет роль инициатора, а полимеризация протекает по механизму координация–внедрение. В ROP молекулы инициатора, содержащие гидроксил, могут служить регуляторами молекулярной массы полимеров. Скорость полимеризации можно контролировать, применяя широкий спектр каталитических систем, таких как октаноат олова и 2-этилгексановая кислота [4].
Для получения биоразлагаемых материалов с заранее заданными свойствами и сроками деградации проводят сополимеризацию нескольких форм лактида между собой, а также сополимеризацию с другими циклическими сложными эфирами: гликолидом, ε-капролактоном, δ-валеролактоном, триметиленкарбонатом и т.д.). Широкое применение нашли в медицине сополимеры лактида с гликолидом (PLGA), они одобрены Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов (Food and Drug Administration – FDA). Как и соответствующие гомополимеры (PLA и PGA), PLGA являются термопластичными сложными полиэфирами, которые синтезируют методом полимеризации с раскрытием цикла:

Из-за различной реакционной способности лактида и гликолида могут быть получены сополимеры с разной микроструктурой. Варьируя химический состав, молекулярную массу и надмолекулярную структуру, можно “управлять” физико-механическими характеристиками материала, его растворимостью, теплофизическими свойствами и сроками биодеградации. Важно подобрать оптимальные условия реакции, позволяющие добиться высокой конверсии и молекулярной массы полимера. Гидрофобность полилактида намного выше, чем у полигликолида, из-за наличия боковой метильной группы, вследствие чего скорость гидролиза основной цепи оказывается гораздо ниже, а растворимость в органических растворителях – выше.
Гомополимеры L-лактида и гликолида – высококристаллические материалы с относительно длительным сроком деградации, однако при их сополимеризации нарушается стереорегулярность цепей и снижается степень кристалличности. Поскольку аморфные области материала существенно доступнее для воды, чем кристаллические, в сополимерах лактида с гликолидом гидролитическая деструкция проходит быстрее, по сравнению с соответствующими гомополимерами. Сополимеры, содержащие более 30% гликолидных звеньев, полностью аморфны. Температура стеклования варьируется от 40 до 60°С и зависит от микроструктуры сополимера и его состава. Скорость биоразложения также зависит от соотношения звеньев, наиболее быстро разлагаются сополимеры с составом лактид : гликолид = = 50 : 50. Благодаря биосовместимости, а также возможности регулирования физико-механических свойств и сроков биодеградации, полимеры и сополимеры лактида с гликолидом широко используются для изготовления шовных материалов для хирургии, крепежных изделий для травматологии и ортопедии, матриксов для регенеративной медицины, систем направленной доставки и пролонгированного высвобождения лекарств. В настоящее время хорошо изучены и коммерчески доступны сополимеры PLGA различного состава (50 : 50, 65 : 35, 75 : 25 и 85 : 15) и молекулярной массы ((10–200) × 103). Большая часть работ сосредоточена на сополимерах двух типов: поли-L-лактид-со-гликолиде (PLGA) и поли-D,L-лактид-со-гликолиде (PDLGA). R.A. Miller с соавторами [13, 14] показали, что PDLGA состава 50 : 50 разлагается примерно за 1–2 месяца, 75 : 25 – за 4–5 месяцев и 85 : 15 – за 5–6 месяцев [15].
PLGA имеет низкую токсичность, превосходную биологическую совместимость и относительно высокую совместимость с другими полимерами и физиологически активными веществами, поэтому его широко используют в медицине, в частности, для создания нано- и микрочастиц, загруженных лекарственными веществами. Разработан ряд методов, позволяющих получать наночастицы PLGA с заданным размером и степенью нагрузки лекарственным соединением. Выбор метода определяется преимущественно типом используемого сополимера и лекарственного вещества и методом введения в организм. Нано- и микрочастицы получают химическими, физическими и физико-химическими методами. Большинство из них представляют собой модификацию трех основных способов: распылительной сушки, фазового разделения (коацервации) и экстракции (испарения) растворителя.
Эмульсионные методы
Простые эмульсии. Метод экстракции/испарения растворителя является наиболее распространенным методом инкапсулирования (рис. 1). Он не требует повышенных температур или введения дополнительных компонентов (например, агентов фазового разделения). Путем тщательного подбора реагентов и условий инкапсулирования, можно получать нано- и микрочастицы регулируемых размеров. Самый простой вариант этого метода включает образование эмульсий “масло в воде” или “вода в масле”.
Рис. 1.
Схема формирования наночастиц путем эмульгирования органического раствора лекарственного вещества с полимером в водной среде с последующей экстракцией/испарением растворителя. Цветные рисунки можно посмотреть в электронной версии.

Данный метод широко применяется для загрузки нерастворимых или плохо растворимых в воде физиологически активных веществ. PLGA растворяют в хорошем, не смешивающемся с водой растворителе (например, дихлорметане, хлороформе, этилацетате) для получения однофазного раствора. Из-за токсичности хлороформ не рекомендуется использовать для получения лекарственных форм. Дихлорметан обладает высокой летучестью и, как сообщается, полученные с его помощью частицы имеют однородную сферическую форму. Этилацетат более предпочтителен, поскольку менее токсичен, но частично смешивается с водой. В связи с этим поиск нетоксичного растворителя с соответствующими свойствами до сих пор остается важной задачей для исследователей, применяющих этот метод. Лекарственное вещество и полимер (например, PLGA) растворяют в органическом растворителе, затем добавляют к воде, которая содержит поверхностно-активное вещество (часто – поливиниловый спирт), при необходимой температуре и режиме перемешивания. Затем органический растворитель постепенно выпаривают или экстрагируют, а частицы PLGA выделяются в виде сфер [16–19]. Первоначально эмульсию выдерживают при пониженном или атмосферном давлении, контролируя скорость перемешивания при испарении растворителя. Позже эмульсию переносят в большой объем воды (с поверхностно-активным веществом или без него) или в другую закалочную среду для удаления остатков органического растворителя. Подготовленные твердые нано- или микрочастицы затем промывают и сушат в подходящих условиях для получения конечного препарата микросфер для инъекций. Варьируя скорость перемешивания, концентрацию поверхностно-активного вещества и химическую структуру PLGA, можно контролировать размер частиц. На сегодняшний день многие гидрофобные препараты разных терапевтических групп были успешно загружены данным методом: антагонисты наркотических анальгетиков (налтрексон, циклазоцин), местные анестетики (лидокаин), противораковые агенты (цисплатин, 5-фтор-урацил), стероиды (прогестерон) и многие другие [20]. Метод испарения растворителя с использованием эмульсии “масло в воде” подходит для инкорпорирования гидрофобных соединений, но он не эффективен для гидрофильных лекарственных средств. Загрузка гидрофильных активных соединений с использованием эмульсии “масло в воде” способствует низкой эффективности загрузки и резким профилям высвобождения. Кроме того, препарат может диффундировать в непрерывную водную фазу во время эмульгирования, что приводит к большой потере лекарства. Для преодоления этих потерь было предложено несколько модифицированных методов, среди которых одним из наиболее эффективных является метод двойной эмульсии “вода в масле/в воде”.
Метод двойных эмульсий. Методы множественных эмульсий (например, “вода в масле/в воде”) хорошо подходят для загрузки гидрофильных соединений и лекарственных препаратов, таких как белки, пептиды и гидрофильные противораковые агенты. В отличие от методов простых эмульсий, которые идеально подходят для не растворимых в воде лекарств, двойные эмульсии лекарственного препарата, подлежащего введению в частицы, обычно получают следующим путем: готовят водный раствор гидрофильного лекарственного вещества и эмульгируют его в органической фазе, представляющей собой раствор полимера в органическом растворителе. Это приводит к образованию первой (или первичной) эмульсии, в которой водные капли, содержащие лекарственное средство, суспендированы в органической фазе (эмульсия типа “вода в масле”). Эмульсию затем диспергируют в основной водной среде, содержащей поверхностно-активные вещества, чтобы предотвратить агрегацию капель, формируя двойную эмульсию “вода/масло/вода”. Размеры получаемых капель можно регулировать, изменяя режимы диспергирования и гомогенизации. Добавление первичной эмульсии к раствору ПАВ и удаление органического растворителя приводит к формированию жесткой полимерной оболочки вокруг гидрофильного лекарственного вещества. Таким способом получают микрочастицы на основе гидрохлорида налтрексона, гидрохлорида псевдоэфедрина, пептида лейпролидацетата, вакцин и других веществ. Частицы на основе PLGA чувствительны к влаге, поэтому их сушат лиофилизацией для длительного хранения, часто вводя криопротекторы (обычно сахарозу или глюкозу) [21–23]. По сравнению с другими методами получения частиц метод испарения растворителя довольно трудоемок. Его продолжительность зависит от скорости испарения растворителя, которая может быть увеличена путем нагревания системы. Таким образом, возникает проблема стабильности лекарственного средства, особенно при работе с термочувствительными веществами. Важно не допустить в конечном продукте остатков токсичных растворителей (метиленхлорида, хлороформа, ацетонитрила, тетрагидрофурана или метанола), что достигается за счет длительной сушки. Очень часто процесс сушки приходится проводить при низких температурах из-за низких температур стеклования применяемых полимеров. Сообщается, что в некоторых случаях сушка может длиться более одной недели. Перечисленные выше недостатки метода испарения растворителя являются аргументом в пользу того, чтобы сосредоточить внимание на других методах приготовления микрочастиц, таких как сушка распылением, которая может обеспечить эффективное инкапсулирование широкого спектра лекарств – как гидрофобных, так и гидрофильных, более короткое время приготовления и более мягкие условия сушки.
Распылительная сушка
Распылительная сушка – сравнительно простой, недорогой и производительный метод получения полимерных микрочастиц с включенным биологически активным веществом. Процесс распылительной сушки является гибким и позволяет получать микрочастицы хорошего качества. Тип лекарственного средства (гидрофобное или гидрофильное) определяет выбор растворителя, который будет использоваться в процессе. Традиционно процесс представляет собой распыление раствора/твердой дисперсии полимера с активным веществом в горячую сухую среду, например в поток нагретого воздуха. На выходе из распылительного сопла поток раствора/дисперсии дробится на мелкие капли, которые в результате быстрого испарения растворителя переходят в твердое высушенное состояние. Это выполняется в три этапа: образование аэрозоля, контакт аэрозоля с теплым воздухом и сушка аэрозоля, а также разделение высушенного продукта и воздуха, насыщенного растворителем. Таким способом получают как микрочастицы с относительно равномерным распределением активного вещества по объему (в данном случае распылительной сушке подвергают совместный раствор полимера и активного вещества в органическом растворителе), так и микрочастицы типа “ядро–оболочка” (в данном случае распылительной сушке подвергают дисперсию вещества, образующего ядро микрочастицы и не растворимого в дисперсионной среде, в концентрированном растворе вещества, образующего оболочку). В литературе описана технология получения микросфер на основе PLGA для доставки диазепама, пироксикама, прогестона, теофиллина, витамина D3 и для инкапсуляции водорастворимых материалов (альбумина и вакцинных антигенов). Размер частиц микросфер, полученных методом распылительной сушки, составлял от нескольких микрон до нескольких десятков микрон и имел относительно узкое распределение [24]. Распылительную сушку раствора PLGA с активным веществом часто проводят в присутствии водного раствора углеводов (маннита, лактозы, трегалозы), кроме того, для снижения склонности к агломерации, вводят стабилизаторы (полиэтиленгликоли, полиэтиленимин и т.д.).
Микрофлюидные технологии
Перспективным подходом при получении полимерных частиц является использование микрофлюидных технологий (рис. 2). Данный метод подразумевает возможность манипулирования потоками жидкости и единичными каплями, что в свою очередь открывает широкие возможности использования микрофлюидных технологий для синтеза и сборки “умных” частиц, создания микрокапсул и полимеросом, биосовместимых клеточных матриц, а также высокопроизводительных методов анализа в экспериментальной биологии. Генерация монодисперсной эмульсии контролируемого размера и морфологии в максимально мягких условиях является одним из основных преимуществ капельной микрофлюидики по сравнению с классическими подходами к эмульсификации, основанными на механическом воздействии. Данный метод позволяет не только принципиально улучшить качество получаемых микро/наночастиц, но и обеспечить получение частиц сложной структуры (многослойных, многоядерных). Преимущество полимерных частиц, полученных в микрореакторах, заключается в более узком распределении частиц по размерам, что достигается за счет строгого и полного контроля условий в микрореакторе (давление, температура и скорость потоков).

Микрожидкостные устройства могут представлять собой капиллярные системы с прямыми потоками, с фокусировкой потока, а также сочетающие оба этих принципа. В капиллярных устройствах с прямоточным потоком водная фаза (например, ПВС в воде) вводится в два боковых канала, а органическая фаза, содержащая лекарство и полимер, направляется в центральный канал устройства с помощью шприцевых насосов с постоянной скоростью. Капли монодисперсной эмульсии непрерывно образуются в точках соединения объединенных микроканалов, используемых в капиллярном устройстве, с прямыми потоками для приготовления микросфер, нагруженных лекарственным препаратом (например, бупивакаином – местным анестетиком). В капиллярных устройствах с фокусировкой потока две жидкие фазы (т.е. органическая фаза, содержащая лекарство и полимер, и непрерывная водная фаза) вводятся с противоположных направлений в микрожидкостную смесительную ячейку. Внутренняя органическая фаза гидродинамически фокусируется потоком внешней водной жидкости через отверстие. Когда органическая фаза попадает в отверстие, она распадается (в условиях капания или недостаточного обдува) с образованием капель монодисперсной эмульсии [25–27]. По сравнению с методом простых эмульсий “масло в воде”, пустые микросферы PLGA, которые были получены методом фокусировки потока, имели более узкое распределение частиц по размерам. Все методы, используемые для получения микрочастиц, могут быть использованы для производства наночастиц PLGA (наносфер и нанокапсул) путем регулирования параметров процесса. За последние два десятилетия было разработано множество лекарственных форм на основе PLGA, которые использовали для лечения различных заболеваний [28–30]. Наиболее популярны PLGA с относительным составом (молярное соотношение лактид : гликолид) 50 : 50 и 75 : 25 [31]. Из-за гидролиза PLGA такие свойства, как температура стеклования Tg, содержание влаги и молекулярная масса, изменяются со временем. Эти изменения свойств PLGA также влияют на скорость высвобождения загруженного лекарственного вещества, которая зависит и от природы лекарства [32].
Перспективным типом полимеров для создания наночастиц являются амфифильные блок-сополимеры на основе лактида и этиленоксида. В отличие от PLGA, которые являются гидрофобными и нуждаются в обязательной стабилизации поверхностно-активными веществами, склонность к агрегации сополимеров лактида и этиленоксида заложена уже в самой их структуре. Отсутствие необходимости в удалении несвязанного с наночастицами стабилизатора дает большое технологическое преимущество.
БЛОК-СОПОЛИМЕРЫ ЛАКТИДА С ПОЛИЭТИЛЕНГЛИКОЛЕМ
Полиэтиленгликоль (PEG) – биосовместимый, нетоксичный, неиммуногенный водорастворимый полимер, широко используемый в фармацевтических и биомедицинских технологиях. Хотя PEG с молекулярной массой ниже определенной не разлагается в биологических средах, он может выводиться из организма через экскрецию. Показано, что PEG с молекулярной массой 4 × 103 на 98% выводится из организма человека [33]. Широкое использование производных PEG в системах направленной доставки лекарств связано с резистентностью PEG к белкам, поскольку образующиеся водородные связи PEG с водой исключают адсорбцию на них белков. Так как PEG является хорошо растворимым, высокополярным полимером, способным к образованию статистического клубка, который может эффективно стерически защищать поверхность различных нано- и микрочастиц, при использовании PEG разной функциональности и молекулярных масс возможно получать блок-сополимеры с различными гидрофобными блоками. Полимеризация лактида с раскрытием цикла – широко используемый метод синтеза двойных и тройных блок-сополимеров с PEG. Полимеризацию, как правило, проводят в блоке при 140–170°С в инертной среде, с использованием катализатора и макроинициатора (монофункциональный метоксиполиэтиленгликоль для синтеза двойных сополимеров или бифункциональный полиэтиленгликоль – для тройных блок-сополимеров) [34]. ROP позволяет синтезировать высокомолекулярные сополимеры в мягких условиях и контролировать молекулярную массу и молекулярно-массовое распределение [4].
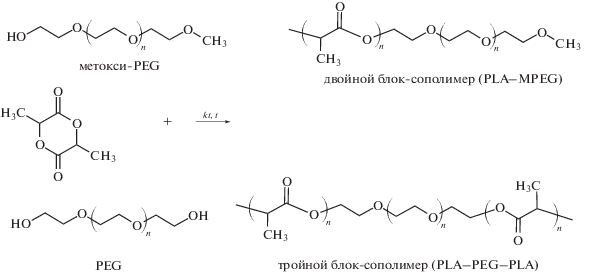
Такие блок-сополимеры возможно модифицировать по концевым гидроксильным группам. Например, H. Asadi с соавторами получали триблок-сополимеры PLA–PEG–PLA с концевыми диакриловыми фрагментами [35]. Также они характеризуются хорошей биосовместимостью и способностью к самоассоциации в водной среде, а наличие концевых двойных связей позволяет проводить фотосшивание этих мицелл, получая полимерные наногели. Металлоорганические соединения широко используются для катализа ROP лактида, а октаноат двухвалентного олова является одним из самых популярных катализаторов полимеризации лактонов в присутствии гидроксилсодержащих соединений [4, 36]. Чистота полимера и его ММР влияет на ожидаемые ключевые характеристики нанообъектов после агрегации (например, дзета-потенциал, гидродинамический диаметр частиц). Кроме того, возможное образование побочного продукта – гомополимера PLA, либо свободных блоков PEG, тоже может влиять на физико-химические характеристики получаемых наночастиц. Например, смешивание гомополимера PLA с сополимерами PLA–PEG изменяет поверхностную плотность PEG на наночастицах PLA–PEG [37, 38]. Как обсуждалось выше, поверхностная плотность PEG является важной характеристикой наночастиц, которая может влиять на коллоидную стабильность, а также на взаимодействие с белками плазмы, включая белки комплемента. Важно контролировать чистоту исходных макроинициаторов, поскольку присутствие примеси PEG-диола в метокси-PEG, используемом для синтеза диблок-сополимеров, может привести к образованию побочного продукта – триблок-сополимера, и, следовательно, к смеси блок-сополимеров разной топологии [39].
Амфифильные блок-сополимеры лактида с полиэтиленгликолем в водной среде способны самоорганизовываться в надмолекулярные структуры – мицеллы. В простейшем случае происходит образование сферических мицелл типа ядро–корона, в которых гидрофобный блок PLA формирует ядро, а гидрофильный PEG – корону. Мицеллы на основе данных блок-сополимеров являются биосовместимыми, биодеградируемыми, нетоксичными, а формирующий корону PEG обеспечивает необходимый стерический барьер и препятствует адсорбции белков плазмы на поверхности частиц, увеличивая тем самым время их циркуляции в организме. Таким образом, варьируя гидрофильно-липофильный баланс амфифильных сополимеров, а также стереоспецифичность и длину составляющих их блоков, можно получать частицы с регулируемым размером и морфологией. Несомненно, такие наночастицы имеют большой потенциал для создания на их основе средств пролонгированной и адресной доставки лекарств.
ЗВЕЗДООБРАЗНЫЕ ПОЛИЛАКТИДЫ
Стремительное развитие синтетических методологий позволяет получать полимеры со сложной архитектурой (звездообразные, сверхразветвленные, дендритные, полимерные щетки и другие), в которой полимерные сегменты разного типа (гидрофобные и/или гидрофильные, аморфные и/или кристаллизующиеся) объединены в одной макромолекуле. Звездообразные полимеры – это простейшие разветвленные полимеры, состоящие из нескольких линейных цепей, связанных с центральным разветвляющим центром. Согласно номенклатуре, рекомендованной IUPAC, звездообразные полимеры характеризуются наличием одной единственной точки разветвления, из которой берут начало линейные цепи. Функциональность этой точки обозначается числом отходящих от нее лучей (линейных цепей). Существует два основных способа синтеза звездообразных полимеров: дивергентный (расходящийся) и конвергентный (сходящийся). В качестве ядра используют низкомолекулярные вещества (например, многоатомные спирты) или макромолекулы с реакционноспособными функциональными группами. Ниже приведены примеры инициаторов для синтеза звездообразных полилактидов.

Звездообразные полимеры обладают уникальными свойствами по сравнению с их линейными аналогами. Благодаря высокой плотности сегментов разветвление приводит к более компактным структурам, что резко влияет на механические, вязкоупругие свойства и характеристики растворов [40]. Изучение этих свойств является предметом многочисленных теоретических и экспериментальных исследований. Для синтеза звездообразных полимеров используют стратегии “сначала ядро”, “сначала луч”, а также различные их сочетания [41–49]. В табл. 1 приведены примеры звездообразных полилактидов, синтезированных в присутствии ядер с различной функциональностью.
Таблица 1.
Примеры звездообразных (3-, 4- и 6-лучевых) PLA-полимеров, синтезированных методом ROP (катализатор Sn(Oct)2)
| Структура ядра | Структура луча | Число лучей PLA | Молекулярная масса полимера Mn × 10–3 | Литература |
|---|---|---|---|---|
| Глицерин | PDLLA | 3 | 4.0–100.0 | [50] |
| Триметилолпропан | PDLLA | 3 | 14.6–75.5 | [50, 51] |
| Пентаэритрит | PDLLA | 4 | 2.7–64.6 | [50, 52, 53] |
| Дипентаэритрит | PDLLA | 6 | 5.9–86.0 | [51, 54, 55] |
| Инозитол | PDLLA | 6 | 4.6–66.8 | [50, 56] |
| Сорбитол | PDLLA | 6 | 1.0–33.6 | [57, 58] |
| Ксилитол | PDLLA | 6 | 2.8 –33.6 | [57] |
| Холевая кислота | PDLLA | 3 | 3.3–23.1 | [50] |
| Триметилолпропан | PLLA | 3 | 2.3–75.5 | [59, 60] |
| Глицерин | PLLA | 3 | 2.8–130.0 | [61, 62] |
| Пентаэритрит | PLLA | 4 | 2.7–320.0 | [59, 63, 64, 68] |
| Дипентаэритрит | PLLA | 6 | 2.3–88.0 | [50, 65, 66] |
| Инозитол | PLLA | 6 | 11.0–34.0 | [50, 56, 67] |
| Сорбитол | PLLA | 6 | 1.0–37.0 | [57, 58] |
| Ксилитол | PLLA | 6 | 1.8–33.6 | [57] |
В случае производных лактида стратегия “сначала ядро” используется более широко, чем стратегия “сначала луч”. Звездообразные биоразлагаемые полиэфиры синтезируют методом ROP в блоке при 140–200°С в инертной среде, с использованием катализатора и многофункционального инициатора, а также в растворителях, например, тетрагидрофуране, м-ксилоле, толуоле, хлористом метилене, хлорбензоле, хлороформе, ДМСО. В то время как линейные полимеры получают ROP с моно- или дифункциональным спиртом, использование полиола приводит к образованию сильно разветвленных (звездообразных) структур.
Обычно количество “лучей” в полимере соответствует числу функциональных групп в инициаторе. Положение гидроксильной группы в ядре и отношение мономера к инициатору определяют, все ли гидроксильные группы инициируют рост цепи PLA. Показано, что первичные гидроксильные группы более эффективны в инициировании полимеризации лактида. Целью исследований [68] была систематическая оценка влияния количества гидроксильных групп инициатора на скорость полимеризации и оптимизация времени полимеризации, необходимого для образования высокомолекулярных полилактидов. Полимеризацию лактида проводили при 200°C (катализатор Sn(Oct)2), используемые инициаторы содержали от 1 до 12 гидроксильных групп. Было показано, что скорость полимеризации возрастает с увеличением количества гидроксильных групп в инициаторе, что видно из анализа кинетических кривых. Для всех инициаторов конверсия лактида составляла около 95%. Из полученных результатов следует, что функциональность инициатора влияет на скорость полимеризации, но не на равновесие мономер–полимер. Использование инициаторов с разным числом гидроксильных групп позволило получить PLLA с разным количеством лучей. Важным условием эффективной инициирующей активности является растворимость инициатора в полимеризационной смеси. Плохой растворимостью характеризуются инициаторы с высокой температурой плавления: пентаэритрит, дипентаэритрит, и β-циклодекстрин, что заметно осложняло полимеризацию, как показано в работе [69]. Используя многофункциональные инициаторы, можно получать различные молекулярные архитектуры со строго определенным числом лучей: от 3 до 32. Например, применяя дендритные инициаторы, полученные из производных 2,2-бис-(гидроксиметил)пропионовой кислоты (bis-MPA), получены звездообразные PDLLA, PLLA и PCL с 2, 4, 6 или 12 лучами, соответствующими количеству гидроксилов, и узким ММР ( менее 1.1). Звезды с большим количеством лучей (до 160) получают, используя сверхразветвленные полимеры и/или дендримеры [44–48, 70, 71]. Более сложные полиолы также могут служить инициаторами, например октафункциональный полиэдрический олигомерный силсесквиоксан (POSS) [72]. Почти во всех работах в качестве катализатора применяют Sn(Oct)2. Также использовали и другие катализаторы, такие как гидрид кальция, ацетилацетонат олова, гексаметилдисилазид калия, тетрафенилолово, ацетат Bi (III) (Bi (OAc)3) и даже фермент липаза. Система на основе Bi(OAc)3 была сопоставима по своей каталитической эффективности с Sn(Oct)2 при 140°C. Публикаций о получении звездообразных полилактидов методом поликонденсации немного. Синтезированные полимеры обычно имели относительно низкие молекулярные массы (Mn < < 4.7 × 103). Полимолочная кислота с более высокой молекулярной массой (Mn ~ 67 × 103) была получена прямой поликонденсацией молочной кислоты, инициированной дипентаэритритом и пентаэритритом, с триоксидом сурьмы и п-толуолсульфоновой кислотой в качестве катализаторов соответственно. Кроме того, звездообразные 3- и 4-лучевые сополимеры PEG–PLA были получены из разветвленного полиэтиленгликоля [73–75]. Также звездообразный полилактид получали путем ROP D,L-лактида в сверхкритическом диоксиде углерода с D-сорбитом в качестве инициатора. Этот процесс выполнялся при значительно более низких температурах (до 80°C) по сравнению с реакциями в блоке (140°C). В результате наблюдалось уменьшение образования побочных продуктов, что привело к более узкой дисперсности полимера и хорошему контролю степени разветвления [76]. Обычно звездообразные полилактиды синтезируют методом ROP, однако известно о применении других подходов, например RAFT и ATRP [77, 78]. Многие звездообразные полимеры на основе лактонов в конечном счете могут использоваться для получения наночастиц для направленной доставки лекарственных препаратов. Недавно было показано, что 3-, 4- и 6-лучевые блок-сополимеры PLGA–PEG (соответственно инициированные триметилолпропаном, пентаэритритом и дипентаэритритом) демонстрируют пониженные значения критических концентраций мицеллообразования более медленное высвобождение инкапсулированного доксорубицина и определенное усиление интернализации в клетках линии HeLa [79]. Эти звездообразные полимеры, вероятно, не образуют мономолекулярные мицеллы: средний размер их агрегатов в воде находится в диапазоне 80–200 нм и увеличивается с ростом степени разветвления. Это свидетельствует о том, что коллоиды образуются посредством агрегации полимера и становятся более стабильными из-за более частых зацеплений в результате наличия точек ветвления (отсюда более низкие значения критической концентрации мицеллообразования). В ряде работ показано, что наночастицы на основе звездообразных полимеров можно нагружать такими препаратами, как индометацин, ибупрофен и другие [72, 80–82].
РАЗВЕТВЛЕННЫЕ ПОЛИМЕРЫ НА ОСНОВЕ ПОЛИГЛИЦЕРИНА
Для получения звездообразных полимеров, число лучей в которых более 10, необходимо использовать инициаторы, содержащие большое количество реакционноспособных групп – сверхразветвленные полимеры и дендримеры. Физико-химические свойства сверхразветвленных полимеров можно менять в достаточно широких пределах, не изменяя существенно исходную макромолекулу, а лишь варьируя набор функциональных групп в концевых цепях и степень их модификации. В последние годы дендримеры, а также сверхразветвленные полимеры привлекают серьезное внимание из-за их уникальных свойств, обусловленных разветвленной архитектурой и большим количеством функциональных групп. Сильно разветвленная и плотная, но нерегулярная, структура сверхразветвленных полимеров приводит к повышенной растворимости по сравнению с линейными аналогами, низкой вязкости раствора и высокой концентрации функциональных концевых групп [83–85].
Исследование звездообразных полимеров со сверхразветвленным высокомолекулярным ядром представляет особый интерес. Сверхразветвленные полимеры являются особым типом дендритных полимеров и имеют очень высокую плотность ветвления с возможностью разветвления в каждом мономерном звене. Их обычно получают одностадийным синтезом, при котором сложно контролировать разветвления и молекулярную массу с высокой точностью, что приводит к продуктам с широким ММР. Это отличает сверхразветвленные полимеры от идеально разветвленных и монодисперсных дендримеров. В литературе имеется большое количество работ, посвященных исследованию сверхразветвленного полиглицерина и его производных в медицинских приложениях [81–89]. Одобренные FDA олиго- и полиглицерины (PG) занимают особое положение в сфере наномедицины благодаря своей топологической однородности, биосовместимости и многофункциональности. Кроме того, PG могут разлагаться ферментами с образованием нетоксичных легко выводимых метаболитов. По этим причинам он нашел множество применений в качестве носителя для доставки лекарств. PG получают полимеризацией с раскрытием цикла. Описано множество подходов к синтезу PG разной структуры, которые предполагают большое разнообразие по степени разветвления, размера, топологии поверхности и химических свойств в целом. Ниже приведена схема синтеза сверхразветвленного полиглицерина из глицидола.

Наряду с синтезом сверхразветвленных полиглицеринов (HBPG) в последнее десятилетие также сообщалось о путях синтеза идеальных дендритных PG, дендронов, микрогелей и гидрогелей. Высокомолекулярный PG обладает высокой гидрофильностью и имеет большое количество концевых гидроксильных групп, которые можно легко модифицировать для получения широкого круга соединений [86].
Модификация гидроксильных групп PG гидрофобными блоками позволяет получать амфифильные системы с гидрофильным ядром и гидрофобной внешней оболочкой, которые проявляют свойства “хозяин–гость”. За последние десятилетия синтезирован широкий ряд PG различной топологии и молекулярных масс. I. Arvanitoyannis с соавторами [87] синтезировали трехлучевой звездообразный PLLA через ROP, используя глицерин в качестве ядра. Синтезированный полиэфир обладал низкой степенью кристалличности, пониженной вязкостью расплава и высокой молекулярной массой. Многолучевые звездообразные блок-сополимеры были получены с использованием олигомеров сверхразветвленных PG в качестве инициаторов. На первом этапе синтезировали сверхразветвленный полиглицерин (ядро), а затем проводили полимеризацию L-лактида на концевых группах HBPG в различных соотношениях, получая линейно-разветвленные сополимеры с разной длиной полилактидного блока [88]. ROP лактида проводили при 115°C в течение 24 ч (катализатор Sn(Oct)2). Аналогичные линейно-дендритные сополимеры могут быть получены химическим связыванием полилактидных блоков карбодиимидным методом с HBPG [89]. Преимущество этого метода состояло в том, что все ветви PLA имели одинаковый молекулярный вес. Установлено, что скорость высвобождения кверцетина (плохо растворимого в воде лекарственного средства) снижалась с увеличением Mn сополимеров HBPG–PLA. Аналогично методом ROP были синтезированы сополимеры с гликолидом. Сополимеры HBPG–PGA были частично кристаллическими со значениями Tg = = 10–18°C и Tm в диапазоне 170–190°C. Агрегация HBPG–PLA в воде приводит к образованию наночастиц со структурой “ядро–корона”. Описан синтез сополимеров HBPG–PLGA карбодиимидным методом. Эти сополимеры могли образовывать наночастицы, функционализированные антителами к трансферрину [90]. В ряде исследований показана возможность регулировать размеры и морфологию наноструктур на основе HBPG–PLA, инкапсулировать в них терапевтические агенты и высвобождать их контролируемым образом. В работах [91–96] показано, что гидрофобная оболочка наночастиц на основе HBPG–PLA способна контролировать скорость высвобождения инкапсулированных водорастворимых лекарств. Чтобы рассмотреть возможность использования HBPG-подобных структур в неврологии, T. Liu с соавторами разработали новую систему доставки лекарств в мозг на основе HBPG-PLGA наночастиц, поверхность которых была функционализирована антителом к трансферрину. Наночастицы получали методом двойной эмульсии [128].
РАЗВЕТВЛЕННЫЕ ПОЛИМЕРЫ НА ОСНОВЕ ПОЛИЭФИРОВ BOLTORN
В связи с широкими возможностями модификации и потенциальным использованием в медицине, в настоящее время уже коммерчески доступны следующие сверхразветвленные полимеры: полиамидоамин (PAMAM), полипропиленимин (PPI), также известный как Astromol, а также полиэфиры марки Boltorn. Одним из перспективных сверхразветвленных полимеров является сверхразветвленный полиэфир, получаемый конденсацией 2,2-бис(метилол)пропионовой кислоты с многоатомными спиртами (торговое название – полиэфир Boltorn). Будучи целиком построенным из биосовместимых биоразлагаемых блоков, данный полиэфир может быть использован в качестве “платформы” для синтеза целого ряда соединений, которые могут найти применение в самых разных отраслях человеческой деятельности, включая целевую доставку лекарственных веществ. При этом чрезвычайно важным является исследование структуры как исходного полиэфира, так и его модифицированных аналогов. Boltorn представляет собой алифатический сверхразветвленный полиэфир, получаемый поликонденсацией 3-гидрокси-2-(гидроксиметил)-2-метилпропионовой кислоты (или 2,2-бис-(метилол)пропионовой кислоты, bis-MPA), которая является мономером типа AB2 и содержит одну карбоксильную (A = –COOH) и две гидроксильные (B = –OH) функциональные группы. Синтез сверхразветвленных полимеров на основе bis-MPA может быть осуществлен как с использованием гидроксилсодержащего “ядра” Bf (f – число функциональных гидроксильных групп), так и без него. Полиэфиры Boltorn марок H20, H30 и H40 представляют собой прогрессивно разветвляющиеся дендритоподобные макромолекулы, содержащие три ветви (в случае разветвляющего центра триметилолпропана) с большим количеством гидроксильных и сложноэфирных групп, они являются биосовместимыми биодеградируемыми полимерами, обладают биодоступностью, биопроницаемостью и низкой токсичностью (ЛД50 = 2000 мг/кг). Полиэфирполиолы данной серии малорастворимы в воде, причем с ростом генерации растворимость в воде снижается, их гидрофобность менее выражена, чем гидрофобность полиолефинов. Известно, что с увеличением генерации сверхразветвленных полимеров возрастает их степень разветвленности, а также размеры и емкость для гостевых молекул.
В структуре полиэфира Boltorn кроме разветвляющего центра (ядра), присутствуют линейные, дендритные и концевые звенья, из соотношения которых может быть рассчитана степень разветвленности:

Активными центрами являются гидроксильные группы, которые расположены в терминальных и линейных единицах. Наличие большого числа свободных гидроксильных групп (как внутри, так и по периферии макромолекул) делает их особо привлекательными для химической модификации с целью применения в биологии и медицине. В результате такой модификации получены сверхразветвленные макромолекулы, содержащие по периферии карбоксильные, амино-, амидо- и силиламинные группы, а также серо-, фосфор- и фторсодержащие группировки. Модифицированные полимеры приобретают способность к самосборке в водных растворах и образованию наноструктур, способных удерживать лекарственные препараты. Показана перспективность использования модифицированных PEG полиэфиров Boltorn для контролируемого высвобождения доксорубицина при терапии рака груди [97, 98].
Для увеличения времени циркуляции и загрузки лекарственным препаратом, в ряде работ Boltorn H40 модифицировали поли-L-лактидом [99], поли-ε-капролактоном [100–102], поли-L-аспартатом) [103] и поли-L-глутаматом [104], после чего гидрофилизировали блоками PEG и получали на их основе наноструктуры.
Синтез многолучевых линейно-дендритных полимеров на основе Boltorn, проводят, как правило, ROP лактонов в блоке, в инертной среде при температурах 140–160°С. Концевые гидроксильные группы модифицируют либо PEG, что позволяет увеличить время циркуляции in vivo, либо фолатами или RGD-пептидами [105–113]. В табл. 2 представлен перечень линейно-дендритных сополимеров, описанных в литературе.
Таблица 2.
Сверхразветвленные полимеры Boltorn-PLA (катализатор Sn(Oct)2)
| Структура ядра | Структура луча | Число лучей PLA | Молекулярная масса сополимера Mn × 10–3 | Литература |
|---|---|---|---|---|
| Boltorn H20 | PLLA | 16 | 17.8–157.7 | [115] |
| Boltorn H20 | PLLA-b-PCL | 16 | 51.8–104.3 | [114] |
| Boltorn H20 | PLLA PLLA-b-PTMC |
16 | 45.4 45.4–56.0 |
[116] |
| Boltorn H40 | PDLA | 64 | 260.0 | [114] |
| Boltorn H40 | PLLA PLLA-b-TPGS |
64 | 26.0 55.0 |
[114] |
| Boltorn H40 | PLLA-b-PEG | 64 | 108.5 | [117] |
| Boltorn H40 | PLLA-b-mPEG PLLA-b-PEG-FA |
20 | 108.5 | [118] |
| Boltorn H40 | PDLLA | 26 | 26.0 | [114] |
В линейно-дендритных блок-сополимерах Boltorn–PLA–PEG полилактидные блоки способствуют загрузке лекарственного средства, как это очевидно, например, когда доксорубицин ковалентно присоединяется к ним через pH-чувствительные связи [110, 119]. Данное преимущество, возможно, менее очевидно в случае физической загрузки лекарств, поскольку гидрофобные соединения, такие как паклитаксел, могут быть физически загружены также в пэгилированном Boltorn H40, не имеющим промежуточных цепей [120]. На основе Boltorn H40 были разработаны амино-функционализированные системы в качестве катионных носителей для нуклеиновых кислот [121], аналогично тому, что было изучено с использованием сверхразветвленных поли(аминоэфиров) [122, 123]. По сравнению с одним из лучших агентов трансфекции на основе сверхразветвленного полиэтиленимина, аминофункциональный Boltorn H40 в целом оказывается менее эффективным, но значительно менее цитотоксичным. Например, он использовался в концентрациях до 1 мг/мл без какой-либо значительной потери жизнеспособности клеток [121]. Для производных Boltorn обычно наблюдаются очень низкие значения критической концентрации агрегации: в диапазоне нескольких мкг/мл [101, 102, 110, 124], и они могут просто отражать недостаточную чувствительность аналитических методов (определяется обычно флуоресценцией пирена), чем реальная деагрегация, которая поддерживала бы концепцию гидрофобных доменов с очень небольшой зависимостью от концентрации. Кроме того, эти системы обычно имеют размеры от нескольких десятков до 100–200 нм, что, тем не менее, предполагает мультимолекулярную природу коллоидных частиц. В работе [125] получены мономолекулярные мицеллярные наноразмерные препараты на основе Boltorn–PCL, модифицированные PEG и яблочной кислотой. Некоторые блоки PEG были модифицированы фолиевой кислотой, а DOX был конъюгирован через pH-чувствительные гидразоновые связи, которые расщепляются в кислой среде [125]. Аналогичным образом октреотид конъюгирован на Boltorn–PLLA–PEG [126, 127], демонстрируя повышенную противораковую активность.
ЗАКЛЮЧЕНИЕ
В последнее десятилетие наблюдается заметный рост интереса к биоразлагаемым полиэфирам для использования в медицине и фармацевтике. Обзор показывает, что в настоящее время разработаны методы синтеза гидрофобных и амфифильных биоразлагаемых полимеров различной топологии: линейных, звездообразных, сверхразветвленных и т.д. Такой широкий ассортимент структур и составов позволяет гибко конструировать системы доставки лекарств, выбирая оптимальный материал для каждого активного агента. Разработка новых катализаторов, а также методов синтеза функциональных биоразлагаемых полимеров с контролируемой топологией и физико-химическими характеристиками является актуальным направлением исследований.
Работа выполнена при частичной финансовой поддержке Российского научного фонда (соглашение с РНФ 18-73-10079 от 01.08.2018 г.). Разделы о полимерах PLGA и материалах на основе полиглицерина подготовлены при поддержке внутреннего гранта НИЦ “Курчатовский институт” (Приказ №1058 от 02.07.2020 г.).
Список литературы
Demina T.S., Gilman A.B., Zelenetskii A.N. // High Energy Chem. 2017. V. 51. № 4. P. 302.
Nair L.S., Laurencin C.T. // Prog. Polym. Sci. 2007. V. 32. P. 762.
Demina T.S., Sotnikova Yu.S., Istomin A.V., Grandfils Ch., Akopova T.A., Zelenetskii A.N. // Adv. Mater. Sci. Eng. 2018. P. 8518016.
Jerome C., Lecomte P. // Adv. Drug Delivery Rev. 2008. V. 60. № 9. P. 1056.
Demina T.S., Akopova T.A., Zelenetskii A.N., Vladimirov L.V., Markvicheva E.A., Grandfils C. // Mater. Sci. Eng. C. 2016. V. 59. P. 333.
Kalinin K.T., Sedush N.G., Dmitryakov P.V., Chvalun S.N. // Chem. Open. 2020. V. 9. P. 1027.
Platel R.H., Hodgson L.M., Williams C.K. // Polym. Rev. 2008. V. 48. P. 11.
Sawyer D.J. // Macromol. Symp. 2003. V. 201. P. 271.
Malafeev K.V., Moskalyuk O.A., Yudin V.E., Sedush N.G., Chvalun S.N., Elokhovskii V.Yu., Popova E.N., Ivan’kova E.M. // Polymer Science A. 2017. V. 59. № 1. P. 53.
Shuklov I.A., Jiao H., Börner A., Schulze J., Tietz W., Kühlein K. // Tetrahedron Lett. 2011. V. 52. № 9. P.1027.
Gomzyak V.I., Demina V.A., Razuvaeva E.V., Sedush N.G., Chvalun S.N. // Fine Chem. Technol. 2017. V. 12. № 5. P. 5.
Penczek S., Cypryk M., Duda A., Kubisa P., Slomkowski S. // Prog. Polym. Sci. 2007. V. 32. P. 247.
Gunatillake P., Mayadunne R., Adhikari R. // Biotechnol. Ann. Rev. 2006. V. 12. P. 301.
Pradip J., Mousumi S., Sneha S., Venkatesan J., Abhimanyu D. // Eur. Polym. J. 2021. V. 142. P. 110155.
Middleton J.C., Tipton A.J. // Biomaterials. 2001. V. 21. № 23. P. 2335.
Chung T.-W., Huang Y.-Y., Liu Y.-Z. // Int. J. Pharm. 2001. V. 212. P. 161.
Almeida A.J., Alpar H., Brown M. // J. Pharm. Pharmacol. 1993. V. 45. P. 198.
Asmatulu R., Zalich M.A., Claus R.O., Riffle J.S. // J. Magn. Magn. Mater. 2005. V. 92. P. 108.
Zielhuis S.W., Nijsen J.F.W., Figueiredo R., Feddes B., Vredenberg A.M., Van Het Schip A.D., Hennink W.E. // Biomaterials. 2005. V. 26. P. 925.
Li M., Rouaud O., Poncelet D. // Int. J. Pharmaceut. 2008. V. 363. P. 26.
Arshady R. // J. Control. Release. 1991. V. 17. P. 1.
Chaisri W., Hennink W.E., Okonogi S. // Curr. Drug Deliv. 2009. V. 6. P. 69.
Mao S., Xu J., Cai C., Germershaus O., Schaper A., Kissel T. // Int. J. Pharm. 2007. V. 334. P. 137.
He P., Davis S., Illum L. // Int. J. Pharm. 1999. V. 187. P. 53.
Perez A., Hernández R., Velasco D., Voicu D., Mijangos C. // J. Colloid Interface Sci. 2015. V. 441. P. 90.
Xu Q., Hashimoto M., Dang Tram T., Hoare T., Kohane D.S., Whitesides G.M., Langer R., Anderson D.G. // Small. 2009. V. 5. № 13. P. 1575.
Petrova E.A., Kedik S.A., Alekseev K.V., Blynskaya E.V., Panov A.V., Suslov V.V., Tikhonova N.V. // Pharmaceut. Chem. J. 2014. V. 48. P. 65.
Makadia H.K., Siegel S.J. // Polymers. 2011. V. 3. P. 1377.
Lee L.Y., Ranganath S.H., Fu Y., Zheng J.L., Lee H.S., Wang C.-H., Smith K.A. // Chem. Eng. Sci. 2009. V. 64. P. 4341.
Alonso-Sande M., des Rieux A., Fievez V., Sarmento B., Delgado A., Evora C., Remuñán-López C., Préat V., J. Alonso M. // Biomacromolecules. 2013. V. 14. P. 4046.
Konan Y.N., Radovan C., Joselyne F., Myriam B., Gurny R., Allémann E. // Eur. J. Pharm. Biopharm. 2003. V. 55. P. 115.
Siegel S.J., Kahn J.B., Metzger K., Winey K.I., Werner K., Dan N. // Eur. J. Pharm. Biopharm. 2006. V. 64. P. 287.
Goddard P., Hutchinson Lusie E., Brown J., Brookman L.J. // J. Control. Release. 1989. V. 10. P. 5.
Zhu K.J., Xiangzhou L., Shilin Y. // J. Appl. Polym. Sci. 1990. V. 39. P. 1.
Asadi H., Rostamizadeh K., Salari D., Hamadi M. // Int. J. Pharm. 2011. V. 416. № 1. P. 356.
Hedrick J.L., Kiesewetter M.K., Shin E.J., Waymouth R.M. // Macromolecules. 2010. V. 43. P. 2093.
Vittaz M., Bazile D., Spenlehauer G., Verrecchia T., Veillard M., Puisieux F., Labarre D. // Biomaterials. 1996. V. 17. P. 1575.
Lee D., Teraoka I., Fujiwara T., Kimura Y. // J. Chromatogr. A. 2002. V. 966. P. 41.
Bouissou C., Rouse J.J., Price R., van der Walle C.F. // Pharm. Res. 2006. V. 23. P. 1295.
Angot S., Murthy K.S., Taton D., Gnanou Y. // Macromolecules. 1998. V. 31. P. 7218.
Yuan W., Zhu L., Huang X., Zheng S., Tang X. // Polym. Degrad. Stab. 2005. V. 87. № 3. P. 503
Li M., Jahed N.M., Min K., Matyjaszewski K. // Macromolecules. 2004. V. 37. P. 2434.
Matyjaszewski K., Miller P.J., Pyun J., Kickelbick G., Diamanti S. // Macromolecules. 1999. V. 32. P. 6526.
Matyjaszewski K., Gaynor S.G. // Macromolecules. 1997. V. 30. P. 7042.
Gao H., Matyjaszewski K. // Progr. Polym. Sci. 2009. V. 34. P. 317.
Gao H., Matyjaszewski K. // Macromolecules. 2006. V. 39. P. 3154.
Gao H., Matyjaszewski K. // Macromolecules. 2007. V. 40. P. 399.
Dong C.-M., Qiu K.-Y., Gu Z.-W., Feng X.-D. // Macromolecules. 2001. V. 34. P. 4691.
Rainbolt E.A., Washington K.E., Biewer M.C., Stefan M.C. // Polym. Chem. 2015. V. 6. № 13. P. 2369.
Zhang W., Zheng S. // Polym. Bull. 2007. V. 58. P. 767.
Yuan M., He Z., Li H., Jiang L. // Polym. Bull. 2014. V. 71. № 6. P. 1331.
Matsumura S., Mabuchi K., Toshima. K. // Macromol. Rapid Commun. 1997. V. 18. P. 477.
Michalski A., Makowski T., Biedron T., Brzezinski M., Biela T. // Polymer. 2016. V. 90. P. 242.
Li Z., Tan B.H., Lin T., He C. // Progr. Polym. Sci. 2016. V. 62. P. 22.
Numata K., Srivastava R.K., Finne-Wistrand A., Albertsson A.C., Doi Y., Abe H. // Biomacromolecules. 2007. V. 8. № 10. P. 3115.
Hao Q., Li F., Li Q., Li Y., Jia L., Yang J., Fang Q., Cao A. // Biomacromolecules. 2005. V. 6. № 4. P. 2236.
Arvanitoyannis I., Nakayama A., Psomiadou E., Kawasaki N., Yamamoto N. // Polymer. 1996. V. 37. № 4. P. 651.
Kundys A., Plichta A., Florjanczyk Z., Zychewicz A., Lisowska P., Parzuchowski P., Wawrzynska E. // Polym. Int. 2016. V. 65. № 8. P. 927.
Kricheldorf H.R., Hachmann-Thiessen H., Schwarz G. // Biomacromolecules. 2004. V. 5. № 2. P. 492.
Tsuji H., Miyase T., Tezuka Y., Saha S.K. // Biomacromolecules. 2005. V. 6 № 1. P. 244.
Socka M., Brzezinski M., Michalski A., Kacprzak A., Makowski T., Duda A. // Langmuir. 2018. V. 34. № 12. P. 3701.
Wang L., Dong C.M. // J. Polym. Sci., Polym. Chem. 2006. V. 44. № 7. P. 2226.
Kim S.H., Han Y.K., Kim Y.H., Hong S.I. // Makromol. Chem. 1992. V. 193. № 7. P. 1623.
Cai C., Wang L., Dong C. // J. Polym. Sci., Polym. Chem. 2006. V. 44. № 6. P. 2034.
Shaver M.P., Cameron D. // Biomacromolecules. 2010. V. 11. № 12. P. 3673.
Meihua Xie, Juan Ge, Yumeng Xue, Yuzhang Du, Bo Lei, Peter X Ma // J. Mech. Behav. Biomed. 2015. V. 51. P. 163.
Asadi H., Rostamizadeh K., Salari D., Hamadi M. // Int. J. Pharm. 2011. V. 416. № 1. P. 356.
Cornelis A.P. Joziasse, Grablowitz H., Albert J. Pennings // Macromol. Chem. Phys. 2000. V. 201. P. 107.
Atthoff B., Trollsås M., Claesson H., Hedrick J.L. // Macromol. Chem. Phys. 1999. V. 200. P. 1333.
Coulembier O., Kiesewetter M.K., Mason A., Dubois P., Hedrick J.L., Waymouth R.M. // Angew. Chem., Int. Ed. 2007. V. 46. P. 4719.
Shi-He Luo, Qun-Fang Wang, Jin-Feng Xiong, Zhao-Yang Wang // J. Polym. Res. 2012. V. 19. P. 9962.
Lele B.S., Leroux J.C. // Polymer. 2002. V. 43. P. 5595.
Kim S.H., Kim Y.H. // Macromol. Symp. 1999. V. 144. P. 277.
Lin Y., Zhang A., Wang L. // J. Appl. Polym. Sci. 2012. V. 124. P. 4496.
Goddard A.R., Pérez-Nieto S., Howdle S.M., Carmichael K., Passos T.M., Quilty B., Irvine D.J. // Green. Chem. 2016. V. 18. P. 4772.
Xiao-Hui Dai, Zhi-Ming Wang, Wei Liu, Chang-Ming Dong, Jian-Ming Pan, Si-Song Yuan, Yong-sheng Yan, Dong-Ming Liu, Lin Sun // Colloid Polym. Sci. 2014. V. 292. P. 2111.
Zhao Y., Shuai X., Chen C., Xi Fu // Macromolecules. 2004. V. 37. P. 8854.
Arvanitoyannis I., Nakayama A., Kawasaki N., Yamamoto N. // Polymer. 1995. V. 36. P. 2947.
Doganci E., Tasdelen M.A., Yilmaz F. // Macromol. Chem. Phys. 2015. V. 216. P. 1823.
Dai X.H., Dong C.M. // J. Polym. Sci., Polym. Chem. 2008. V. 46. P. 817.
Gou P.-F., Zhu W.-P., Shen Z.-Q. // Biomacromolecules. 2010. V. 11. P. 934.
Wang F., Rauh R.D., Bronich T.K., Kabanov A.V., Roovers J. // Bioconjug. Chem. 2008. V.19. P. 1423.
Voit B.I. // J. Polym. Sci., Polym. Chem. 2000. V. 38. P. 2505.
Jikei M., Kakimoto M. // J. Polym. Sci., Polym. Chem. 2004. V. 42. P. 1293.
Lim M.P.A., Lee W.L., Widjaja E., Chye S., Loo J. // Biomater. Sci. 2013. V. 1. P. 486.
Burgath A., Sunder A., Neuner I., Mülhaupt R., Frey H. // Macromol. Chem. Phys. 2000. V. 201. P. 792.
Gottschalk C., Wolf F., Frey H. // Macromol. Chem. Phys. 2007. V. 208. P. 1657.
Xiujun Gao, Xinge Zhang, Xuejiao Zhang, Yanxia Wang, Lei Sun, Chaoxing Li // J. Pharm. Pharmacol. 2011. V. 63. P. 757.
Wolf F.K., Fischer A.M., Frey H. // Beilstein J. Org. Chem. 2010. V. 6. P. 67.
Mohiuddin M.C., Quadir A., Sunil Q., Sharma K., Haag R. // Adv. Mater. 2010. V. 22. P. 190.
Deng Y., Saucier-Sawyer J.K., Hoimes C.J., Zhang J., Seo Y.-E., Andrejecsk J.W., Saltzman W.M. // Biomaterials. 2014. V. 35. P. 6595.
Vijayakumar M.R., Muthu M.S., Singh S. // Expert Opinion on Drug Delivery. 2013. V. 10. P. 529.
Mehrabadi S.F., Hirsch O., Zeisig R., Posocco P., Laurini E., Pricl S., Haag R., Kemmner W., Calderón M. // RSC Adv. 2015. V. 5. P. 78760.
Gao X., Zhang X., Wu Z., Zhang X., Li C. // J. Control. Release. 2009. V. 140. P. 141.
Hecht S., Vladimirov N., Fréchet J.M.J. // J. Am. Chem. Soc. 2001. V. 123. P. 18.
Biela T., Duda A., Rode K., Pasch H. // Polymer. 2003. V. 44. P. 1851.
Zeng X., Morgenstern R., Nystrom A.M. // Biomaterials. 2014. V. 35. № 4. P. 1227.
Bao J., Chang X., Xie Q., Yu C., Shan G., Bao Y., Pan P. // Macromolecules. 2017. V. 50. № 21. P. 8619.
Chen S., Zhang X.-Z., Cheng S.-X., Zhuo R.-X., Zhong W.-G. // Biomacromolecules. 2008. V. 9. P. 2578.
Aryal S., Prabaharan M., Pilla S., Gong S. // Int. J. Biol. Macromol. 2009. V. 44. P. 346.
Wang D., Chen H., Su Y., Qiu F., Zhu L., Huan X., Zhu B., Yan D., Guo F., Zhu X // Polym. Chem. 2013. V. 4. № 1. P. 85.
Prabaharan M., Grailer J.J., Pilla S., Steeber D.A., Gong S. // Biomaterials. 2009. V. 30. № 29. P. 5757.
Xiao Y., Hong H., Javadi A., Engle J.W., Xu W., Yang Y., Zhang Y., Barnhart T., Cai W., Gong S. // Biomaterials. 2012. V. 33. № 11. P. 3071.
Corneillie S., Smet M. // Polym. Chem. 2014. V. 6. № 6. P. 850.
Lukowiak M.C., Thota B.N.S., Haag R. // Biotechnol. Adv. 2015. V. 33. № 6. P. 1327.
Prabaharan M., Grailer J.J., Pilla S., Steeber D.A., Gong S. // Biomaterials. 2010. V. 30. № 16. P. 3009.
Yang H., Zhao X., Zhang X., Ma L., Wang B., Wei H. // J. Polym. Sci., Polym. Chem. 2018. V. 56. № 13. P. 1383.
Istratov V.V., Krupina T.V., Vasnev V.A., Gomzyak V.I. // High Performance Polymers. 2017. V. 29. № 6. P. 708.
Sun C., Sze R., Zhang M. // J. Biomed. Mater. Res. A. 2006. V. 78. № 3. P. 550.
Istratov V.V., Gomzyak V.I., Krupina T.V., Vasnev V.A., Chvalun S.N. // Polymer Science B. 2017. V. 59. № 6. P. 730.
Li X., Qian Y., Liu T., Hu X., Zhang G., You Y., Liu S. // Biomaterials. 2011. V. 32. № 27. P. 6595.
Wu Y., Xu G., Jin X., Zhu X. // Sci. China Mater. 2018. V. 61. № 11. P. 144.
Rezaei S.J.T., Abandansari H.S., Nabid M.R., Niknejad H. // J. Colloid Interface Sci. 2014. V. 425. P. 27.
Zhang W., Zheng S. // Polym. Bull. 2007. V. 58. P. 767.
Coady D.J., Engler A.C., Yang Y.Y., Hedrick J.L. // Polym. Chem. 2011. V. 2. № 11. P. 2619.
Suneel K.S., Buzza D.M.A., Groves D.J., McLeish T.C.B., Parker D., Keeney A.J., Feast W.J. // Macromolecules. 2002. V. 35. № 25. P. 9605.
Smart Polymer Materials for Biomedical Applications / Ed. by S. Li, A. Tiwari, M. Prabaharan, S. Aryal. New York: Nova Sci. Publ., 2010.
Yang X., Grailer J.J., Pilla S., Steeber D.A., Gong S. // Bioconjug. Chem. 2010. V. 21. № 3. P. 496.
Kedik S.A., Zhavoronok E.S., Sedishev I.P., Panov A.V., Suslov V.V., Petrova E.A., Sapelnikov M.D., Shatalov D.O., Eremin D.V. // Res. Development of New Drug Prod. 2013. V. 2. № 3. P. 18.
Chen H., Li G., Chi H., Wang D., Tu C., Pan L., Zhu L., Qiu F., Guo F., Zhu X. // Bioconjug. Chem. 2012. V. 23. P. 1915.
Pang Y., Zhu Q., Liu J., Wu J., Wang R., Chen S., Zhu X., Yan D., Huang W., Zhu B. // Biomacromolecules. 2010. V. 11. № 3. P. 575.
Wu D., Liu Y., Jiang X., He C., Goh S.H., Leong K.W. // Biomacromolecules. 2006. V. 7. № 6. P. 1879.
Ornatska M., Peleshanko S., Genson K.L., Rybak B., Bergman K.N., Tsukruk V.V. // J. Am. Chem. Soc. 2004. V. 126. № 31. P. 9675.
Ma G., Zhang C., Zhang L., Sun H., Song C., Wang C., Kong D. // J. Mater. Sci. Mater. Med. 2016. V. 27. № 1. P. 17.
Indoria S., Singh V., Hsieh M.-F. // Int. J. Pharmaceut. 2020. V. 582. P. 119314.
Jaskula-Sztul R., Xu W., Chen G., Harrison A., Dammalapati A., Nair R., Cheng Y., Gong S., Chen H. // Biomaterials. 2016. V. 91. P. 1.
Zeng X., Zhang Y., Wu Z., Lundberg P., Malkoch M., Nystrom A.M. // J. J. Polym. Sci., Polym. Chem. 2012. V. 50. № 2. P. 280.
Bao H., Jin X., Li L., Lv F., Liu T. // J. Mater. Sci. Mater. Med. 2012. V. 23. № 8. P. 1891.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)