

Высокомолекулярные соединения (серия Б), 2021, T. 63, № 5, стр. 354-362
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ЛИГНИНА В МАТЕРИАЛАХ РАСТИТЕЛЬНОГО ПРОИСХОЖДЕНИЯ С ПОМОЩЬЮ ТВЕРДОТЕЛЬНОЙ СПЕКТРОСКОПИИ ЯМР 13С
С. Г. Кострюков a, *, П. С. Петров a, В. А. Калязин a, Ю. Ю. Мастерова a, В. С. Тезикова a, Н. А. Хлучина a, Л. Я. Лабзина a, Д. Х. Алалван a
a Национальный исследовательский Мордовский государственный университет им. Н.П. Огарева
430005 Саранск, ул. Большевистская, 68, Россия
* E-mail: kostryukov_sg@mail.ru
Поступила в редакцию 01.03.2021
После доработки 27.05.2021
Принята к публикации 29.06.2021
Аннотация
Показаны возможности твердотельной ЯМР 13С спектроскопии (СP-MAS) для определения содержания лигнина в большом наборе лигноцеллюлозных материалов: древесины различных пород деревьев и растительных отходов. Анализ основан на использовании зависимостей интегральной интенсивности сигналов ЯМР 13С ароматических или метоксильных атомов углерода лигнина от его процентной концентрации в смесях с целлюлозой. Содержание лигнина, полученное методом спектроскопии ЯМР 13С, хорошо согласуется с литературными данными и результатами, полученными с помощью кислотного гидролиза соответствующих образцов лигноцеллюлозных биоматериалов.
В настоящее время во всем мире производится огромное количество различных отходов, значительная часть которых является отходами растительного происхождения. Практически все растительные отходы – это лигноцеллюлозная биомасса, которая, как известно, представляет собой важный возобновляемый углеродсодержащий ресурс, широко используемый для производства биоэнергии и биоматериалов [1–5].
В последние годы появилось много различных методов (биологических и химических) для преобразования биомассы в топливо или химические вещества. В настоящее время наиболее разработаны термохимические способы, включающие газификацию, пиролиз, сжигание и т.д., многие из которых характеризуются широким ассортиментом перерабатываемого сырья и высокой эффективностью [6]. Среди всех термохимических методов наиболее перспективным является пиролиз, так как позволяет превращать биомассу в жидкие, твердые и газообразные продукты [7–11]. Кроме того, в последние годы большое внимание привлекает использование микроволнового излучения в процессе переработки растительного сырья [12].
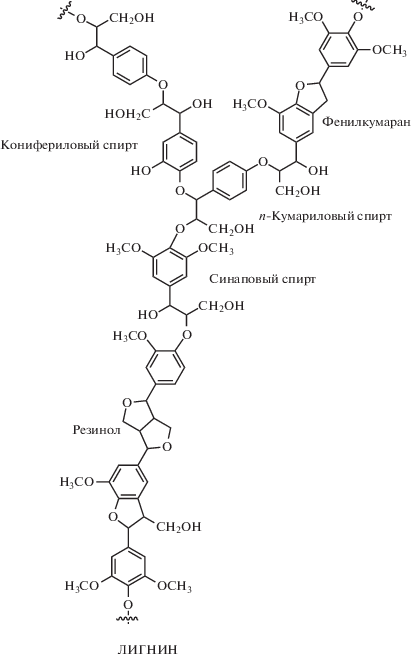
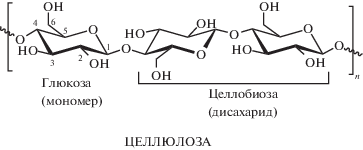
Однако использование лигноцеллюлозного сырья сталкивается с рядом ограничений, которые нужно преодолеть, чтобы снизить стоимость конечных продуктов. В частности, необходимо частично или полностью разрушить сложную сеть лигноцеллюлозной структуры с помощью химических, механических или биологических методов [13, 14]. Стенка растительной клетки в лигноцеллюлозной биомассе состоит из трех основных компонентов: целлюлозы, гемицеллюлозы и лигнина, связанных между собой в устойчивую структуру, отвечающую за механическую прочность растений и обеспечивающую транспорт воды и питательных веществ [8, 9]. Целлюлоза, представляющая собой линейный полимер, мономерным звеном которого является D-глюкоза, отвечает за механическую прочность. В свою очередь, гемицеллюлоза, состоящая из различных моносахаридов, служит связующим звеном между целлюлозой и лигнином [11]. Лигнин – это трехмерный полимер, отвечающий за связывание клеток и поддержку целлюлозной сети [8]. Он имеет сложную структуру, состоящую в основном из трех различных фенилпропановых единиц: п-гидроксифенил, гваяцил и сирингил, мономерами которых соответственно являются п-кумариловый, конифериловый и синаповый спирты [13]. Ниже представлено строение основных компонентов лигноцеллюлозных материалов.

Лигнин широко используется в качестве топлива в бумажной промышленности и на биоперерабатывающих заводах. Кроме того, он применяется как добавка в пластмассы для увеличения биодеградируемости, как энтеросорбент в медицине, как полимерная матрица для контролируемого высвобождения пестицидов [15]. Лигнин можно использовать в качестве недорогого сырья для производства химических веществ и добавки к материалам для механического армирования, улучшения антиоксидантной и антимикробной активности, защиты от ультрафиолета, а также в биомедицинских целях [16, 17]. Наличие химических связей между углеводными фракциями и лигнином – основной барьер для деградации лигноцеллюлозной структуры и ее превращения в топливо или химическое сырье [1, 4, 18]. Знание исходного содержания лигнина и углеводов в лигноцеллюлозных материалах имеет первостепенное значение для мониторинга изменений, происходящих при их переработке [19, 20]. Большинство аналитических методов определения содержания лигнина и углеводов трудоемки, для их проведения необходима деструкция материала, приводящая к большому количеству химических отходов; кроме того, указанные методы не всегда дают достоверные результаты [16]. Следовательно, крайне желательно разработать простые и быстрые методы определения состава полимерных компонентов, и среди таких методов твердотельная спектроскопия ЯМР 13С представляется одной из наиболее привлекательных альтернатив [21].
Твердотельная спектроскопия ЯМР 13С широко используется для изучения химического состава лигноцеллюлозной биомассы и анализа изменений, происходящих в результате процессов выделения лигноцеллюлозных компонентов или при химической/термической обработке [22–26]. Основные проблемы, затрудняющие количественный анализ лигноцеллюлозных материалов с помощью твердотельной спектроскопии ЯМР 13С, заключаются в перекрывании сигналов, длительной спин-решеточной релаксации ядер 13С в экспериментах с количественной прямой поляризацией (DP) и неоднородном переносе поляризации в 1Н-13С кросс-поляризационных экспериментах (CP) [25–27]. Тем не менее, некоторые исследования показывают принципиальную возможность использования твердотельной спектроскопии ЯМР 13С в качестве быстрого метода определения содержания лигнина в биомассе по анализу интенсивности сигналов лигнина в спектре [25–31].
Впервые использование спектроскопии ЯМР 13С для определения содержания лигнина в образцах древесины было выполнено в 1984 г. и основывалось на использовании в расчетах средней молекулярной формулы лигнина [28]. Другие авторы использовали калибровочную кривую, построенную по интенсивности ароматических сигналов в твердотельных спектрах ЯМР 13С чистого лигнина, чтобы определить его содержание в природных образцах [25, 27] или же калибровочную кривую, построенную по интенсивности сигналов атомов углерода метоксильных фрагментов [30]. Кроме того, был предложен метод, основанный на использовании внутреннего стандарта (натриевой соли 3-(триметилсилил)пропановой кислоты) [29]. Хотя эти исследования демонстрируют потенциал использования твердотельной спектроскопии ЯМР 13С для количественного анализа содержания лигнина в лигноцеллюлозных материалах, некоторые вопросы все еще остаются открытыми, например, выбор адекватного стандарта лигнина. Поскольку его химический состав и структура могут варьироваться в зависимости от различных типов растительных материалов, универсального стандарта лигнина не существует [31]. Кроме того, количество различных типов лигноцеллюлозных материалов, охватываемых этими исследованиями, весьма ограничено.
В настоящей работе для оценки содержания лигнина в лигнинсодержащих биоматериалах была применена твердотельная спектроскопия ЯМР 13С с использованием техники кросс-поляризации и вращения образца под магическим углом (СР-MAS). Объектами исследования служили различные образцы древесины, скорлупа плодов и другие растительные материалы – в общей сложности, 16 образцов. Содержание лигнина, полученного методом ЯМР, сравнивали с литературными данными и результатами, полученными с помощью стандартного метода количественного определения лигнина, основанного на кислотном гидролизе углеводов [31, 32]. Данное исследование позволило установить возможности и ограничения твердотельной спектроскопии ЯМР 13С для количественного определения лигнина в различных материалах растительного происхождения.
МАТЕРИАЛЫ И МЕТОДЫ ИССЛЕДОВАНИЯ
В работе использовали микрокристаллическую целлюлозу и крафт-лигнин (оба “Sigma-Aldrich”). Стандартные образцы, имитирующие лигноцеллюлозные материалы, готовили смешиванием целлюлозы и лигнина в массовом соотношении 90 : 10, 80 : 20; 70 : 30, 60 : 40; 50 : 50; 40 : 60.
Исследовали несколько образцов древесины и различные сельскохозяйственные отходы, информация о которых представлена в табл. 1, где также приводятся полученные результаты по определению содержания лигнина, которые обсуждаются в дальнейшем.
Таблица 1.
Общая характеристика исследованных образцов лигноцеллюлозной биомассы
| Образец | Содержание лигнина (%) | |||
|---|---|---|---|---|
| ЯМР 13С | кислотный гидролиз | лит. данные | ||
| Саром. | СMeO | |||
| Древесина ели (1) | 27.2 | 29.5 | 28.7 | 27.9 [33] |
| Древесина сосны (2) | 19.7 | 20.1 | 20.1 | 20 [33] |
| Древесина лиственницы сибирской (3) | 28.1 | 29.2 | 29.4 | 29.2 [34] |
| Древесина осины (4) | 18.2 | 20.6 | 21.5 | 19.5 [35] |
| Древесина тополя (5) | 15.0 | 16.2 | 16.4 | 15.5–16.3 [33] |
| Древесина березы (6) | 22.7 | 16.2 | 22.1 | 22.4 [36] |
| Древесина дуба (7) | 24.3 | 24.2 | 24.5 | 24.1 [33] |
| Древесина липы (8) | 23.6 | 22.8 | 23.5 | 28.3 [37] |
| Ячменная солома (9) | 7.8 | 7.4 | 8.3 | 6.3–9.8 [33] |
| Пивная дробина (10) | 3.3 | 5.4 | 5.5 | 16.9–27.8 [35] |
| Скорлупа семян подсолнечника (11) | 9.5 | 13.1 | 13.0 | 7.72–13.4 [35] |
| Скорлупа семян тыквы (12) | 46.8 | 60.3 | 47.5 | – |
| Скорлупа грецкого ореха (13) | 22.5 | 28.0 | 54.1 | 53.5 [35] |
| Скорлупа арахиса (14) | 33.0 | 40.2 | 32.5 | 28 [35] |
| Скорлупа фисташки (15) | 6.5 | 8.0 | 30.2 | 29.4 [35] |
| Скорлупа миндаля (16) | 22.2 | 27.5 | 28.1 | 36 [35] |
Сбор образцов осуществляли с живых деревьев, произрастающих в лесопарковой зоне г. Саранска. Предпочтение отдавалось деревьям возраста 60–80 лет. Для взятия образцов древесины использовали возрастные буры Haglof (d = = 5.15 мм, l = 100 мм), при помощи которых высверливали радиальные керны. Образцы древесины высверливали перпендикулярно продольной оси ствола на высоте примерно 1 м от поверхности земли. Ячменная солома сорта ячменя ярового Маргрет, тыквенная скорлупа (сорт Витаминная), подсолнечная лузга (сорт Родник) были предоставлена местными сельскохозяйственными производителями. Скорлупа грецкого ореха (сорт Чандлер), арахиса (сорт Вирджиния), фисташки (сорт Узун), миндаля (сорт Монтерей) взята с орехов, приобретенных на рынке. Пивная дробина предоставлена саранским филиалом пивоваренной компании “AB InBev Efes”. Предварительное измельчение материалов до фракции 0.5 мм проводили на роторно-ножевой мельнице РМ-120 (фирма “Вибротехник”) и модернизированном измельчителе ИЗ-14М (Общество с ограниченной ответственностью “Уралспецмаш”). Наиболее твердые материалы, такие как скорлупа грецкого ореха и фисташки измельчали с помощью полуавтоматической вибрационной мельницы “Herzog HP-M 100” в течение 40 с. Размер частиц определяли на виброанализаторе А-20 с набором сит фирмы “Вибротехник”. Для спектроскопии ЯМР использовали материал с размером частиц 100–300 мкм. Перед всеми исследованиями образцы сушили до постоянной массы в сушильном шкафу при 100 ± 3°С в течение 2 ч.
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ЛИГНИНА ХИМИЧЕСКИМ МЕТОДОМ
Для сравнительного анализа определяли содержание лигнина с помощью сернокислотного гидролиза [32]. Для этого измельченные и высушенные образцы первоначально экстрагировали ацетоном и высушивали. Коэффициент экстрагирования Кэ находили по формуле
Здесь Э – масса экстрактивных веществ, m – масса образца.
Затем воздушно-сухие обессмоленные (обработанные ацетоном) образцы подвергали гидролизу 72%-ным раствором H2SO4 при 25°C в течение 2.5 ч при перемешивании; после чего гидролизат разбавляли водой до концентрации 7% и кипятили при перемешивании с обратным холодильником в течение 2 ч. Фильтрование лигнина проводили на следующий день, чтобы частицы достаточно укрупнились. Фильтр с лигнином сушили при 100°С в сушильном шкафу до постоянной массы. Массовую долю лигнина в % от исходного (необессмоленного) материала рассчитывали по формуле
где l – масса лигнина, mз – масса золы.Массу золы mз определяли следующим образом: навеску исходного образца m прокаливали в муфельной печи при 575 ± 25°С в течение 5 ч, после охлаждения взвешивали и прокаливали в тех же условиях еще 1 ч, контролируя массу. Прокаливание завершали, когда масса образца переставала меняться.
ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ЛИГНИНА ТВЕРДОТЕЛЬНОЙ СПЕКТРОСКОПИЕЙ ЯМР 13С
Твердотельные спектры ЯМР 13C регистрировали на спектрометре “JEOL JNM-ECX400 (9.39 Т, 100.5 MГц) при комнатной температуре с применением техники кросс-поляризации и вращения под магическим углом (CP-MAS эксперимент) с частотой вращения 10 кГц в 4 мм роторах из диоксида циркония [38]. Важно было добиться высокой однородности частиц исследуемого объекта по размеру, чтобы ротор был хорошо отцентрован с целью достижения необходимой скорости вращения. Магический угол вращения образца (MAS) настраивали при скорости вращения 6 кГц с использованием стандартного образца KBr. Настройку разрешения прибора проводили по адамантану, он же служил реперным образцом (29.5 м.д.). Все CP-MAS-эксперименты выполняли при комнатной температуре; протонную развязку осуществляли с помощью двойной импульсной модуляции фазы (TPPM). Для оптимизации процесса регистрации спектра, с целью получения количественных зависимостей интегральных интенсивностей сигналов от количества углеродных атомов на примере образца целлюлозы было подобрано наиболее оптимальное время задержки между импульсами. На рис. 1 приведены спектры целлюлозы, полученные при различных временах задержки (интегральная интенсивность атомов С-1 принята за единицу).
Рис. 1.
CP-MAS ЯМР 13С спектры целлюлозы, полученные при времени задержки между импульсами 1 (1), 3 (2), 5 (3) и 7 с (4). Цветные рисунки можно посмотреть в электронной версии.

Таким образом, при времени задержки между импульсами 7 с интегральная интенсивность атомов углерода С-1 : С-4 : С-2, 3, 5 : С-6 соотносится как 1 : 1 : 3 : 1, что свидетельствует о сопоставимом отклике всех атомов углерода целлюлозы и возможности проведения количественных измерений. Длительность 90°-ного импульса для ядер 13С составила 2.93 мкс. Общее количество сканов 1024. Спектры записывали с использованием программы Delta 4.3.6, обрабатывали с помощью ACD/NMR Processor Academic Edition, Ver. 12.01 [39].
Твердотельные спектры ЯМР 13С некоторых из исследованных лигноцеллюлозных материалов представлены на рис. 2. Во всех спектрах идентифицируются сигналы, связанные с тремя основными компонентами лигноцеллюлозной структуры – лигнином (L), целлюлозой (C) и гемицеллюлозой (H) [29, 30]. Наиболее интенсивные пики в спектрах принадлежат углеводам и содержат перекрывающиеся сигналы атомов углерода целлюлозы и гемицеллюлозы. Наложение данных сигналов затрудняет правильное определение содержания каждого из этих компонентов. Что касается лигнина, то сигналы, обусловленные метоксильными и ароматическими атомами углерода, обнаруживаются в спектральных областях, не содержащих сигналов атомов углерода полисахаридов. Таким образом, эти сигналы лигнина потенциально полезны для определения содержания лигнина в материалах биомассы [26]. Удобство использования для аналитических целей сигналов ароматических атомов углерода состоит в том, что количество ароматических колец для лигнинов различного происхождения практически постоянно. Недостаток же состоит в том, что ароматические сигналы менее интенсивны и слабо разрешены по сравнению с метоксильными, что особенно заметно в спектрах образцов с низким содержанием лигнина. Сигнал атомов углерода метоксильных групп (52–56 м.д.), как видно из рис. 2, более узкий, но частично перекрывается с сигналами атомов С-6 углеводов. В данном случае интегрирование можно проводить по правой половине сигнала, умножая полученное значение на два. Таким образом, метоксильный сигнал также может быть использован для количественного определения содержания лигнина в исследуемых образцах. Кроме того, известно, что лигнин разного происхождения отличается соотношением п-гидроксифенильных, гваяцильных и сирингильных фрагментов, что приведет к различному количеству метоксильных групп в молекулярной структуре [40, 41], а это ставит вопросы о точности и пределах корректности данного метода количественного анализа.
Рис. 2.
CP-MAS ЯМР 13С спектры древесины осины (1), древесины березы (2), ячменной соломы (3), пивной дробины (4), скорлупы семян подсолнечника (5) и скорлупы грецкого ореха (6).

Содержание лигнина в образцах находили по калибровочным графикам, полученным на основе спектров искусственных смесей целлюлоза–лигнин. Калибровочные графики отражают зависимость интегральных интенсивностей соответствующих сигналов от содержания лигнина в смеси в массовых процентах (рис. 3).
Рис. 3.
Калибровочные графики зависимости содержания лигнина от интегральных интенсивностей ароматических углеродов в диапазоне 142–160 м.д. (а) и метокси-групп в диапазоне 52–56 м.д. (б).

Для стандартизации определения интенсивности анализируемых спектральных диапазонов поступали следующим образом: первоначально проводили коррекцию базовой линии, затем спектральный диапазон 100–112 м.д., принадлежащий ацетальным атомам углерода (С-1), приравнивался к единице, а уже потом определяли интенсивность спектральных диапазонов 142–160 м.д. (Саром.) и 52–56 м.д. (ОСН3).
На рис. 4 в качестве примера представлены CP-MAS ЯМР 13C спектры образцов лигнина, целлюлозы, смеси целлюлозы (70%) и лигнина (30%) и скорлупы тыквенных семечек, как примера лигноцеллюлозного материала.
Рис. 4.
CP-MAS ЯМР 13С спектры лигнина (1), целлюлозы (2), смеси целлюлозы (70%) и лигнина (30%) (3) и скорлупы тыквенных семечек (4).

При сравнении результатов определения лигнина, приведенных в табл. 1, наблюдается хорошее согласие данных, полученных разными методами, за исключением скорлупы грецкого ореха и миндаля, для которых химический анализ дает завышенные результаты. Как показали CP-MAS ЯМР 13C спектры лигнинов, полученных из этих образцов, гидролиз углеводов не был полным. В то же время результаты, полученные для пивной дробины (11) и скорлупы фисташки (15) сильно отличаются от литературных данных, что мы объясняем происхождением и сортовыми отличиями образцов. Достоверность результатов для образцов (11) и (15) подтверждается тем, что содержание лигнина, определенное на основе ЯМР и кислотного гидролиза, практически не различается. Таким образом, если не принимать во внимание отдельные отклонения, то корреляция результатов, полученных разными методами, хорошая, что подтверждает надежность разработанного метода количественной оценки содержания лигнина в различных биоматериалах.
Список литературы
Somerville C., Youngs H., Taylor C., Davis S.C., Long S.P. // Science. 2010. V. 329. № 5993. P. 790.
Wang H., Pu Y., Ragauskas A., Yang B. // Bioresource Technol. 2018. V. 217. P. 449.
Amaral H.R., Cipriano D.F., Santos M.S., Schettino M.A., Ferreti J.V.T., Meirelles C.S., Pereira V.S., Cunha A.G., Emmerich F.G., Freitas J.C.C. // Carbohydr. Polymers. 2019. V. 210. P. 127.
Long H., Li X., Wang H., Jia J. // Renewable Sustainable Energy Revs. 2013. V. 26. P. 344.
Limayem A., Ricke S.C. // Progr. Energy Combustion Sci. 2012. V. 38. № 4. P. 449.
Burra K.G., Gupta A.K. Thermochemical Reforming of Wastes to Renewable Fuels (Book Chapter). Green Energy and Technology. 2018. P. 395.
Solarte-Toro J.C., González-Aguirre J.A., Poveda Giraldo J.A., Cardona Alzate C.A. // Renewable Sustainable Energy Revs. 2021. V. 136. № 110376.
Liu C., Wang H., Karim A.M., Sun J., Wang Y. // Chem. Soc. Revs. 2014. V. 43. № 22. P. 7594.
Dai L., Wang Y., Liu Y., He C., Ruan R., Yu Z., Jiang L., Zeng Z., Wu Q. // Sci. Total Environment. 2020. V. 749. № 142386.
Kim H., Lee S., Lee B., Park J., Lim H., Won W. // Fuel. 2021. V. 287. № 119369.
Zhou C.-H., Xia X., Lin C.-X., Tong D.-S., Beltramini J. // Chem. Soc. Revs. 2011. V. 40. P. 5588.
Маркин В.И., Чепрасова М.Ю., Базарнова Н.Г. // Химия растительного сырья. 2014. № 4. С. 21.
Singh R., Shukla A., Tiwari S., Srivastava M. // Renewable Sustainable Energy Revs. 2014. V. 32. P. 713.
Gao Y., Xu J., Zhang Y., Yu Q., Yuan Z., Liu Y. // Bioresource Technol. 2013. V. 144. P. 396.
Zobiole L.H.S., dos Santos W.D., Bonini E., Ferrarese-Filho O., Kremer R.J., de Oliveira R.S., Constantin J. Lignin: Properties and Applications in Biotechnology and Bioenergy/ Ed. by R.J. Paterson. Nova Science. 2012. P. 419.
Tian X., Fang Z., Smith R.L., Wu Z., Liu M. Production of Biofuels and Chemicals from Lignin / Ed. by Z. Fang, R.L. Smith. Springer, 2016. P. 3.
Kai D., Tan M.J., Chee P.L., Chua Y.K., Yap Y.L., Loh X.J. // Green Chem. 2016. V. 18. № 5. P. 1175.
Nanda S., Mohammad J., Reddy S.N., Kozinski J.A., Dalai A.K. // Biomass Conversion Biorefinery. 2014. V. 4. № 2. P. 157.
Rezende C., de Lima M., Maziero P., deAzevedo E., Garcia W., Polikarpov I. // Biotechnol. Biofuels. 2011. V. 4. № 54.
Qin Z., De Wang X., Liu H.M., Wang D.M., Qin G.Y. // Bioresource Technol. 2018. V. 262. P. 212.
Кострюков С.Г., Петров П.С. // Химия растительного сырья. 2020. № 4. С. 7.
Freitas J.C.C., Bonagamba T.J., Emmerich F.G. // Carbon. 2001. V. 39. № 4. P. 535.
Perras F.A., Luo H., Zhang X., Mosier N.S., Pruski M., Abu-Omar M.M. // J. Phys. Chem. A. 2017. V. 121. № 3. P. 623.
Ghaffar S.H., Fan M. // Biomass Bioenergy. 2013. V. 57. P. 264.
Cipriano D.F., Gonçalves G.R., Cunha A.G., Schettino M.A., Chinelatto L.S., Menezes S.M.C., De Freitas J.C.C. // Revista Virtual de Quimica. 2020. V. 12. № 3. P. 639.
Fu L., McCallum S.A., Miao J., Hart C., Tudryn G.J., Zhang F., Linhardt R.J. // Fuel. 2015. V. 141. P. 39.
Sievers C., Marzialetti T., Hoskins T.J.C., Valenzuela Olarte M.B., Agrawal P.K., Jones C.W. // Bioresource Technol. 2009. V. 100. № 20. P. 4758.
Haw J.F., Maciel G.E., Schroeder H.A. // Analyt. Chem. 1984. V. 56. № 8. P. 1323.
Gao X., Laskar D.D., Zeng J., Helms G.L., Chen S. // ACS Sustainable Chem. Eng. 2015. V. 3. № 1. P. 153.
Cipriano D.F., Chinelatto L.S., Nascimento S.A., Rezende C.A., de Menezes S.M.C., Freitas J.C.C. // Biomass Bioenergy. 2020. V. 142. № 105792.
Hatfield R., Fukushima R.S. // Crop Science. 2005. V. 45. № 3. P. 832.
Tappi. T222 Om-02 Lignin in Wood and Pulp. TAPPI Test Methods. 2006.
Liu R., Chen Y., Cao J. // RSC Adv. 2015. V. 5. № 94. P. 76708.
Park Y., Jang S.-K., Park J.-H., Yang S.-Y., Chung H., Han Y., Chang Y.-S., Choi I.-G., Yeo H. // J. Wood Sci. 2017. V. 63. № 6. P. 635.
Kumar B., Bhardwaj N., Agrawal K., Chaturvedi V., Verma P. // Fuel Proc. Technol. 2020. V. 199. № 106244.
Borrega M., Nieminen K., Sixta H. // Bioresource Technol. 2011. V. 102. № 22. P. 10724.
Gallina G., Cabeza Á., Grénman H., Biasi P., García-Serna J., Salmi T. // J. Supercritical Fluids. 2018. V. 133. P. 716.
Кострюков С.Г., Арасланкин С.В., Петров П.С. // Химия растительного сырья. 2017. № 4. С. 31.
ACD/NMR Processor Academic Edition (n.d.), www.acdlabs.com.
Kolodziejski W., Frye J.S., Maclel G.E. // Analyt. Chemy. 1982. V. 54. № 8. P. 1419.
Ralph J., Lundquist K., Brunow G., Lu F., Kim H., Schatz P.F., Marita J.M., Hatfield R.D., Ralph S.A., Christensen J.H., Boerjan W. // Phytochem. Revs. 2004. V. 3. № 1–2. P. 29.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)