

Высокомолекулярные соединения (серия Б), 2021, T. 63, № 5, стр. 307-316
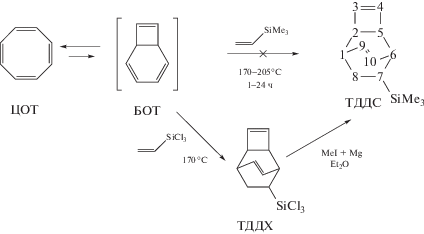
СИНТЕЗ И МЕТАТЕЗИСНАЯ ПОЛИМЕРИЗАЦИЯ НОВОГО МОНОМЕРА 7-ТРИМЕТИЛСИЛИЛТРИЦИКЛО[4.2.2.02,5]ДЕКА-3,9-ДИЕНА
В. А. Жигарев a, *, М. Л. Грингольц a, М. П. Филатова a, Е. Ш. Финкельштейн a
a Институт нефтехимического синтеза им. А.В. Топчиева Российской академии наук
119991 Москва, Ленинский пр., 29, Россия
* E-mail: zhigarev@ips.ac.ru
Поступила в редакцию 27.05.2021
После доработки 01.07.2021
Принята к публикации 14.07.2021
Аннотация
Впервые изучена реакция 1,3,5,7-циклооктатетраена с кремнийзамещенными этиленами. Показано, что винилтриметилсилан неактивен в реакции, а винилтрихлорсилан образует 7-трихлорсилил-трицикло[4.2.2.02,5]дека-3,9-диен с выходом до 12%. Метилированием хлораддукта синтезирован новый кремнийзамещенный мономер 7-триметилсилилтрицикло[4.2.2.02,5]дека-3,9-диен, содержащий 92% эндо-изомера. Исследована метатезисная полимеризация синтезированного мономера на Ru-катализаторах Граббса первого и второго поколений. С выходами 90–96% получен новый поли(7-триметилсилилтрицикло[4.2.2.02,5]дека-3,9-диен), содержащий объемный бициклический фрагмент и преимущественно транс-двойные связи в основной цепи (до 94%). Полимер характеризуется наиболее высокой температурой стеклования (187°С) в ряду монотриметилсилил-замещенных полинорборненов. Присутствующие в мономерном звене двойные связи открывают перспективы дальнейшей модификации.
ВВЕДЕНИЕ
Напряженные циклоолефины являются важными синтонами в органическом и полимерном синтезе, благодаря высокой реакционной способности эндоциклической двойной связи С=С в их молекулах [1–5]. Популярными соединениями такого рода являются замещенные бицикло [2.2.1]гепт-2-ены (далее норборнены), легко синтезируемые по реакции диеновой конденсации циклопентадиена с замещенными олефинами [5–7], а также трицикло[4.2.1.02,5]нон-7-ены (далее трициклононены), получаемые реакцией [2σ + + 2σ + 2π]-циклоприсоединения квадрициклана к олефинам [8, 9]. Наличие напряженной двойной связи в их молекулах обеспечивает высокую реакционную способность в полимеризационных процессах метатезисной и аддитивной полимеризации [5, 6, 10–13]. Введение разного рода заместителей в норборнены и трициклононены можно назвать инструментом макромолекулярного дизайна материалов с заданными свойствами [6, 11, 14, 15]. Эффективным подходом в этом направлении стало введение в полинорборнены и политрициклононены кремний- и фторсодержащих заместителей, что значительно улучшило газоразделительные, адгезионные, диэлектрические и другие свойства полимеров [6, 16–23].
Интересными мономерами являются описанные в литературе трициклические диены – трицикло[4.2.2.02,5]дека-3,9-диены (ТДД) [24–33], потенциал которых в синтезе полимеров, на наш взгляд, изучен недостаточно. Привлекательность ТДД заключается в существовании одностадийного пути их синтеза из олефинов и 1,3,5,7-циклооктатетраена (ЦОТ), который способен при температуре более 100°С претерпевать термическую изомеризацию в бицикло[4.2.0]окта-2,4,7-триен (БОТ) с цисоидной конформацией двух сопряженных двойных связей [24, 27, 28, 33]. Последнее обстоятельство способствует участию БОТ в [4π + 2π]-циклоприсоединении активных диенофилов с образованием ТДД [24, 27, 28, 33, 34]. Осуществлен диеновый синтез ЦОТ с кислород- и азотсодержащими диенофилами, главным образом, производными малеинового ангидрида – N-замещенными малеимидами [28, 35], а также имеются публикации по диеновому синтезу ЦОТ с фторзамещенными диенофилами [34–39]. На примере взаимодействия ЦОТ с замещенными N-фенилмалеимидами показано, что бòльший выход продукта реакции наблюдается для диенофилов с менее электрононасыщенной и менее стерически затрудненной двойной связью и составляет от 30 до 85% [35]. Образующиеся замещенные ТДД содержат двойные связи в четырехчленном и шестичленном циклах. Циклобутеновые двойные связи активно участвуют в метатезисной полимеризации, протекающей с раскрытием напряженного четырехчленного фрагмента, в то время как шестичленный цикл, будучи неактивным в полимеризационном циклораскрывающем метатезисе, остается без изменений [27, 37–47]. В результате метатезисной полимеризации ТДД зачастую удается получить полимеры с ММР, близким к единице, что указывает на протекание полимеризации по механизму “живых цепей” [35, 43, 47]. В недавней публикации Р. Граббса и сотрудников [48] методом спектроскопии ЯМР показано, что близкое расположение двойных связей после раскрытия циклобутенового кольца в ходе метатезисной полимеризации ТДД способствует образованию хелатированного Ru-карбенового активного центра. Это приводит к понижению скорости роста цепи относительно скорости инициирования, что обеспечивает протекание контролируемой/“живой” полимеризации ТДД. С использованием способности производных ТДД полимеризоваться по механизму “живых” цепей, синтезированы сополимеры различной архитектуры: блок-, графт-, градиентные, дендримерные и другие [43–45, 47, 49, 50]. Таким образом, анализ литературы показывает, что ТДД представляют интерес в области синтеза новых полимеров, и их метатезисная полимеризация изучается достаточно активно.
Вместе с тем, практически отсутствуют исследования кремнийсодержащих производных ТДД. Насколько нам известно, осуществлена метатезисная полимеризация единственного представителя кремнийзамещенных ТДД – 7,8-бис-(((трет-бутилдиметилсилил)окси)метил)трицикло[4.2.2.02,5]дека-3,9-диена [47]. Причем его получали взаимодействием трет-бутилдиметилхлорсилана с 7,8-бис-((гидроксиметил)трицикло [4.2.2.02,5]дека-3,9-диеном. Сведений о реакции ЦОТ с кремнийсодержащими диенофилами найти не удалось. Представляется интересным получение новых кремнийзамещенных мономеров на основе ТДД и исследование их возможностей в макромолекулярном дизайне в сравнении с кремнийзамещенными полинорборненами и поли-трициклононенами. Наличие в метатезисных поли(трициклодекадиенах) (ПТДД) эндоциклической незамещенной двойной связи в бициклическом мономерном звене, делает их потенциально способными к участию в полимераналогичных превращениях и, следовательно, введению большего числа заместителей.
В настоящей работе впервые изучена реакция ЦОТ с кремнийсодержащими диенофилами, синтезирован новый мономер 7-триметилсилилтрицикло[4.2.2.02,5]дека-3,9-декадиен (ТДДC), исследована его метатезисная полимеризация, изучены термические свойства нового поли(7-триметилсилилтрицикло[4.2.2.02,5]дека-3,9-декадиена) (ПТДДC).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реагенты
1,3,5,7-Циклооктатетраен (98%), ионол и 2,2'-метилен бис-(6-трет-бутил-4-метилфенол) в качестве ингибиторов окисления, катализаторы Граббса первого поколения [бис-(трициклогексилфосфин) бензилиден рутений дихлорид, Г1] и второго поколения [(1,3-бис-(2,4,6-триметилфенил)-2-имидазолидинилиден)(трициклогексилфосфин) бензилиден рутений дихлорид, Г2], приобретенные в “Sigma-Aldrich”, метилйодид (99%, “КемикалЛайн”), винилэтиловый эфир (99%, “Fluka”) использовали без дополнительной очистки. н-Гексан (“Экос-1”), винилтриметилсилан (97%, “Sigma-Aldrich”), винилтрихлорсилан (97%, “Sigma-Aldrich”) и метилбензоат (99%, “Sigma-Aldrich”) перегонкой (20 мм рт.ст.) очищали в атмосфере аргона. Мелкодисперсный порошок Mg прогревали перед применением. Толуол, ТГФ и диэтиловый эфир абсолютировали над металлическим Na. Хлороформ абсолютировали над CaH2.
Методы измерений
Спектры ЯМР регистрировали для растворов образцов в дейтерохлороформе при комнатной температуре на спектрометре “Bruker Avance III”. Рабочая частота прибора составляла 500.13 МГц (ЯМР 1H), 125.76 МГц (ЯМР 13С), 99.36 МГц (ЯМР 29Si). Химические сдвиги на спектрах ЯМР 1H и ЯМР 13C определяли с точностью не менее 0.001 м.д. относительно остаточного сигнала внутреннего стандарта хлороформа. Для спектра ЯМР 29Si в качестве внутреннего стандарта использовали SiMe4.
Комбинированный газохроматографический и масс-спектрометрический анализ проводили с использованием хроматомасс-спектрометра “Thermo Focus DSQ II” с квадрупольным масс-анализатором, энергия электронов 70 эВ, напряжение на электронном умножителе 1244 В, температура источников ионов и интерфейса 280°С, детектирование в режиме регистрации полного ионного тока SIM (selected ion monitoring). Капиллярная колонка со слабополярной неподвижной жидкой фазой DB–5MS 15 м × 0.25 мм × × 0.25 мкм, газ-носитель гелий, скорость газа-носителя 1 мл/мин, температура инжектора 280°С, объем пробы 0.5 мкл. Режим анализа: начальная температура 40°С, через 5 мин повышение температуры со скоростью 8 град/мин до 280°С, общее время анализа 42 мин.
Молекулярно-массовые характеристики определяли методом ГПХ на жидкостном хроматографе “Waters” с рефрактометрическим детектором и системой последовательно соединенных колонок WAT054460 (“Waters”) и G3000HHR (“TosohBiosep”) со сшитым ПС в качестве наполнителя, элюент ТГФ, скорость 1 мл/мин, температура колонки 25°C, концентрация образца 1 мг/мл, объем пробы 100 мкл, калибровка по ПС-стандартам (“PolymerLabs”).
Термические свойства полимеров определяли методами дифференциальной сканирующей калориметрии и термогравиметрического анализа. Термограммы ДСК получали на приборе “MettlerToledo DSC823e”. Нагревание и охлаждение образцов осуществляли со скоростью 20 град/мин в атмосфере аргона со скоростью потока 70 мл/мин в интервале 20–150°C. Представительными считали результаты второго прогревания. ТГА полимеров проводили в атмосфере аргона на приборе “MettlerToledo TGA/DSC-1” со скоростью измерения 10 град/мин и скоростью потока 10 мл/мин при нагревании от 30 до 1000°C. Образцы с массой 10 мг предварительно термостатировали при 30°C.
Синтез 7-трихлорсилилтрицикло [4.2.2.02,5]дека-3,9-диена
Все опыты по синтезу мономеров, полимеров и их модификации проводили на стандартной вакуумной установке и линии Шленка в атмосфере аргона.
В круглодонную стеклянную ампулу объемом 20 мл загружали 46 мг ионола – ингибитора окисления. Добавляли 2.2 мл (2.1 г, 0.020 моля) ЦОТ и 3.8 мл (4.7 г, 0.030 моля) винилтрихлорсилана. Дегазировали смесь 3 раза, используя цикл замораживание жидким азотом–вауумирование– размораживание. Затем запаивали ампулу в вакууме (0.09 мм рт.ст.) и выдерживали 29 ч при 170°C. Для контроля степени протекания реакции 0.2 мл смеси запаивали в вакууме в ампулу-вкладыш, которую прогревали одновременно с большой ампулой. Периодически вкладыш помещали в стандартную ампулу ЯМР с CDCl3, выполнявшего роль внешнего стандарта, и регистрировали спектры ПМР. После окончания реакции отгоняли целевую фракцию с Ткип = 120–121°C (7 мм рт.ст.). Получили 0.6 г (0.0023 моля) 7-трихлорсилилтрицикло[4.2.2.02,5]дека-3,9-диена (ТДДХ) (выход 12%).
Спектр ЯМР 1H (500 МГц, СDCl3; δН, м.д.): 5.74–5.55 (м, 4H, HC3,4,9,10); 2.67, 2.45, 1.54–1.38 (м, 7Н, НC1,2,5,6,7, Н2С8).
Спектр ЯМР 29Si (99 МГц, CDCl3; δSi, м.д.): 13.05 (м, эндо-ТДДХ, 92%); 11.12 (м, экзо-ТДДХ, 8%).
Синтез 7-триметилсилилтрицикло [4.2.2.02,5]дека-3,9-диена
Синтез осуществляли метилированием ТДДХ по методике, аналогичной описанной в [51]. В трехгорлой колбе объемом 250 мл, снабженной магнитной мешалкой, капельной воронкой и обратным водяным холодильником готовили реактив Гриньяра прикапыванием раствора CH3I (4.2 мл, 0.067 моля) в 4.6 мл диэтилового эфира к суспензии из 1.7 г (0.070 моля) мелкодисперсного магния в 23 мл диэтилового эфира и последующим кипячением реакционной смеси в течение 40 мин. К полученному реактиву Гриньяра прикапывали раствор 3.9 г (0.015 моля) ТДДХ в 3.8 мл диэтилового эфира, после чего реакционную смесь кипятили еще 9 ч. Эфир отгоняли при постепенном нагревании куба до 120°C. Продукт экстрагировали гексаном (5 раз по 10 мл). Экстракт фильтровали, пропуская через 1 см (ø5 мм) слой силикагеля. Целевой 7-триметилсилилтрицикло[4.2.2.02,5]дека-3,9-диен (ТДДC) выделяли разгонкой, Ткип = 85–86°С (3 мм рт.ст.), выход 65% (2.0 г , 0.0098 моля).
Спектр ЯМР 1H (500 МГц, СDCl3; δН, м.д.): 6.14 (м, 0.1H, экзо-HC3,4); 5.95–5.88 (м, 1.9H, эндо-HC3,4); 5.88–5.80 (м, 2Н, HC9,10); 2.72–2.63 (м, 2Н, HC2,5); 2.63–2.58 (м, 0.2Н, экзо-HC1,6); 2.58–2.49 (м, 1.8Н, эндо-HC1,6); 1.66–1.60 (м, 0.9Н, эндо-Н2С8), 1.55–1.49 (м, 0.1Н, экзо-H2C8); 1.35–1.32 (м, 0.1Н, экзо-H2C8); 1.32–1.26 (м, 0.9Н, эндо-H2C8); 0.73–0.68 (м, 0.1Н, экзо-HC7); 0.68–0.63 (м, 0.9Н, эндо-HC7); 0.05 (с, 1.2Н, экзо-Si(CH3)3); –0.08 (м, 7.8Н, эндо-Si(CH3)3).
Спектр ЯМР 13C (126 МГц, CDCl3; δС, м.д.): 138.84 (с, экзо-НС3); 138.54 (с, эндо-НС3); 138.20 (с, эндо-НС4); 138.00 (с, экзо-НС4); 133.25 (с, экзо-НС9,10); 130.17 (с, эндо-НС9,10); 129.09 (с, эндо-НС9,10); 128.24 (с, экзо-НС9,10); 49.57 (с, эндо-НС2,5); 46.52 (с, экзо-НС2,5); 45.93 (с, эндо-НС2,5); 44.31 (с, экзо-НС2,5); 35.77 (с, эндо-НС1,6); 35.74 (с, экзо-НС1,6); 35.22 (с, экзо-НС1,6); 35.12 (с, эндо-НС1,6); 26.85 (с, эндо-Н2С8); 25.97 (с, экзо-Н2С8); 24.26 (с, экзо-НС7); 24.14 (с, эндо-НС7); –1.40 (с, 1.2С, экзо-Si(CH3)3); –2.09 (м, 7.8С, эндо-Si(CH3)3).
Спектр ЯМР 29Si (99 МГц, CDCl3; δSi, м.д.): 3.40 (c, эндо-ТДДC, 89%); 2.54 (с, экзо-ТДДC, 11%).
Газохроматографический и масс-спектрометрический анализ (эндо-ТДДC) (94%): 204 [M] + + (3%), 130 [М–SiMe3] + (27%), 73 [М–SiMe3–C4H4] + (100%).
Газохроматографический и масс-спектрометрический анализ (экзо-ТДДC) (6%): 204 [М] + (2%), 130 [М–SiMe3] + (17%), 73 [M–SiMe3–C4H4] + (100%).
Термическая олигомеризация 1,3,5,7-циклооктатетраена
В стеклянный вкладыш для ЯМР-ампулы объемом 0.3 мл, загрузили 0.1 мл (0.1 г, 0.91 ммоля) ЦОТ. Дегазировали мономер 3 раза, используя цикл замораживание жидким азотом–вауумирование– размораживание. Затем запаивали вкладыш в вакууме (0.07 мм рт.ст.) и выдерживали 8 ч при 200°C. Вкладыш помещали в стандартную ЯМР-ампулу с CDCl3, выполнявшего роль внешнего стандарта, и регистрировали ПМР-спектр.
Метатезисная полимеризация 7-триметилсилилтрицикло [4.2.2.02,5]дека-3,9-диена
В круглодонной двугорлой колбе объемом 100 мл, снабженной магнитной мешалкой, смешивали 0.41 г (0.48 мл, 2.0 ммоля) ТДДC и 0.27 мл хлороформа. Затем добавляли отдельно приготовленный раствор 0.5 мг (0.00064 ммоля) катализатора Граббса второго поколения (Г2) в 0.53 мл хлороформа. Перемешивали реакционную смесь при комнатной температуре 21.5 ч. Реакцию останавливали введением 0.2 мл винилэтилового эфира и 15 мл хлороформа, через 30 мин добавляли 24 мг ингибитора окисления. Разбавив реакционную смесь 16 мл хлороформа, выделяли поли(7-триметилсилилтрицикло[4.2.2.02,5]дека-3,9-диен) (ПТДДC) осаждением в этиловый спирт, сушили в вакууме до постоянной массы. В результате с выходом 95% получали 0.39 г ПТДДC.
Спектр ЯМР 1H (500МГц, СDCl3; δH, м.д.): 6.39 (ш.м, 0.2H, цис-HC3,4); 6.17 (ш.м, 1.8Н, транс-HC3,4); 5.03 (ш.м, 2Н, HC9,10); 2.84 (ш.м., 1.4Н, HC1,6); 2.60–2.20 (ш.м, 2.4Н, HC1,2,5,6); 1.78 (ш.м, 0.8Н, H2C8); 1.45 (ш.м, 0.4Н, H2C8, HC7); 1.16 (ш.м, 1Н, H2C8); 0.88 (ш.м, 0.9Н, HC7); 0.58 (ш.м, 0.1Н, H2C7); 0.16–0.13 (ш.м, 1.3Н, экзо-Si(CH3)3); –0.10 (ш.м, 7.7Н, эндо-Si(CH3)3).
Спектр ЯМР 13C (126 МГц, СDCl3; δC, м.д.): 137.16 (ш.м, цис-HC3,4); 133.53 (ш.м, HC3,4); 131.70 (ш.м, HC9,10); 52.34 (ш.м, HC1,2,5,6); 46.44 (ш.м, HC1,6); 41.70 (ш.м, HC1,6); 37.99 (ш.м, HC2,5); 28.86 (ш.м, H2C8); 25.60 (ш.м, HC7); –1.55 (м, 0.4С, экзо-Si(CH3)3); –2.19 (м, 2.6С, эндо-Si(CH3)3).
Спектр ЯМР 29Si (99 МГц, CDCl3; δSi, м.д.): 2.93 (м, Si(CH3)3).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Синтез мономера
Синтез трицикло[4.2.2.02,5]дека-3,9-диенов (ТДД) из 1,3,5,7-циклооктатетраена протекает через изомеризацию ЦОТ в бицикло[4.2.0]окта-2,4,7-триен (БОТ) и последующее [4 + 2]-циклоприсоединение к диенофилу. Известны методы направленного синтеза БОТ [52–55], однако получение ТДД из него ограничено нестабильностью БОТ [52, 56, 57]. Более удобным является прямой синтез ТДД из ЦОТ. Как правило, взаимодействие ЦОТ с диенофилами проводят при повышенной температуре, так как изомеризация ЦОТ в БОТ происходит при температуре выше 100°С [24, 35, 43]. Конкурентным побочным процессом является олигомеризация ЦОТ, протекающая по механизму [4 + 2]-циклоприсоединения через взаимодействие молекул БОТ [24]:

В настоящей работе было изучено взаимодействие ЦОТ с винилтриметилсиланом и винилтрихлорсиланом:
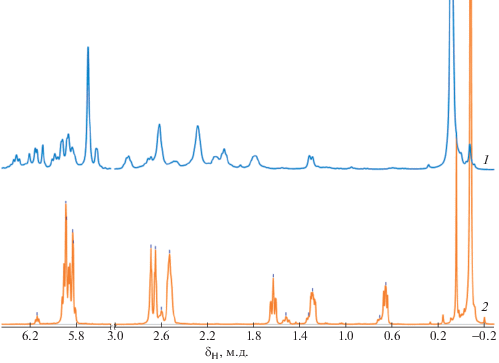
Контроль степени протекания реакции осуществляли методом спектроскопии ЯМР 1Н. Варьировали соотношение реагентов, температуру и время реакции. Смеси винилтриметилсилана (ВТМС) с ЦОТ в мольных соотношениях 1.0 : 1.3 и 1.5 : 1.0 выдерживали при Ткомн в течение 16 ч, нагревали 1 ч при 170°С, и 24 ч при 200–210°С. На рис. 1 представлены спектры реакционной смеси, полученной после длительного нагрева при 200–210°С, и спектр целевого ТДДC. Наиболее заметным, не перекрывающимся другими сигналами, должен быть сигнал ТДДC c δH = 0.66 м.д., который отсутствовал на спектрах реакционной смеси при всех исследованных условиях. Новые сигналы, появляющиеся при нагревании реакционной смеси при 200°С и выше, относятся к продуктам олигомеризации ЦОТ, что было продемонстрировано специальными экспериментами по нагреванию ЦОТ при 200°С. Разгонка реакционной смеси ЦОТ и ВТМС подтвердила данные мониторинга ЯМР 1Н: ожидаемый продукт реакции (ТДДC) отсутствовал.
Рис. 1.
Спектры ЯМР 1H реакционной смеси ЦОТ и ВТМС (1/1.5, моль/моль) после нагревания при 200–210°С в течение 24 ч (1) и ТДДC (2).
Аналогичные эксперименты были проведены с более активным диенофилом винилтрихлорсиланом (ВТХС), содержащим Cl3Si-электроноакцепторный заместитель. Реакцию исследовали при мольных соотношениях ЦОТ : ВТХС = 1 : (1.3–1.5), различной температуре и времени реакции: 16 ч при Ткомн, 13 ч при 140°С, и 18 ч при 200°С. Анализ спектров ЯМР 1Н показал, что реакция ЦОТ с ВТХС практически не протекала при температуре ниже 140°С. Наибольшее количество образовавшихся продуктов наблюдалось при 170°С, а при более высокой температуре (200°С) состав смеси не изменялся. Добавление в реакционную смесь ингибитора ионола не приводило к заметным изменениям состава продуктов. К сожалению, четко выделить сигналы ТДДХ на ЯМР-спектрах реакционной смеси не удалось, так как большинство сигналов целевого ТДДХ (δН = 2.67, 2.45, 1.54–1.38 м.д.; рис. 2, спектр 3) перекрывались сигналами продуктов олигомеризации ЦОТ (рис. 2, спектр 4). Тем не менее по характеру сигналов в указанных областях спектра можно было наблюдать изменения состава реакционной смеси. В качестве режима синтеза ТДДХ были выбраны условия нагревания при 170°С в течение 29 ч. Из реакционной смеси с выходом 12% был выделен ТДДХ. Интересно, что, согласно спектру ЯМР 29Si, ТДДХ содержал 92% эндо- и 8% экзо-изомера. В то же время в литературе указывается, что реакция ЦОТ с производными малеинового ангидрида протекает с образованием исключительно эндо-изомера [35]. Возможно, проведение реакции диеновой конденсации ЦОТ с ВТХС в более жестких условиях, по сравнению с производными малеинового ангидрида, способствует образованию экзо-изомера. Увеличение выхода экзо-5-триметилсилил-2-норборнена с повышением температуры наблюдали ранее в реакции диеновой конденсации циклопентадиена/дициклопентадиена с ВТХС и ВТМС [16].
Рис. 2.
Спектры ЯМР 1H исходной смеси ЦОТ с винилтрихлорсиланом (1.3–1.5/1, моль/моль) (1), реакционной смеси после нагревания при 170°С в течение 24 ч (2), ТДДХ (3) и реакционной смеси после нагревания ЦОТ (4).

Дальнейшим метилированием хлораддукта ТДДХ реактивом Гриньяра был получен целевой мономер ТДДC с выходом 65%. Строение ТДДC определено методами газохроматографического и масс-спектрометрического анализа, спектроскопии ЯМР 1Н, ЯМР 13С и ЯМР 29Si. Отнесение сигналов в ЯМР-спектрах осуществлено с помощью JMODECHO-ЯМР 13C и двумерных корреляций С-Н и Н-Н HMQC и COSY соответственно. Согласно газохроматографическому и масс-спектрометрическому анализу ТДДC состоит из двух изомеров в соотношении 94 : 6, что можно отнести к наличию эндо- и экзо-изомеров. Это подтверждается также данными спектра ЯМР 29Si, на котором наблюдаются два сигнала атомов кремния в эндо- и экзо-звеньях ТДДC в соотношении 89 : 11, а также в спектрах ЯМР 1Н и ЯМР 13С. Указанное соотношение в пределах погрешности соответствует содержанию экзо- и эндо-изомеров в ТДДХ.
Таким образом, исследование взаимодействия ЦОТ с кремнийолефинами ВТМС и ВТХС продемонстрировало ряд закономерностей, характерных для реакции диеновой конденсации: меньшую активность ВТМС по сравнению с ВТХС из-за наличия в последнем электроноакцепторного Cl3Si-заместителя, активирующего двойную связь диенофила; образование продукта реакции преимущественно в виде эндо-изомера, а также тенденцию к формированию экзо-изомера в более жестких условиях реакции. Низкий выход ТДДХ, возможно, является следствием стерических затруднений, возникающих в ходе диенового синтеза, вызываемых объемным Cl3Si-заместителем в диенофиле ВТХС. В литературе [35] отмечают существенное влияние стерического фактора, выражавшееся в понижении выхода аддукта циклоприсоединения ЦОТ к N-фенилмалеимидам от 85 до 35% при введении заместителей в орто-положение фенильного заместителя. Стоит отметить, что другие пути синтеза ТДД и ТДТ являются многостадийными [52–57], а каталитические превращения ЦОТ сопровождаются скелетными перегруппировками и приводят к продуктам другого строения [58].
Метатезисная полимеризация 7-триметилсилилтрицикло[4.2.2.02,5]дека-3,9-диена
Полимеризацию ТДДC проводили по схеме метатезиса в присутствии Ru-карбеновых катализаторов Г1 и Г2. Реакция протекает с раскрытием напряженного циклобутенового фрагмента, не затрагивая двойную связь неактивного в реакции метатезиса олефинов шестичленного цикла:
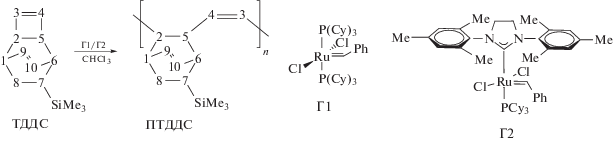
В присутствии катализатора Г1, при мольном соотношении [ТДДC] : [Г1] = 600 : 1 практически с количественным выходом был получен поли(7-триметилсилилтрицикло[4.2.2.02,5]дека-3,9-диен) (ПТДДC) с Mw = 107 × 103 и Ðм = 1.3 (табл. 1, опыт 1). Увеличением мольного соотношения до [ТДДC] : [Г1] = 3430 : 1 и концентрации реакционной смеси до 1.5 моль/л удалось достичь Mw = = 217 × 103 (опыт 3). Для получения более высокомолекулярного продукта был использован более активный катализатор Г2. В условиях, подобранных для катализатора Г1, на Г2 синтезирован ПТДДC с ММ почти на порядок выше, чем на Г1 (опыт 4).
Cтруктура впервые синтезированного полимера ПТДДC была доказана с использованием спектров ЯМР 1Н, ЯМР 13С JMODECHO (рис. 3), ЯМР 29Si, а также двумерных спектров COSY и HMQC. Содержание звеньев с заместителем SiMe3 в экзо-положении удается определить из спектров ЯМР 1Н и ЯМР 13С по сигналам протонов и углеродов группы –SiMe3: оно составило 13%, что примерно соответствует содержанию экзо-ТДДC в мономере. Cоотношение цис-/транс- двойных связей С = С основной цепи ПТДДC, определенное из спектра ЯМР 1Н по сигналам 6.39 (цис-HC3,4) и 6.17 м.д. (транс-HC3,4), составило 8/92. Спектры полимеров, синтезированных на катализаторах Г1 и Г2, были практически идентичны.
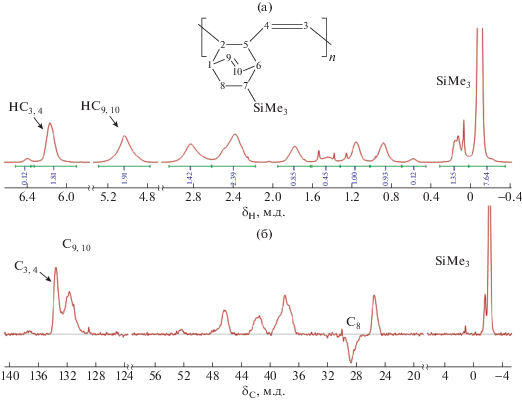
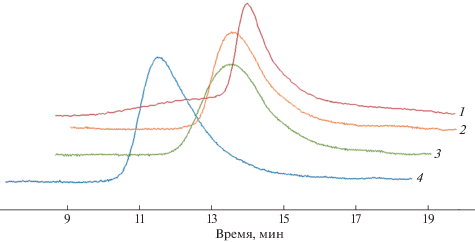
Сравнение условий метатезисной полимеризации ТДДC, характеристик и свойств полученного полимера с ранее изученными норборненом и его кремнийпроизводными 5-триметилсилилнорборненом (НБС) и 3-триметилсилилтрициклононеном (ТЦНС) показывает, что новый мономер демонстрирует активность, сопоставимую с норборненами, образуя ПТДДC c близкими выходами (табл. 1, опыты 1, 5–7). Вместе с тем, в схожих условиях формируется полимер ПТДДC с меньшей ММ, по сравнению с полинорборненами. Получаемый ПТДДC по характеристикам и микроструктуре ближе к ПТЦНС, демонстрируя относительно узкое ММР (рис. 4) и меньшее содержание цис-звеньев по сравнению с ПНБ и ПНБС. Возможная причина более узкого ММР в наличии объемного бициклического мономерного звена в ПТДДC и ПТЦНС, что затрудняет межцепную передачу цепи, обычно приводящую к увеличению ММР. Бòльшая однородность микроструктуры ПТДДC и ПТЦНС, выражающаяся в меньшей доле цис-звеньев, может быть обусловлена стерически более объемным строением молекул мономеров ТДДC и ТЦНС. Возможно, определенное значение имеет то, что ТДДC практически на 92% состоит из эндо-изомера, ТЦНС представляет собой исключительно экзо-изомер, а НБС состоит из почти эквимольной смеси экзо- и эндо-изомеров. Отличие строения циклического фрагмента мономерного звена ПТДДC в значительной степени сказывается на его термических свойствах: он обладает наибольшей температурой стеклования, превышающей на 100 и более градусов температуры стеклования приведенного ряда полинорборненов, что свидетельствует о бóльшей жесткости его цепи (табл. 1). Согласно данным ТГА термическая стабильность ПТДДC ниже, чем стабильность полинорборненов (табл. 1). Температура разложения описанных в литературе ПТДД, в основном производных малеинового ангидрида, имеет значения 300–344°С [35]. После гидрирования двойных связей основной цепи их температура разложения возрастает примерно на 100°С, что объясняют возможным значительным влиянием жесткости основной цепи ПТДД на их термостабильность.
Таблица 1.
Метатезисная полимеризация ТДДC в сравнении с норборненом, 5-триметилсилилнорборненом (НБС) и 3-триметилсилилтрициклононеном (ТЦНС) в растворе CHСl3, при Ткомн, в присутствии катализатора Г1
| Опыт, № | М | М/кат, м/м | [C], моль/л | Время, ч | Выход, % | Mw × 10–3 | Ɖм | Цис-/транс- С=С основной цепи | Тс, °С | Tразлож,°C (5%) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Воздух | Аргон | ||||||||||
| 1 | ТДДC | 600 | 0.4 | 19.3 | 96 | 107 | 1.3 | – | – | – | – |
| 2 | ТДДC | 1420 | 1.5 | 20.4 | 92 | 164 | 1.6 | – | – | – | – |
| 3 | ТДДC | 3430 | 1.5 | 20.4 | 89 | 217 | 1.9 | 8/92 | 187 | 266 | 261 |
| 4* | ТДДC | 3150 | 1.6 | 21.5 | 95 | 1575 | 2.7 | 8/92 | 204 | – | – |
| 5 | Норборнен | 710 | 0.8 | 1.5 | 92 | 330 | 2.8 | 12/88 | 42 | 415 | 421 |
| 6 | [23, 59] НБС [23, 59] | 500 | 0.4 | 24 | 92 | 200 | 2.0 | 28/72 | 103 | 318 | 406 |
| 7 | ТЦНС [60] | 3000 | 0.4 | 24 | 98 | 634 | 1.5 | 10/90 | 104 | – | – |
Рис. 4.
Кривые гель-проникающей хроматографии ПТДДС, номера кривых (1–4) соответствуют номерам опытов в табл. 1.
Ранее мы наблюдали повышение Тст в ряду полинорборненов при введении в них Me3Si-заместителей и с изменением строения основной цепи от метатезисных к аддитивным полинорборненам, что, за редким исключением, сопровождалось увеличением коэффициентов газопроницаемости полимеров [16, 59, 60]. Поэтому можно ожидать, что большая жесткость цепи ПТДДС, а также присутствие Ме3Si-заместителя будут способствовать хорошим газоразделительным свойствам полимера.
Исследование выполнено при финансовой поддержке Российского фонда фундаментальных исследований в рамках научного проекта № 20-33-90158. Строение полученных соединений изучено с использованием научного оборудования Центра коллективного пользования ИНХС РАН “Аналитический центр проблем глубокой переработки нефти и нефтехимии” и Центра исследования строения молекул ИНЭОС РАН.
Авторы выражают благодарность сотрудникам ИНХС РАН Р.С. Борисову, Я.И. Дерикову, Г.А. Шандрюку за помощь в анализе полимеров, а также А.С. Перегудову (ИНЭОС РАН) за съемку ЯМР 13C, ЯМР 29Si и двумерных ЯМР-спектров.
Список литературы
Elling B.R., Su J.K., Xia Y. // Acc. Chem. Res. 2021. V. 54. № 2. P. 356.
Grubbs R.B., Grubbs R.H. // Macromolecules. 2017. V. 50. № 18. P. 6979.
Mol J. // J. Mol. Catal. A. Chem. 2004. V. 213. № 1. P. 39.
Yamazaki M. // J. Mol. Catal. A. Chem. 2004. V. 213. № 1. P. 81.
Janiak C., Lassahn P.G. // J. Mol. Catal. A. Chem. 2001. V. 166. № 2. P. 193.
Flid V.R., Gringolts M.L., Shamsiev R.S., Finkelshtein E.S. // Russ. Chem. Rev. 2018. V. 87. № 12. P. 1169.
Farquhar A.H., Brookhart M., Miller A.J.M. // Polym. Chem. 2020. V. 11. № 14. P. 2576.
Petrov V., Vasil’ev N. // Curr. Org. Synth. 2006. V. 3. № 2. P. 215.
Finkelshtein E.S., Chapala P.P., Gringolts M.L., Rogan Y.V. // Polymer Science C. 2019. V. 61. № 1. P. 17.
Slugovc C. Industrial Applications of Olefin Metathesis Polymerization // Olefin Metathesis / Ed. K. Grela Hoboken: Wiley, 2014. P. 329.
Bermeshev M.V., Chapala P.P. // Prog. Polym. Sci. 2018. V. 84. P. 1.
Kovačič S., Slugovc C. // Mater. Chem. Front. 2020. V. 4. № 8. P. 2235.
Blank F., Janiak C. // Coord. Chem. Rev. 2009. V. 253. № 7. P. 827.
Handbook of Metathesis / Eds by R.H. Grubbs, E. Khosravi. Weinheim, 2015. V. 3.
Bermeshev M.V., Finkelshtein E.S. // Ineos Open. 2018. V. 1. № 1. P. 39.
Finkelshtein E.S., Gringolts M.L., Bermeshev M.V., Chapala P.P., Rogan Y.V. // Membrane Materials for Gas and Vapor Separation. Chichester: Wiley, 2017. P. 143.
Yampolskii Y., Starannikova L., Belov N., Bermeshev M., Gringolts M., Finkelshtein E. // J. Membr. Sci. 2014. V. 453. P. 532.
Alentiev D.A., Egorova E.S., Bermeshev M.V., Starannikova L.E., Topchiy M.A., Asachenko A.F., Gribanov P.S., Nechaev M.S., Yampolskii Y.P., Finkelshtein E.S. // J. Mater. Chem. A. 2018. V. 6. № 40. P. 19393.
Karpov G.O., Bermeshev M.V., Borisov I.L., Sterlin S.R., Tyutyunov A.A., Yevlampieva N.P., Bulgakov B.A., Volkov V.V., Finkelshtein E.S. // Polymer. 2018. V. 153. P. 626.
Karpov G.O., Borisov I.L., Volkov A.V., Finkelshtein E.S., Bermeshev M.V. // Polymers. 2020. V. 12. № 6. P. 1282.
Karpov G.O., Alentiev D.A., Wozniak A.I., Bermesheva E.V., Lounev I.V., Gusev Y.A., Shantarovich V.P., Bermeshev M.V. // Polymer. 2020. V. 203. P. 122759.
Bermesheva E.V., Alentiev D.A., Moskalets A.P., Bermeshev M.V. // Polymer Science B. 2019. V. 61. № 3. P. 314.
Morontsev A.A., Zhigarev V.A., Nikiforov R.Y., Belov N.A., Gringolts M.L., Finkelshtein E.S., Yampolskii Y.P. // Eur. Polym. J. 2018. V. 99. P. 340.
Reppe W., Schlichting O., Klager K., Toepel T. // Justus Liebigs Ann. Chem. 1948. V. 560. № 1. P. 1.
Avram M., Nenitzescu C.D., Marica E. // Chem. Ber. 1957. V. 90. № 9. P. 1857.
Miller R.D., Dolce D. // Tetrahedron Lett. 1972. V. 13. № 44. P. 4541.
Paquette L.A., Oku M., Heyd W.E., Meisinger R.H. // J. Am. Chem. Soc. 1974. V. 96. № 18. P. 5815.
Dauben W.G., Rivers G.T., Twieg R.J., Zimmerman W.T. // J. Org. Chem. 1976. V. 41. № 5. P. 887.
Osawa E., Aigami K., Inamoto Y. // J. Org. Chem. 1977. V. 42. № 15. P. 2621.
Miller R.D., Dolce D.L., Merritt V.Y. // J. Org. Chem. 1976. V. 41. № 7. P. 1221.
Albert B., Heller C., Iden R., Martin G., Martin H.-D., Mayer B., Oftring A. // Isr. J. Chem. 1985. V. 25. № 1. P. 74.
Avram M., Sliam E., Nenitzescu C.D. // Justus Liebigs Ann. Chem. 1960. V. 636. № 1. P. 184.
Huisgen R., Mietzsch F. // Angew. Chemie. 1964. V. 76. № 1. P. 36.
Liu R.S.H., Krespan C.G. // J. Org. Chem. 1969. V. 34. № 5. P. 1271.
Charvet R., Novak B.M. // Macromolecules. 2001. V. 34. № 22. P. 7680.
Edwards J.H., Feast W.J. // Polymer. 1980. V. 21. № 6. P. 595.
Edwards J.H., Feast W.J., Bott D.C. // Polymer. 1984. V. 25. № 3. P. 395.
Feast W.J., Taylor M.J., Winter J.N. // Polymer. 1987. V. 28. № 4. P. 593.
Jones C.A., Lawrence R.A., Martens J., Friend R.H., Parker D., Feast W.J., Lögdlund M., Salaneck W.R. // Polymer. 1991. V. 32. № 7. P. 1200.
Park L.Y., Schrock R.R., Stieglitz S.G., Crowe W.E. // Macromolecules. 1991. V. 24. № 12. P. 3489.
Charvet R., Acharya S., Hill J.P., Akada M., Liao M., Seki S., Honsho Y., Saeki A., Ariga K. // J. Am. Chem. Soc. 2009. V. 131. № 50. P. 18030.
Fischer W., Stelzer F., Heller C., Leising G. // Synth. Met. 1993. V. 55. № 2–3. P. 815.
Kim K.O., Choi T.-L. // Macromolecules. 2013. V. 46. № 15. P. 5905.
Yoon K.-Y., Shin S., Kim Y.-J., Kim I., Lee E., Choi T.-L. // Macromol. Rapid Commun. 2015. V. 36. № 11. P. 1069.
Charvet R., Novak B.M. // Macromolecules. 2004. V. 37. № 23. P. 8808.
Stelzer F., Brunthaler J.K., Leising G., Hummel K. // J. Mol. Catal. 1986. V. 36. № 1–2. P. 135.
Kim K.O., Shin S., Kim J., Choi T.L. // Macromolecules. 2014. V. 47. № 4. P. 1351.
Song J.-A., Park B., Kim S., Kang C., Lee D., Baik M.-H., Grubbs R.H., Choi T.-L. // J. Am. Chem. Soc. 2019. V. 141. № 25. P. 10039.
Shin S., Yoon K.-Y., Choi T.-L. // Macromolecules. 2015. V. 48. № 5. P. 1390.
Rajaram S., Choi T.-L., Rolandi M., Fréchet J.M.J. // J. Am. Chem. Soc. 2007. V. 129. № 31. P. 9619.
Zhigarev V.A., Morontsev A.A., Nikiforov R.Y., Gringolts M.L., Belov N.A., Komalenkova N.G., Lakhtin V.G., Finkelshtein E.S. // Polymer Science C. 2019. V. 61. № 1. P. 107.
Adam W., Cueto O., De Lucchi O. // J. Org. Chem. 1980. V. 45. № 25. P. 5220.
Huisgen R., Boche G. // Tetrahedron Lett. 1965. V. 6. № 23. P. 1769.
Kelebekli L., Kara Y., Balci M. // Carbohydr. Res. 2005. V. 340. № 12. P. 1940.
Karanfil A., Şahin E., Kelebekli L. // Tetrahedron. 2020. V. 76. № 11. P. 131000.
Vogel E., Kiefer H., Roth W.R. // Angew. Chemie. 1964. V. 76. № 10. P. 432.
Zimmerman H.E., Iwamura H. // J. Am. Chem. Soc. 1970. V. 92. № 7. P. 2015.
Dyakonov V.A., Kadikova G.N., Gazizullina G.F., Khalilov L.M., Dzhemilev U.M. // Tetrahedron Lett. 2015. V. 56. P. 2005.
Morontsev A.A., Gringolts M.L., Filatova M.P., Finkelshtein E.S. // Polymer Science B. 2016. V. 58. № 6. P. 695.
Gringolts M.L., Bermeshev M.V., Syromolotov A.V., Starannikova L.E., Filatova M.P., Makovetskii K.L., Finkelshtein E.S. // Pet. Chem. 2010. V. 50. № 5. P. 352.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)