

Высокомолекулярные соединения (серия Б), 2021, T. 63, № 5, стр. 332-339
ФЕРМЕНТАТИВНОЕ РАЗРУШЕНИЕ КАПСУЛ НА ОСНОВЕ ПОЛИЭЛЕКТРОЛИТНОГО КОМПЛЕКСА ПОЛИПЕПТИД–ПОЛИСАХАРИД С РЕГУЛИРУЕМЫМ ВЫСВОБОЖДЕНИЕМ ДНК
Т. Н. Бородина a, *, Д. А. Шепеленко a, Д. Б. Трушина a, b, В. В. Артемов a, Т. В. Букреева a, c
a Федеральный научно-исследовательский центр “Кристаллография и фотоника” Российской академии наук
119333 Москва, Ленинский пр., 29, Россия
b Первый Московский государственный медицинский университет им. И.М. Сеченова
119991 Москва, ул. Большая Пироговская, 2, Россия
c Национальный исследовательский центр “Курчатовский институт”
123098 Москва, пл. Академика Курчатова, 1, Россия
* E-mail: borodina@crys.ras.ru
Поступила в редакцию 26.04.2021
После доработки 15.06.2021
Принята к публикации 29.06.2021
Аннотация
Получены и охарактеризованы содержащие ДНК биодеградируемые микрокапсулы на основе поликристаллических частиц ватерита CaCO3 и полиэлектролитного комплекса полиаргинин–сульфат декстрана. Методами сканирующей электронной микроскопии, лазерной конфокальной микроскопии и спектрофотометрии исследована ферментативная деструкция микрокапсул под действием проназы. Показано, что микрокапсулы, оболочка которых сформирована полиэлектролитными комплексами полипептид–полисахарид, чувствительны к ферментативному гидролизу под действием проназы, что приводит к высвобождению инкапсулированной ДНК.
ВВЕДЕНИЕ
Интерес к изучению микро- и наноносителей, существенно выросший за последние несколько десятилетий, связан с широкими возможностями применения наноструктурированных материалов практически во всех сферах жизни [1]. В настоящее время наноматериалы и нанотехнологии относятся к приоритетным направлениям развития науки и производства. Особую актуальность приобретает технология капсулирования, к преимуществам которой можно отнести защиту лабильных биологически активных веществ (таких как ферменты, вакцины, витамины) от воздействий внешней среды, разделение реагирующих или несовместимых между собой веществ в составе одного препарата, а также обеспечение контролируемого пролонгированного их высвобождения.
С этой точки зрения метод послойной (Layer-by-Layer, LbL) адсорбции полиэлектролитов из водных растворов на поверхности неорганических коллоидных микрочастиц имеет ряд преимуществ. Технология LbL основана на обусловленной электростатическими взаимодействиями поочередной адсорбции поликатионов и полианионов на подложке. Решающим фактором для формирования многослойной структуры является образование интерполиэлектролитных комплексов (ИПК) в результате взаимодействия полианионов и поликатионов. Образование ИПК с точки зрения свободной энергии электростатических взаимодействий ионных групп полиэлектролитов заключается в существовании солевых связей между звеньями противоположно заряженных цепей в них. Каждая из таких связей сравнительно слаба, но суммарная энергия всех связей в комплексе велика, поэтому ИПК крайне устойчивы [2].
Для эффективного применения в качестве матрицы для инкапсулирования полимеры должны отличаться высокой чистотой и гомогенностью, не оказывать раздражающего, токсического или иного побочного действия, подвергаться биодеструкции и выводиться из организма, не образовывая токсичных веществ [3]. Для создания биосовместимых биодеградируемых микрокапсул в качестве полианионов, входящих в состав ИПК, используют как природные полимеры (альгинаты, камеди, каррагинаны, пектин, декстрансульфокислоты и другие), так и синтетические (поли-L-аспарагиновая кислота, поли-L-глутаминовая кислота, полимеры и сополимеры акриловой и метакриловой кислот и другие) [4–8]. Поликатионным компонентом ИПК часто служат природные полимеры желатин и хитозан, а также синтетические полимеры поли-L-лизин, поли-L-аргинин, поли-L-орнитин и прочие.
ИПК на основе биосовместимых и биодеградируемых полимеров можно назвать перспективными материалами для применения в технологии микрокапсулирования. Это связано прежде всего с тем, что разрушение оболочек полимерных капсул может быть реализовано за счет биодеградации полимеров, входящих в состав оболочки капсул, под действием соответствующих ферментов [9]. Так, в работе [10] изучена биодеградируемость микрокапсул, оболочка которых включает полилизин, под действием фермента трипсина с целью анализа возможности доставки биологически активных веществ через желудок в область двенадцатиперстной кишки. В работе [11] на примере альбумина показана возможность пролонгированного высвобождения инкапсулированного вещества из микрокапсул на основе хитозана и сульфата декстрана в результате их разрушения под действием хитозаназы.
E.R. Simons с соавторами [12] установили возможность гидролизовать широкий ряд макромолекул с пептидными связями, таких как полилизин, полиаспаргиновая кислота, полиаргинин и даже полипролин, с помощью проназы. В работе [13] изучено влияние концентрации проназы и количества полиэлектролитных слоев на биоразложение капсул, содержащих в оболочке не только биодеградируемые (полиаргинин и полиаспарагиновая кислота), но и синтетические полимеры (полистиролсульфонат натрия и полиаллиламин гидрохлорид). По сравнению с другими доступными протеазами, проназа вызывает полное разложение полипептидов до индивидуальных аминокислот. Это вызвано в бόльшей степени действием различных протеолитических ферментов в проназе, которую выделяют из штамма бактерий Streptomyces griseus. Важным преимуществом ферментативно разлагаемых капсул является то, что они не требуют внешнего триггера для деградации. Следует отметить, что формирование микрокапсул с использованием полипептидов открывает дополнительные возможности для транспортировки биологически активных веществ в живую клетку [14].
В данной работе показана возможность ферментативного разрушения капсул из полиэлектролитного комплекса полиаргинина (PАrg) и сульфата декстрана (DS) под действием проназы, способствующей контролируемому высвобождению модельного инкапсулированного соединения. Перспективы применения полиэлектролитных капсул на основе PArg и DS основаны на способности данных полимеров к биодеструкции под действием ферментов, присутствующих в организме. Так, полиаргинин гидролизуется под действием трипсина, вырабатываемого поджелудочной железой, а декстраны расщепляются под действием таких ферментов, как амилаза и альфа-глюкозидаза. В полученные микрокапсулы была включена дезоксирибонуклеиновая кислота (ДНК), которая не подвержена деструкции под влиянием проназы [15]. В работе исследовано влияние концентрации неспецифического фермента на скорость разрушения полимерной оболочки и скорость высвобождения ДНК.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Хлорид натрия и хлорид кальция (“Acros organics”, Бельгия), карбонат натрия безводный (“AppliChem–Panreac”, Германия), натриевую соль сульфата декстрана (“Fluka”, США), поли-L-аргинин гидрохлорид (“Sigma”, Германия), двухцепочечную ДНК (тип IV: из семенников сельди) (“Sigma”, Германия), проназу из Streptomyces griseus (“Fluka”, St. Louis, MO), этилендиаминтетрауксусную кислоту (ЭДТА) (“Sigma”, Германия) и деионизованную воду использовали без дополнительной очистки.
Микрочастицы CaCO3 получали в соответствии с реакцией CaCl2 + Na2CO3 = CaCO3↓ + 2NaCl. Для чего к 2 мл 0.33 моль/л раствора карбоната натрия добавляли 2 мл 0.33 моль/л водного раствора хлорида кальция при непрерывном перемешивании на магнитной мешалке (IKA RH digital KT/C, 500 об/мин). Спустя 30 с перемешивания суспензию образовавшихся сферических микрочастиц оставляли на 5 мин для созревания осадка поликристаллических частиц карбоната кальция. После завершения процесса частицы СаCO3 трижды промывали от не вступивших в реакцию ионов деионизованной водой.
ДНК включали в частицы CaCO3 методом сорбции. Для этого микрочастицы CaCO3 ресуспендировали в 1 мл раствора ДНК (5 мг/мл) в деионизованной воде. После инкубации на шейкере (1000 об/мин) (IKA MS 3 basic, Германия) в течение 2 ч микрочастицы осаждали центрифугированием (3 мин, 3000 об/мин) (“Eppendorf”, Германия) и отделяли супернатант. Далее частицы дважды промывали водой (1 мл), проводя ресуспендирование и центрифугирование (3 мин, 3000 об/мин) (“Eppendorf”).
Частицы CaCO3, содержащие ДНК, получали также методом соосаждения. При непрерывном перемешивании с помощью магнитной мешалки (IKA RH digital KT/C, 500 об/мин) к 2.5 мл раствора ДНК (2 мг/мл) последовательно приливали 0.615 мл 1 моль/л CaCl2 и 0.615 мл 1 моль/л Na2CO3. Суспензию перемешивали в течение 30 с при комнатной температуре. Далее осадок микрочастиц отделяли от супернатанта центрифугированием (3 мин, 3000 об/мин) (“Eppendorf”). Микрочастицы с ДНК дважды промывали деионизованной водой, проводя центрифугирование и ресуспендирование. Супернатант после центрифугирования отбирали для определения содержания ДНК с помощью спектрофотометрического исследования.
Микрокапсулы получали с помощью последовательной адсорбции противоположно заряженных полиэлектролитов на поверхность микрочастиц с последующим растворением карбонатной матрицы. Микрочастицы CaCO3/ДНК ресуспендировали сначала в 1 мл водном растворе поли-L-аргинина (2 мг/мл), содержащем 0.15 моль/л NaCl, и инкубировали на шейкере (1000 об/мин; IKA MS 3 basic, Германия) в течение 15 мин. Затем частицы трижды промывали от не адсорбировавшихся молекул полиэлектролита. Далее осуществляли адсорбцию противоположно заряженного полиэлектролита – сульфата декстрана в тех же условиях. После нанесения шести слоев полиэлектролитов микрочастицы CaCO3/ДНК(PArg/DS)3 инкубировали с 2 мл 0.2 моль/л раствора динатриевой соли ЭДТА при интенсивном перемешивании на шейкере (1000 об/мин, IKA MS 3 basic, Германия) и осаждали центрифугированием (3 мин, 3000 об/мин). Процесс растворения СаСО3 повторяли несколько раз до полного растворения карбонатных частиц, затем полученные частицы трижды промывали деионизованной водой и хранили в виде суспензии при 4°С.
Для нахождения скорости высвобождения ДНК из микрокапсул под действием проназы к аликвоте микрокапсул с инкапсулированной ДНК (100 мкл) добавляли 100 мкл раствора проназы (5 и 20 мг/мл). Образцы в пробирках инкубировали на шейкере (1000 об/мин, IKA MS 3 basic, Германия) для предотвращения седиментации. Через определенные промежутки времени (5, 15, 30, 60 и 120 мин) пробирки снимали с шейкера, центрифугировали (1 мин, 5000 об/мин), супернатант отбирали для спектрофотометрического исследования.
Размер, форму и морфологию поверхности частиц капсул анализировали на сканирующем электронном микроскопе (СЭМ) “JSM-7401F” (“JEOL”, Япония), изображения получали при ускоряющем напряжении 1 кВ в режиме регистрации вторичных электронов. Распределение частиц по размерам и дзета-потенциал образцов определяли на анализаторе “Malvern Zetasizer ZS” (“Malvern Instruments Ltd”, Великобритания). Спектрофотометрические измерения проводили на двухлучевом сканирующем УФ-спектрофотометре “Lambda-650” (“Perkin Elmer”, США) в кварцевых кюветах. Количество ДНК, включенного в частицы СаСО3, находили с помощью предварительно построенной калибровочной прямой по разнице концентрации исходного раствора ДНК и супернатанта. Оптическую плотность раствора ДНК измеряли на длине волны 260 нм, соответствующей максимуму поглощения нуклеиновых кислот. Эффективность включения ДНК в частицы определяли по формуле (К0 – Ксн) × 100%, где К0 – исходное количество ДНК, Ксн – количество ДНК в супернатанте.
По полученной калибровочной прямой и измеренным значениям оптической плотности супернатантов устанавливали также количество не включившейся в микрокапсулы ДНК и время высвобождения ДНК при деструкции капсул под действием фермента.
Для визуализации капсул применяли оптический лазерный конфокальный флуоресцентный микроскоп “Leica TCS SPE” (“Leica Mycrosystems”, Германия). Визуализацию осуществляли для капсул, сформированных из поли-L-аргинина, меченого флуоресцеин изоцианатом (FITС), FITC–PArg.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Микрочастицы ватерита представляют большой интерес для использования в качестве носителя лекарственных веществ благодаря своей развитой поверхности и высокой пористости (рис. 1) [16]. Эти качества позволяют не только производить эффективную адсорбцию целевых биологически активных молекул на поверхность частицы, но и формировать на ней многослойное покрытие с помощью нанесения противоположно заряженных молекул полиэлектролитов.

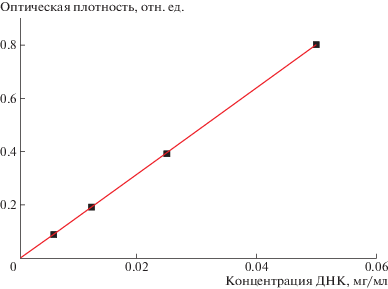
Включение ДНК в частицы ватерита было проведено методом сорбции, заключающимся в физической адсорбции вещества в поры предварительно синтезированных частиц ватерита, а также методом соосаждения. При соосаждении формирование микрочастиц CaCO3 при взаимодействии ионов хлорида кальция и карбоната натрия происходит в присутствии ДНК, так что ДНК распределяется по всему объему кристаллизующихся частиц ватерита. Эффективность включения ДНК в частицы ватерита была исследована спектрофотометрически, для чего была построена калибровочная прямая (рис. 2). Как в случае сорбции, так и при соосаждении в реакцию вводили 5 мг ДНК. При этом методом адсорбции в порах удалось достигнуть включения 2 мас. % (при эффективности инкапсулирования 22%), в то время как метод соосаждения способствовал инкапсулированию 8 мас.% (при эффективности капсулирования 95%).
Поверхность полученных микрочастиц CaCO3 с ДНК имеет небольшой отрицательный заряд (дзета-потенциал составляет –11.5 мВ), в связи с чем в качестве первого слоя для адсорбции был выбран положительно заряженный полиэлектролит PArg. Движущей силой LbL-процесса является электростатическое взаимодействие между противоположно заряженными полимерами. Данное взаимодействие обусловлено наличием нескомпенсированных зарядов макромолекул полиэлектролита, адсорбированного на нерастворимой подложке, что связано со строением полиэлектролитного комплекса, образующегося при контакте противоположно заряженных полимеров. Именно за счет этого избыточного заряда и появляется возможность наносить разноименные полиэлектролиты последовательно, что способствует созданию многослойной полиэлектролитной оболочки.
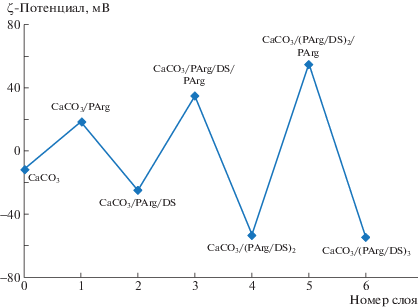
Формирование полимерной оболочки контролировалось путем измерения дзета-потенциала поверхности микрочастиц после адсорбции каждого слоя полиэлектролита. Смена знака заряда после нанесения каждого последующего полимерного слоя свидетельствовала об успешном образовании (рис. 3). После адсорбции шести полимерных слоев и растворения карбонатной матрицы был измерен дзета-потенциал полых микрокапсул, который составил –54.6 мВ. Данное значение свидетельствует о высокой стабильности полученного образца.
Электронно-микроскопические изображения показывают, что микрокапсулы (PArg/DS)3 с ДНК сохраняют сферическую форму после растворения частиц СаСО3 и имеют диаметр 3–5 мкм (рис. 4). При этом толщина полимерной оболочки составляет ~90–100 нм.
На рис. 5 представлено распределение микрокапсул по размерам, установленное методом динамического рассеяния света. Распределение имеет одномодальный характер, средний размер капсул составляет 3.5–6.5 мкм. Коэффициент полидисперсности образцов равен 0.3. Полученные данные сопоставимы с результатами определения размеров микрокапсул из СЭМ-изображений.

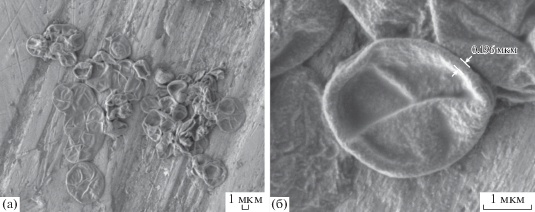
В ходе предварительной оценки воздействия растворов проназы на микрокапсулы состава (PArg/DS)3, проведенной с помощью оптической микроскопии, было обнаружено, что при концентрации фермента 20 мг/мл капсулы перестают визуализироваться через 5 ч, в то время как при концентрации проназы 5 мг/мл капсулы не растворяются полностью даже через двое суток инкубации. Методом конфокальной флуоресцентной микроскопии показано, что через 2 ч инкубации в растворе проназы с концентрацией 20 мг/мл большинство микрокапсул разрушено, вследствие чего можно наблюдать отдельные фрагменты полиэлектролитных мембран (рис. 6).
Рис. 6.
Конфокальные изображения микрокапсул (FITC–PArg/DS)3 сразу после добавления проназы (20 мг/мл) (а) и через 2 ч инкубации в растворе фермента (б).

На рис. 7 представлены электронно-микроскопические изображения капсул после 30 мин инкубации в присутствии проназы (20 мг/мл). Размер капсул не изменился по сравнению с интактными образцами (см. рис. 4), однако видно, что оболочка капсул стала более тонкой из-за ферментативной деструкции слоев полиаргинина.

Обнаружено, что модуль дзета-потенциала капсул, находившихся под действием проназы, уменьшается с 54.6 до 34.1 и 15.7 мВ за 10 и 30 мин инкубации соответственно. Эти изменения свидетельствуют об ухудшении коллоидной стабильности полимерных микрокапсул и их агрегировании, что подтверждается отсутствием четкой границы отдельных оболочек капсул на СЭМ-изображениях (рис. 7).
Спектрофотометрически была измерена оптическая плотность супернатантов на характеристической для ДНК длине волны 260 нм после инкубации капсул с ферментом в концентрации 5 мг/мл. Показано, что в течение первых 60 мин воздействия проназы происходит возрастание оптической плотности, а значит, высвобождение ДНК из микрокапсул (рис. 8). Далее процесс выходит на насыщение – дальнейшее увеличение времени инкубации не приводит к возрастанию концентрации высвободившегося вещества, при увеличении времени действия фермента с 60 до 120 мин количество ДНК в супернатанте даже немного снижается. Это может быть связано с процессом обратной адсорбции ДНК на фрагменты полимерной оболочки вследствие их неполного разрушения. При увеличении концентрации фермента с 5 до 20 мг/мл процесс высвобождения ДНК становится более интенсивным: через 30 мин инкубации капсул в растворе проназы концентрация ДНК повышается с 0.025 до 0.031 мг/мл.

Отметим, что в течение всего эксперимента образцы хранились при температуре 2–4°C и в отсутствие проназы спонтанного высвобождения ДНК не происходило.
ЗАКЛЮЧЕНИЕ
Получены биодеградируемые полимерные капсулы, сформированные за счет электростатическоой адсорбции поли-L-аргинина и сульфата декстрана на поверхности пористых микросферолитов карбоната кальция. Сравнение методов включения ДНК в частицы-матрицы показало, что присутствие ДНК во время синтеза частиц CaCO3 (метод соосаждения) способствует более эффективному инкапсулированию вещества (8 мас. %) по сравнению с методом физической сорбции на предварительно синтезированные частицы (2 мас. %). Биодеструкция полимерных капсул с оболочками состава (PArg/DS)3 может быть достигнута под действием проназы, которая катализирует гидролиз пептидных связей полипептидного компонента оболочки исследуемых объектов. В результате, сначала уменьшается толщина оболочки капсулы за счет частичного разложения и растворения поликатионных слоев. Происходящая перестройка оболочки снижает устойчивость капсул к агрегации. При дальнейшем гидролизе капсулы начинают разрушаться, однако в растворе присутствуют еще фрагменты полиэлектролитной оболочки. Затем наблюдается полное растворение оболочек. Постепенная биодеградация полимерной оболочки способствует высвобождению инкапсулированного компонента – ДНК. В работе продемонстрировано, что скорость разрушения оболочки и скорость выхода ДНК можно регулировать путем изменения концентрации фермента. Полученные результаты свидетельствуют о том, что капсулы на основе полиэлектролитного комплекса полипептид–полисахарид можно рассматривать в качестве перспективного средства доставки биологически активных веществ с регулируемым высвобождением инкапсулируемого материала.
Работа выполнена при финансовой поддержке Министерства науки и высшего образования в рамках работ по Государственному заданию Федерального научно-исследовательского центра “Кристаллография и фотоника” РАН с использованием оборудования Центра коллективного пользования ФНИЦ “Кристаллография и фотоника” РАН в части получения микрокапсул и их характеризации, при финансировании НИЦ “Курчатовский институт” (№ 1058 от 02.07.2020 г.) в части изучения биодеградации капсул под действием фермента.
Список литературы
Головин Ю.И. // Справочник. Инженерный журнал, Приложение № 1. 2006. С. 1.
Biesheuvel P.M., Stuart M.A.C. // Langmuir. 2004. V. 20. P. 2785.
Piskin E. // J. Biomater. Sci. Polym. Ed. 1995. V. 6. № 9. P. 775.
Tiourina O.P., Sukhorukov G.B. // Int. J. Pharm. 2002. V. 242. № 1–2. P. 155.
Sukhorukov G.B., Donath E., Moya S., Susha A.S., Voigt A., Hartmann J., Möhwald H. // J. Microencapsul. 2000. V. 17. № 2. P. 177.
Berth G., Voigt A., Dautzenberg H., Donath E., Mohwald H. // Biomacromolecules. 2002. V. 3. № 3. P. 579.
Балабушевич Н.Г., Ларионова Н.И. // Биохимия. 2004. Т. 69. № 7. С. 930.
Платэ Н.А. // Наука в СССР. 1986. Т. 1. С. 2.
Кленова Н.А., Макурина О.Н. Химия белка и ферментов. Самара: Самарский ун-т, 2015.
Borodina T.N., Rumsh L.D., Kunizhev S.M., Sukhorukov G.B., Vorozhtsov G.N., Feldman B.M., Markvicheva E.A. // Biochem. Suppl. Ser. Biomed. Chem. 2. 2008. V. 53. № 5. P. 557.
Itoh Y., Matsusaki M., Kida T., Akashi M. // Biomacromolecules. 2006. V. 7. № 10. P. 2715.
Simons E.R., Blout E.R. // Biochim. Biophys. Acta – Spec. Sect. Enzymol. Subj. 1964. V. 92. № 1. P. 197.
Marchenko I., Yashchenok A., Borodina T., Bukreeva T., Konrad M., Möhwald H., Skirtach A. // J. Controlled Release. 2012. V. 162. № 3. P. 599.
De Geest B.G., Vandenbroucke R.E., Guenther A.M., Sukhorukov G.B., Hennink W.E., Sanders N.N., Demeester J., De Smedt S.C. // Adv. Mater. 2006. V. 18. № 8. P. 1005.
Sweeney P.J., Walker J.M. // Methods Mol. Biol. 1993. V. 16. P. 271.
Sukhorukov G.B., Volodkin D.V., Günther A.M., Petrov A.I., Shenoy D.B., Möhwald H. // J. Mater. Chem. 2004. V. 14. P. 2073.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)