

Высокомолекулярные соединения (серия Б), 2022, T. 64, № 2, стр. 85-106
УСПЕХИ В ОБЛАСТИ СИНТЕЗА ОЛИГОМЕРНЫХ ЭПОКСИФОСФАЗЕНОВ ПОНИЖЕННОЙ ГОРЮЧЕСТИ
В. В. Киреев a, *, Ю. В. Биличенко a, И. С. Сиротин a, С. Н. Филатов a
a Российский химико-технологический университет им. Д.И. Менделеева
125047 Москва, Миусская пл., 9, Россия
* E-mail: kireev@muctr.ru
Поступила в редакцию 14.12.2021
После доработки 23.12.2021
Принята к публикации 10.01.2022
- EDN: QBWRCQ
- DOI: 10.31857/S230811392202005X
Аннотация
Рассмотрены основные способы синтеза фосфазенсодержащих эпоксидных олигомеров реакциями функциональных производных циклофосфазенов с глицидолом или эпихлоргидрином, а также окислением двойных связей в соединенных с атомами фосфора органических радикалах. Сопоставлены основные закономерности перечисленных процессов и показаны возможности регулирования свойств фосфазенсодержащих эпоксидных олигомеров, которые после отверждения обычными для эпоксидных смол отвердителями образуют огнестойкие или полностью негорючие композиции.
Эпоксидные олигомеры (ЭО) находят все возрастающее применение в различных областях техники в качестве связующих полимерных композиционных материалов, лакокрасочных покрытий, диэлектрических материалов и для других целей [1–3]. Одним из недостатков этих олигомеров является горючесть, которую уменьшают введением в их состав вспомогательных веществ – антипиренов или включением в структуру молекул ЭО атомов или групп атомов, затрудняющих горение. Применение антипиренов (чаще оксидов и галогенидов металлов) обычно приводит к ухудшению механических характеристик материалов на основе ЭО и ограничивает их применение.
Наличие в молекулах ЭО галогенов и атомов фосфора приводит к пониженной горючести, однако это требует использования соответствующих исходных веществ, синтез и тем более производство которых могут быть сложными и затратными.
В последние годы успешно развивается направление, связанное с синтезом, ограничено горючих и даже полностью негорючих фосфазенсодержащих эпоксидных олигомеров (ФЭО), содержащих в составе молекул группы ≡P=N–. Цель настоящей работы – рассмотрение в основном публикаций последних 20 лет в данной области, выявление наиболее перспективных типов ФЭО в части доступности исходных соединений, простоты и эффективности методов получения указанных эпоксидов.
СИНТЕЗ ФЭО НА ОСНОВЕ ГЛИЦИДОЛА
Еще с середины ХХ века были сделаны попытки синтеза фосфазенсодержащих соединений реакцией гексахлорциклотрифосфазена с глицидолом [4, 5]

Однако в связи с неоднозначным протеканием указанной реакции и нестабильностью образующихся глицидилоксифосфазенов, в частности фосфазен-фосфазановой перегруппировки [6] и других возможных превращений с участием эпоксидных групп, указанное направление не получило дальнейшего развития. Тем не менее, в последние 10 лет М. Gouri и сотрудниками продолжили исследование продуктов реакции (1) и сообщили об образовании соединения I с выходом 78% за 45–48 ч в среде толуола при комнатной температуре в присутствии триэтиламина [7–13]. К сожалению, приведенный в работе [8] спектр ЯМР 31Р соединения I содержит лишь незначительный по интенсивности синглетный сигнал δр = 9.32 м.д., относящийся к соединению I. Основные сигналы на этом спектре расположены в области δр = –5…+5 м.д. и свидетельствуют о деградации фосфазенового цикла с образованием, вероятно, связей Р=О, Р–ОН и Р–NH, и это через 48 ч реакции при комнатной температуре! Возможно, деструкция начинается именно с фосфазен-фосфазеновой перегруппировки [6]. Логично допустить, что при хранении продуктов реакции указанные превращения будут протекать более глубоко. Тем не менее, эти олигомеры, содержание эпоксидных групп в которых авторы [7, 8] не приводят, хорошо совместимы с обычными эпоксидными смолами (DER-331, Epon-828, DER-732) и после отверждения 4,4′-диаминодифенилметаном образуют композиции с пониженной горючестью (UL-94–V0), чему, по-видимому, способствует введение фосфор-азотсодержащих соединений. Это предположение согласуется с данными анализа состава газов и твердого остатка, образующихся при нагревании композиции до 600°С [10].
По данным динамического ТГА наличие в композициях 5–20 мас. % продуктов реакции (1) существенно не влияет на массу твердого остатка при 600°С. Более того, при нагревании понижается температура начала потери массы, видимо, за счет деструкции менее стабильных фосфоразотсодержащих модификаторов. Незначительно изменяется и температура стеклования отвержденных образцов с указанным количеством добавок PN-компонента [10], равно как и величина модуля упругости [11]. Некоторое улучшение механических и диэлектрических свойств композиций с 5–10 мас. % продуктов реакции (1) авторы [12] связывают с оптимальной совместимостью органической смолы и фосфорсодержащих добавок.
При использовании в реакции с глицидолом вместо гексахлорциклотрифосфазена его производного с двумя хлорфосфазеновыми циклами Cl5P3N3OC6H4C(CH3)2C6H4OP3N3Cl5 [13] образуется соединение с 10 эпоксидными группами и эпоксидным эквивалентом EEW = 628 г/экв (вычислено 613 г/экв). Строение этого соединения подтверждено данными спектроскопии ЯМР 1Н, ЯМР13 С и ЯМР31 Р, однако проблема его стабильности во времени в работе [13], к сожалению, не рассмотрена.
СИНТЕЗ ФЭО ОКИСЛЕНИЕМ ДВОЙНЫХ СВЯЗЕЙ В ОРГАНИЧЕСКИХ РАДИКАЛАХ, СВЯЗАННЫХ С АТОМАМИ ФОСФОРА
Известным способом синтеза эпоксидных соединений является окисление олефиновых связей надуксусной, надбензойной и другими надкислотами, а также перекисью водорода. Особенно легко подвергаются окислению аллильные группы органических радикалов, связанных с атомами фосфора в органофосфазенах [14]. Из-за невысокой стабильности алкоксифосфазенов для синтеза эпоксидных PN-содержащих олигомеров преимущественно используют арилоксифосфазены, в частности эвгенольные производные гексахлорциклотрифосфазена и высших циклов. Так, в работе [15] синтезировано гексааллильное соединение
 ,
,
которое при обработке м-хлорнадбензойной кислотой в CH2Cl2 при комнатной температуре в течение 48 ч с выходом 74% образует белый твердый гексаэпоксид
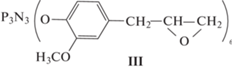
Оба соединения детально охарактеризованы методами спектроскопии ИК, ЯМР 1Н и ЯМР 31Р. Позже соединение II было выделено в кристаллическом виде с Тпл = 82 ± 1°С; его эпоксидирование в указанных выше условиях привело к образованию гексаэпоксида III в смеси с его димерным производным [16, 17] предполагаемого строения

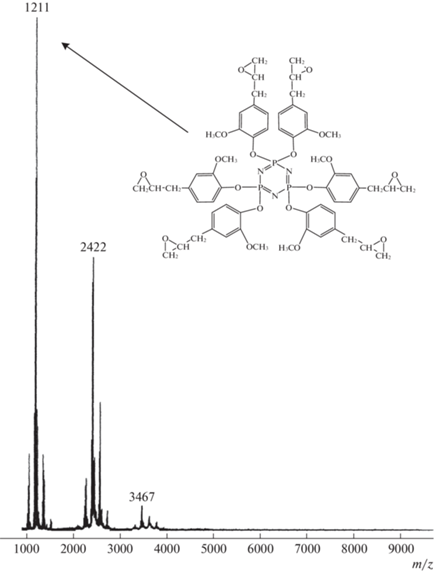
Как считают авторы [16, 17], соединение IV образуется при взаимодействии енольной формы эпоксидной группы одной молекулы III с эпоксидной группой другой такой молекулы. Образование димера IV подтверждает его MALDI-TOF масс-спектр (рис. 1), содержащий наряду с пиком m/z = 1211 второй пик с удвоенным значением m/z = 2422. По соотношению интенсивностей этих пиков, а также по найденному значению эпоксидного числа 20.1% (расчет для соединения III – 21.3%) содержание димера в продукте эпоксидирования составляет ~30%.
Рис. 1.
MALDI-TOF масс-спектр продукта эпоксидирования гексакис-(4-аллил-2-метоксифенокси)циклотрифосфазена.
Примечательно, что в более поздней работе [18] также приведен лазерный масс-спектр соединения III с основным пиком m/z = 1210, однако область с m/z > 1400 на рис. 5 этой статьи не показана.
В противоположность III при синтезе его тетрамерного аналога эпоксидированием

наряду с основным октаэпоксидом VI (Тпл = 84°С) с m/z = 1468

на MALDI-TOF масс-спектре (рис. 2) появляются дополнительные пики других соединений [19]. Их интенсивность зависит от избытка надкислоты: при мольном соотношении V : м-хлорнадбензойная кислота, равном 1 : 12, в составе продукта присутствуют соединения с m/z = 1582 и 1598, содержащие соответственно две и одну неокисленные аллильные группы. Пики с m/z = 1636 и 1649 относятся к продуктам присоединения одной или двух молекул воды к молекуле VI, а пики m/z ≥ ≥ 1768 соответствуют продуктам взаимодействия м-хлорбензойной кислоты с эпоксигруппами:
Рис. 2.
Масс-спектры MALDI-TOF октакис-(4-аллил-2-метоксифенокси)циклотетрафосфазена (а) и продуктов его эпоксидирования в присутствии 12 (б) и 16 (в) моль м-хлорнадбензойной кислоты на 1 моль циклотетрафосфазена. 154 – молекулярная масса присоединившейся к эпоксиду м-хлорбензойной кислоты.


Показана возможность использования для синтеза эпоксипроизводных смеси хлорциклофосфазенов [PNCl2]3–7 [19]: на MALDI-TOF масс-спектрах зафиксированы пики всех эвгенольных и эпоксидных гомологов. Однако попытка получить указанные соединения непосредственно в реакционной смеси после синтеза хлорциклофосфазенов в хлорбензоле без удаления последнего привела к образованию эпоксифосфазенов с одной или двумя группами Р–ОН, образующихся вследствие частичного гидролиза атомов хлора следовыми количествами воды в исходной надкислоте [19].
Для синтеза ФЭО окислением двойных аллильных связей использован циклоспиротрифосфазен, в котором четыре атома хлора геминально замещены пирокатехином, а два других – эвгенолом [20]. Полученный окислением этого соединения диэпоксид использован для смешения с полиамидом-6 или полибутилентерефталатом; образующийся композит обладает улучшенными реологическими, морфологическими и механическими свойствами.
ФЭО, полученные окислением эвгенольных производных хлорциклофосфазенов, содержат до 15–20% эпоксидных групп, отверждаются обычными для органических эпоксидов отвердителями с образованием самозатухающих или негорючих композиций. Однако практическая реализация этого способа затруднена из-за многостадийности процесса, его продолжительности и необходимости использования нестабильных окислителей.
ФЭО, СИНТЕЗИРУЕМЫЕ С ИСПОЛЬЗОВАНИЕМ ЭПИХЛОРГИДРИНА
Реакция эпихлоргидрина (ЭХГ) с соединениями, содержащими подвижный атом водорода (чаще фенолы и амины), лежит в основе наиболее распространенных методов синтеза эпоксидных олигомеров

(здесь и ниже Х=О или NH).
Применительно к ФЭО исходными для их получения в реакции с ЭХГ чаще являются гидроксиарилоксифосфазены или их аминосодержащие аналоги

В случае наиболее доступного и дешевого гексахлорциклотрифосфазена при его взаимодействии с дифенололами вследствие высокой функциональности хлорфосфазена для исключения гелеобразования используют три основных подхода: временную блокировку одной группы ОН дифенола с ее регенерацией после образования арилоксифосфазена; понижение функциональности гексахлорциклотрифосфазена замещением части атомов хлора на инертные радикалы; использование избытка дифенола.
Первый подход реализован на примере п-метоксифенола [21] и п-гидроксибензальдегида [22–24]
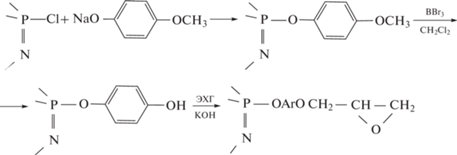
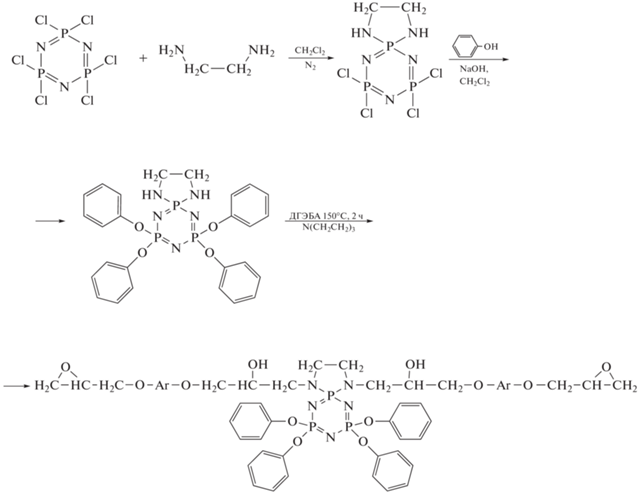
В случае п-гидроксибензальдегида синтез ФЭО осуществлен по трем направлениям (схема (3) ): через метилольные производные феноксифосфазенов (путь А) [25], через гидроксиарилоксиазометиновые соединения (путь В) и эпоксидированием карбоксифеноксициклофосфазенов (путь С) [23, 24, 26].

Здесь и ниже Ar =  , ДГЭБА (диглицидиловый эфир бисфенола А)
, ДГЭБА (диглицидиловый эфир бисфенола А) 
Синтезированные в работах [22–26] ФЭО и их композиции охарактеризованы спектроскопией ИК и ЯМР; к сожалению, в них не приведены значения эпоксидного эквивалента, по которому авторы рассчитывали необходимое количество отвердителей. Отвержденные ароматическими диаминами ФЭО образуют самозатухающие композиции с огнестойкостью V-0 (UL-94) и LOI 32-34.
В случае необходимости функциональность образующегося гексакис-(п-гидроксифенокси)циклотрифосфазена понижают предварительным взаимодействием, например, с бис-(этокси)хлор фосфатом [21]
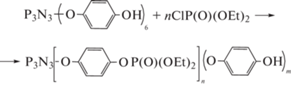
Однако чаще синтез гидроксиарилоксифосфазенов и ФЭО на их основе осуществляют предварительным частичным замещением атомов хлора в гексахлорциклотрифосфазене на инертные радикалы. Например, при взаимодействии тетрафеноксидихлорциклотрифосфазена с гидрохиноном [27] или бисфенолом А [28, 29] образуются тетрафеноксидигидроксиарилоксициклотрифосфазены, эпоксидированием которых получены соответствующие диэпоксиды
 ,
,
где Ar =  или
или  .
.
Как полагают авторы [27–29], эти продукты содержат до 90% соединений приведенной выше формулы и имеют значениями EEW от 500 до 600 г/экв. К сожалению, исходный P3N3(OPh)4Cl2 в работах [27–29] не охарактеризован по содержанию в нем соединений с различной степенью замещения атомов хлора, соответственно не определен и точный состав образующихся ФЭО.
В работах [30, 31] проведен анализ MALDI-TOF масс-спектров продуктов, образующихся на всех стадиях процесса по схеме

Здесь Ar =  .
.
Все ФЭО, образующиеся при различном мольном соотношении х = гексахлорциклотрифосфазен : п-хлорфенол, содержат по нескольку соединений, при этом основными (40–46%) являются задаваемые указанным соотношением (рис. 3). При этом наиболее сложным по составу является ФЭО, полученный при х = 2, который включает смесь соединений, содержащих от одной до четырех эпоксидных групп (табл. 1).
Рис. 3.
MALDI-TOF масс-спектры: а, б, в – хлорфеноксигидроксиарилоксициклотрифосфазены P3N3(OС6Н4Сl)a(OArOH)b; г, д, е – хлорфенокси-гилицидилоксиарилоксициклотрифосфазены P3N3(OС6Н4Сl)a(OArOH)b . Мольное соотношение гексахлорциклотрифосфазен : NaOС6Н4Сl при синтезе исходных P3N3(OС6Н4Сl)aCl6 – a : х = 2 (а, г), 3 (б, д) и 4 (в, е).
. Мольное соотношение гексахлорциклотрифосфазен : NaOС6Н4Сl при синтезе исходных P3N3(OС6Н4Сl)aCl6 – a : х = 2 (а, г), 3 (б, д) и 4 (в, е).
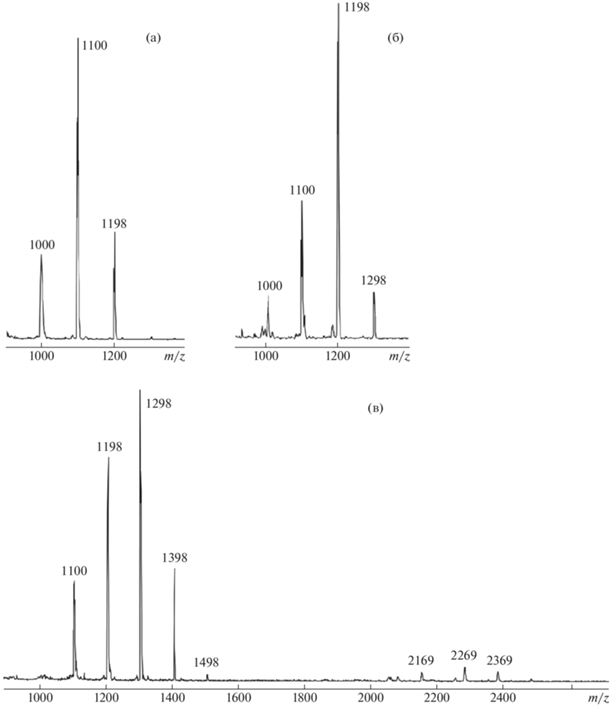
Рис. 3.
Окончание.

Таблица 1.
Содержание индивидуальных соединений в эпоксифосфазенах 
| Соединение со значением m/z | а : b : c | Содержание (%) соединений в ФЭО, полученных при различных x | ||
|---|---|---|---|---|
| x = 2 | x = 3 | x = 4 | ||
| 1056 | 5:0:1 | – | – | 23 |
| 1156 | 4:1:1 | 8 | – | 25 |
| 1212 | 4:0:2 | 12 | 27 | 46 |
| 1311 | 3:1:2 | 40 | 29 | 6 |
| 1366 | 3:0:3 | 13 | 44 | – |
| 1466 | 2:1:3 | 14 | – | – |
| 1522 | 2:0:4 | 8 | – | – |
| 1622 | 1:1:4 | 5 | – | – |
| Эпоксидное число, % (вычислено/найдено) | 9.7/9.1 | 7.1/6.8 | 6.3/5.9 | |
Столь сложный состав ФЭО на основе частично замещенных арилоксихлорциклотрифосфазенов не влияет на их способность к отверждению, но, естественно, скажется на параметрах образующейся трехмерной сетки.
Оригинальный метод получения ФЭО, предложенный в работах [32, 33], представлен на приведенной ниже схеме
Строение конечного диэпоксида подтверждено спектрами ЯМР 1Н, ЯМР 13С, ЯМР 31Р и функциональным анализом: EEW = 674 г/экв (вычислено 642 г/экв). Тем не менее, авторы не исключают наличия в олигомере некоторого количества соединений с двумя или тремя спироциклическими фосфазеновыми фрагментами. У отвержденных ароматическими диаминами или новолаком композиции указанного диэпоксида Тс находится в пределах 157–164°С, кислородный индекс (LOI) 31–32 и индекс негорючести V-0 (по стандарту UL-94) [32]. Нанокомпозиты на его основе, содержащие 0.125–0.175 об. % графена, имеют повышенные механические свойства, обладают электрической проводимостью до 10–3 См/м и являются самозатухающими [33].
С целью повышения содержания фосфора и понижения вязкости ФЭО для их синтеза вместо бисфенола А используют резорцин [34].
При взаимодействии избытка резорцина и гексахлорциклотрифосфазена образующаяся смесь была очищена от избытка дифенола многократной экстракцией водой, а полученный гексакис-(м-гидроксифенокси)циклотрифосфазен охарактеризован спектроскопией ЯМР 1Н и ЯМР 31Р (δр = 9.8 м.д.), а также лазерными масс-спектрами (m/z = 790) [34]. Реакция этого полифенола в среде избытка ЭХГ в присутствии спиртового растовора щелочи приводит к образованию соответствующего гексаэпоксида (m/z = 1126) с примесью не более 5% пентаэпоксидного соединения.
Эпоксидное число данного олигомера составляет ~20, а содержание в нем фосфора около 8%. При отверждении его диаминами или ангидридами образуются полностью негорючие композиции.
Хотя большинство ФЭО синтезируют на основе гексахлорциклотрифосфазена, в работе [35] для их получения использован линейный полидихлорфосфазен, превращенный в полиэпоксид через последовательные стадии
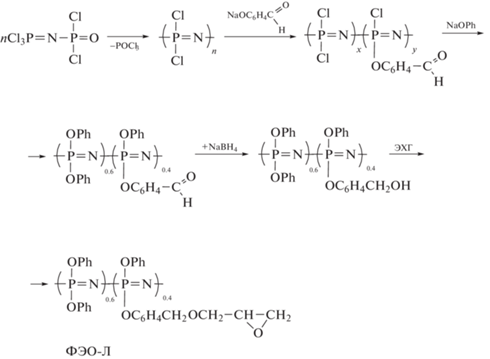
По данным ГПХ продукт ФЭО-Л имел Мn = = 186 × 103, Мw = 374 × 104 и эпоксидный эквивалент EEW = 671 г/экв, что при указанном соотношении звеньев (0.6 и 0.4) не соответствует расчетному значению 264 г/экв.
Вызывает удивление наличие двух сигналов в области δр = –17 и –20 м.д. на ЯМР 31Р-спектре ФЭО-Л; маловероятно, что наличие удаленного от атомов фософора глицидильного фрагмента в фенокси-радикалах сильно скажется на энергии перехода между уровнями магнитной энергии ядер атомов фосфора с различными арилоксизаместителями. Необычной представляется также эволюция спектров ЯМР 31Р полидихлорфосфазена (δр = ~18.2 м.д.) при частичном замещении в нем атомов хлора на п-формилфениленокси-радикалы. На спектре частично замещенного полихлорфосфазена проявляются концевые группы Cl3Р= и РОCl2, концентрация которых, судя по интенсивности сигналов, достаточно высока и не соответствует заявленной авторами молекулярной массе исходного полидихлорфосфазена 101 × 103. Возможно, при реакции последнего с фенолятом п-формилфенола протекают побочные деструктивные процессы.
Тем не менее ФЭО-Л оказались эффективными модификаторами обычных олигоэпоксидов; так, при введении ФЭО-Л в количестве 10–30 мас. % в ДГЭБА в 2.5 раза повышаются максимальная скорость отверждения композиции, более чем в 2 раза ударная прочность, хотя при этом понижается прочность на разрыв и изгиб. Значения LOI при содержании ФЭО-Л 30% увеличивается до 32, а композиция является сомозатухающей (V-0 по UL-94).
Рассмотренные выше методы синтеза фосфазенсодержащих эпоксидных олигомеров, как правило, многостадийные, что затрудняет их практическую реализацию. В связи с этим по аналогии с промышленными эпоксидными олигомерами, синтезируемыми с использованием в основном трех исходных компонентов – полифенолов или ароматических диаминов, эпихлоргидрина и щелочи, представлялось интересным распространить данный подход на получение ФЭО с включением в состав исходных смесей кроме трех указанных соединений еще и гексахлорциклотрифосфазен. Были использованы три основных схемы синтеза, представленные ниже в виде превращений D, E и F:
где Ar =
 или
или  , Ar′ = Ph, C6H4X (X-галоген).
, Ar′ = Ph, C6H4X (X-галоген).
Основным в этой схеме является синтез промежуточных гидроксиарилоксифосфазенов, для образования которых требуется значительный избыток дифенолов как для достижения максимального замещения атомов хлора в гексахлорциклотрифосфазене, так и для исключения гелеобразования в случае превращений E и F.
При использовании монофенолята бисфенола А (путь D) процесс осложняется наличием в феноляте равновесных количеств дифенола и его дифенолята, что приводит к образованию гидроксиарилоксифосфазенов (ГАРФ), содержащих наряду с основным количеством P3N3(OArOH)6 (m/z = = 1500 на рис. 4) и его катионизированной ионом Na формы (m/z = 1522), а также димера с (m/z = = 2766) и аналогов последнего с неполностью замещенными атомами хлора m/z ≤ 2542 [36]. Несмотря на относительно невысокое содержание димерных соединений (менее 20%) получаемые гидроксиарилоксифосфазены являются высоковязкими веществами. Их эпоксидирование в избытке ЭХГ в присутствии спиртового раствора КОН приводит к образованию полутвердых ФЭО с эпоксидным числом до 12%, что ниже расчетного для гексаэпоксида и свидетельствует о незавершенном эпоксидировании гидроксиарилоксифосфазена, полученного через монофенолят.
Рис. 4.
MALDI-TOF масс-спектр гидроксиарилоксифосфазенов, синтезированных реакцией фенолятов дифенилолпропана с гексахлорциклотрифосфазеном в мольном соотношении 12 : 1 (Na : дифенол = 0.8 : 1.0).

Рис. 5.
Масс-спектр MALDI-TOF фосфазенового продукта реакции гексахлорциклотрифосфазена с избытком дифенилолпропана в расплаве при 170°С в присутствии K2CO3.
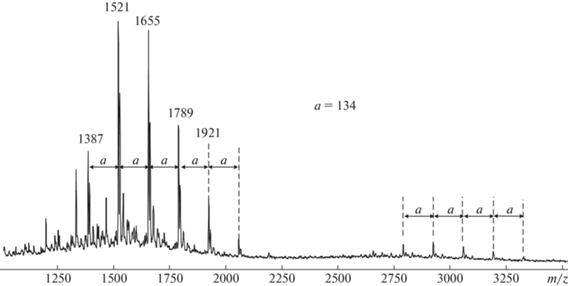
ГХФ и бисфенол А в расплаве и в растворе не реагируют до 200°С [37], однако в присутствии карбоната калия при 170°С реакция завершается за 2–3 ч, при этом наряду с целевым P3N3(OArOH)6 (m/z = 1521) (рис. 5), в составе ГАРФ появляется продукт алклирования гексаэпоксида п-изпропенилфенолом; последний образуется при разложении бисфенола А или (в меньшей степени) связанных с атомами фосфора фосфазенового цикла фрагментов бисфенола А:
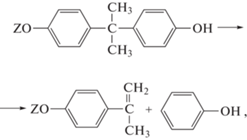
где Z = H или  =N–.
=N–.
Как следует из рис. 5, алкилированию подвергаются от 1 до 4 связанных с атомами фосфора радикалов бисфенола, о чем свидетельствуют: пики с m/z = 1655, 1921 и 2055, разница между которыми (Δm/z = 134) соответствует молекулярной массе присоединившихся молекул п-изопропенилфенола.
Алкилирование арилоксирадикалов в образующихся гидроксиарилоксифосфазенах имеет место и при осуществлении взаимодействия гексахлорциклотрифосфазена с избытком бисфенола А в среде пиридина при 110°С (рис. 6), хотя и в меньшей степени.
Рис. 6.
Масс-спектр MALDI-TOF продукта взаимодействия гексахлорциклотрифосфазена с избытком дифенилолпропана в среде пиридина при 110°С.

При эпоксидировании этого гидроксиарилоксифосфазена в среде ЭХГ формируется ФЭО с преимущественным содержанием гексаэпоксида с m/z = 1836 (рис. 7).

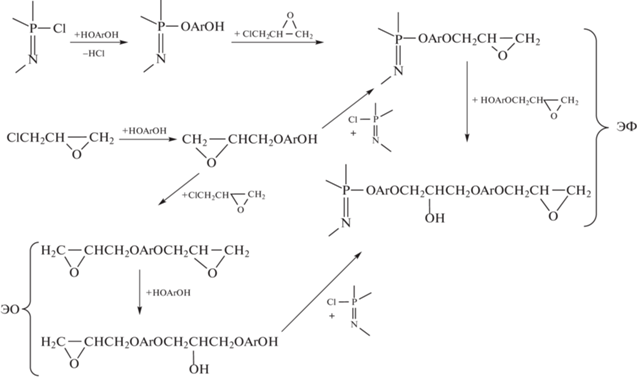
С целью максимального упрощения синтеза ФЭО была использована одностадийная схема получения смеси обычного и фосфазенового эпоксидов в избытке эпихлоргидрина – условно названная однореакторной [38, 39].
При наличии в исходной смеси четырех исходных компонентов (бисфенол, эпихлоргидрин, КОН и гексахлорциклотрифосфазена) возможно одновременное протекание следующих реакций, приводящих к образованию ФЭО, состоящего из двух фракций – фосфазеновой (ЭФ) и органической (ЭО):
В выбранных условиях (температура 100°С, продолжительность 10 ч) независимо от избытка дифенола в интервале от 6 до 16 молей на моль гексахлорциклотрифосфазена образующаяся фосфазеновая фракция (ЭФ) по данным MALDI-TOF-спектрометрии содержит преимущественно три соединения, которым соответствуют пики с m/z = = 1338, 1588 и 1872, формулы этих соединений представлены на рис. 8.
Рис. 8.
MALDI-TOF масс-спектры фосфазенсодержащих эпоксидных олигомеров на основе гексахлорциклотрифосфазена и бисфенола А. х – Мольное соотношение гексахлорциклотрифосфазен : бисфенол А в реакционной смеси равно 6 (а), 8 (б), 10 (в), 12 (г) и 16 (д).
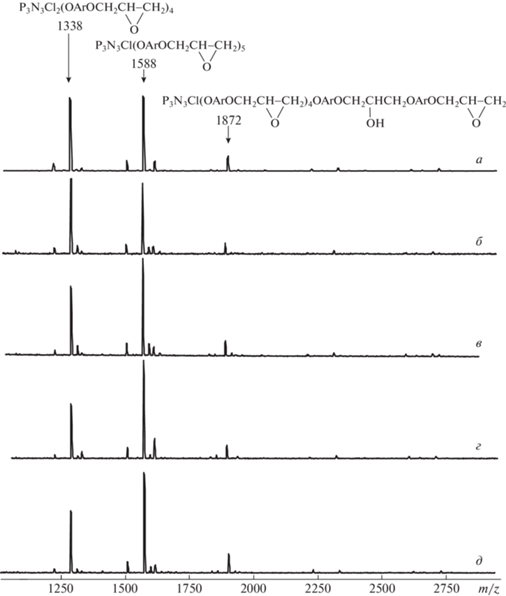
Как видно, по мере увеличения избытка дифенола наблюдается незначительный рост содержания монохлорпентаглицидилоксиарилоксициклотрифосфазена с m/z = 1588 за счет частичной конверсии одного из двух оставшихся атомов хлора в арилоксипроизводное, однако полного замещения хлора в цикле не происходит. При этом в составе ФЭО за счет повышения содержания органического эпоксида уменьшается доля фосфазеновой фракции (табл. 2), а вместе с ней содержание фосфора, хлора и, естественно, падает и огнестойкость (табл. 3). В то же время эпоксидное число ФЭО с увеличением доли органического эпоксида растет.
Таблица 2.
Содержание и состав фосфазеновых фракций в смеси эпоксидных олигомеров, полученных однореакторным методом
| Образец, № | Значение х (число молей бисфенола А на 1 моль ГХФ) | Количество ЭФ-фракции (мас. %), рассчитанное | |||
|---|---|---|---|---|---|
| по данным ГПХ | по содержанию | ||||
| фосфора | хлора | эпоксидных групп | |||
| 1 | 8 | 49 | 49 | 60 | 60 |
| 2 | 10 | 41 | 32 | 50 | 41 |
| 3 | 12 | 36 | 30 | 41 | 33 |
Таблица 3.
Огнестойкость отвержденных эквивалентным количеством метилтетрагидрофталевого ангидрида композиций на основе фосфазенсодержащих эпоксидных олигомеров (ЭО + ЭФ), определенная по ГОСТ 28157-89
| Образец*, № | Горизонтально закрепленный образец | Вертикально закрепленный образец | ||
|---|---|---|---|---|
| время горения, с | скорость горения, мм/мин | время горения (с) после приложения пламени (первого/второго) | наличие горящих капель | |
| 1 | 13 | 0 | 13/22 | нет |
| 2 | 22 | 2 | 20/26 | нет |
| 3 | 60 | 10 | 150/– | нет |
* По табл. 2.
Синтез ФЭО однореакторным (one-spot) методом на основе резорцина приводит к образованию фосфазеновой фракции [40], состав которой также зависит от мольного соотношения гексахлорциклотрифосфазен : резорцин. Так, при указанном соотношении 1 : 24 образующийся фосфазеновый компонент по данным MALDI-TOF содержит соединения общей формулы
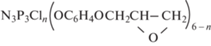 ,
,
в которой n = 2 (20–30% тетраэпоксида), n = 1 (более 50% пентаэпоксида) и всего лишь 2–5% гексаэпоксида (n = 0). При этом в составе фосфазенсодержащих фракций присутствуют заметные количества хлоргидринных групп, свидетельствующих о незавершенном дегидрохлорировании.
Второй особенностью резорциновых ФЭО является наличие в них значительного количества соединений, молекулы которых содержат по два и даже по три фосфазеновых цикла, соединенных м-диоксифениленовыми фрагментами. Но при мольном соотношении гексахлорциклотрифосфазен : резорцин ≥24 содержание данных олигомеров уменьшается и в состав ЭФ входят в основном указанные выше тетра- и пентазамещенные циклофосфазены. Эти ФЭО содержат 3–5% фосфора, 29–30% эпоксидных групп и имеют вязкость при 70°С близкую к вязкости ДГЭБА и в 10–15 раз ниже, чем у ФЭО на основе бисфенола А.
Сравнение ФЭО, синтезированных one-spot методом на основе бисфенола А и резорцина (табл. 4) показывает, что резорциновые олигомеры содержат больше фосфора и хлора и имеют большие значения эпоксидного числа.
Таблица 4.
Сравнительный анализ ФЭО, синтезированных one-spot методом на основе бисфенола А и резорцина
| Мольное соотношение ГХФ : дифенол | Выход, % | Найдено, % | |||
|---|---|---|---|---|---|
| общий | фосфазеновой фракции* | фосфор | хлор | эпоксигруппы** | |
| ФЭО на основе бисфенола А [17] | |||||
| 1:6 | 65 | 39*** | 2.4 | 2.2 | 14.1 |
| 1:8 | 83 | 55 | 3.1 | 2.7 | 17.1 |
| 1:10 | 85 | 41 | 2.3 | 1.9 | 19.0 |
| 1:12 | 86 | 33 | 1.8 | 1.5 | 20.0 |
| 1:16 | 88 | 29 | 1.5 | 1.2 | 21.4 |
| ФЭО на основе резорцина [40] | |||||
| 1:8 | 61 | 73 | 6.8 | 10.8 | 5.5 |
| 1:10 | 71 | 51 | 4.8 | 8.3 | 14.5 |
| 1:12 | 77 | 42 | 4.0 | 4.4 | 21.0 |
| 1:16 | 89 | 32 | 3.0 | 2.4 | 28.6 |
| 1:24 | 90 | 21 | 2.0 | 1.9 | 29.6 |

Путь F в схеме (5) , используемый для понижения функциональности гексахлорциклотрифосфазена путем частичного замещения в нем атомов хлора на инертные ароматические радикалы [30], также был исследован в рамках однореакторного метода синтеза ФЭО. В исходную смесь гексахлорциклотрифосфазена, дифенола, избытка ЭХГ и акцептора НCl в качестве регулятора функциональности смеси вводили фенол [41].
Были осуществлены два варианта этого процесса: вариант А – одновременное введение всех четырех реагентов и использование КОН в качестве акцептора НCl; в варианте В на первой стадии к раствору гексахлорциклотрифосфазена в ЭХГ добавляли фенол и К2СО3, а на второй бисфенол и твердый КОН [41].
Вариант А
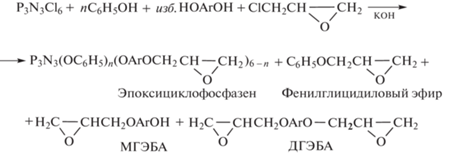
Вариант В

(МГЭБА – моноглицидиловый эфир бисфенола А).
В обоих вариантах синтеза с выходом 70–75% образуются ФЭО, содержание фосфазеновой фракции в которых зависит от соотношения гексахлорциклотрифосфазен : фенол : бисфенол и изменяется в пределах 40–50%.
По данным спектроскопии ЯМР 31Р смешанные феноксиглицидилоксициклотрифосфазены содержат по меньшей мере по одному остаточному атому хлора (табл. 5), при этом число таких соединений и их молекулярная масса уменьшаются с увеличением мольного соотношения гексахлорциклотрифосфазен : фенол : бисфенол (рис. 9).
Таблица 5.
Состав синтезированных по вариантам А и Б фосфазеновых фракций по данным MALDI-TOF-спектрометрии (мольное соотношение гексахлорциклотрифосфазен : фенол : бисфенол А = 1 : 3 : 5)
| m/z | Формула соединения* | Относительное содержание соединения (мас. %) в продуктах, полученных: | |
|---|---|---|---|
| по варианту А | по варианту В | ||
| 636 | P3N3Cl(OPh)5 | 12.3 | 3.6 |
| 654 | P3N3Cl4(OPh)(OArOGly) | – | 3.0 |
| 768 | P3N3Cl2(OPh)3(OArOGly) | 1.6 | 7.2 |
| 826 | P3N3Cl(OPh)4(OArOGly) | 14.0 | 8.6 |
| 958 | P3N3Cl(OPh)3(OArOGly)(OArOGly′) | – | 5.0 |
| 960 | P3N3Cl(OPh)3(OArOH)(OArOGly) | – | 8.5 |
| 1016 | P3N3Cl(OPh)3(OArOGly)2 | 22.2 | 9.2 |
| 1150 | P3N3Cl(OPh)2(OArOH)(OArOGly)2 | – | 3.0 |
| 1206 | P3N3Cl(OPh)2(OArOGly)3 | 25.0 | 19.3 |
| 1397 | P3N3Cl(OPh)(OArOGly)4 | 16.7 | – |
| 1587 | P3N3Cl(OArOGly)5 | 1.0 | – |


Рис. 9.
MALDI-TOF масс-спектры эпоксициклофосфазенов, полученных по варианту А при мольных соотношениях гексахлорциклотрифосфазен : фенол : бисфенол А = 1 : 2 : 5 (а), 1 : 2 : 6 (б), 1 : 4 : 3 (в) и 1 : 4 : 4 (г).

Как следует из табл. 5, при мольном соотношении гексахлорциклотрифосфазен : фенол : бисфенол = 1 : 3 : 5 образуются более однородные по составу ЭФ, полученные по варианту А, хотя основными в обоих вариантах являются ди- и триглицидиловые эфиры арилокситрифосфазенов – 47 и 31% соответственно в вариантах А и В. Особенностью ФЭО пониженной функциональности, синтезируемх по вариантам А и В, является наличие во всех составляющих фосфазеновую фракцию соединениях остаточного хлора.
Органическая фракция ФЭО, синтезированных при указанном мольном соотношении реагентов, по данным хромато-масс-спектрометрии [41] содержит до восьми соединений, основным среди которых является диглицидиловый эфир бисфенола А в количестве 50–60 мас. %.
Другими наиболее значимыми соединениями в этой фракции являются предшественник ДГЭБА – моноглицидиловый эфир бисфенола А, а также продукты побочных превращений, например, с деметилированной изопропильной группой.
Существенным преимуществом ФЭО пониженной функциональности, получаемых на основе смесей фенола и бисфенола, является их пониженная вязкость, приближающаяся при температуре 70°С к вязкости обычных эпоксидных олигомеров (ДГЭБА, эпоксидиановая смола).
Изложенное выше позволяет сделать следующие заключения. Наиболее простые и удобные методы синтеза ФЭО – эпоксидирование гидроксиарилоксифосфазенов, осуществляемое постадийно (синтез гидроксиарилоксифосфазенов и их последующее взаимодействие с эпихлоргидрином или непосредственно в одну стадию в одном реакторе).
Образующаяся в первом случае фосфазеновая фракция ФЭО в зависимости от условий (растворитель, избыток бисфенола, температура и продолжительность) может не содержать остаточного хлора, в то время как при одностадийном синтезе его доля в фосфазеновой фракции составляет 2.2–2.7% при содержании фосфора 4–5%.
Наличие этих двух элементов в составе ФЭО обеспечивает их пониженную горючесть (V-1 или V-0 по UL-94). Важным фактором при практическом применении является возможность регулирования вязкости ФЭО частичной заменой бисфенола А на фенол или полной заменой бисфенола А на резорцин. С использованием полученных результатов [38–41] создана опытная установка синтеза различных ФЭО производительностью 1 кг/сутки [42].
Работа выполнена при финансовой поддержке Российского научного фонда (проект 19-73-10204).
Список литературы
Potter W.G. Epoxy Resins. New York: Springer-Verlag, 1970. P. 92.
Handbook of Composites / Ed. by G. Lubin. New York: Van Nostrand Reinhold, 1982. P. 57.
Lee H., Neville K. Handbook of Epoxy Resins. New York: McGraw-Hill, 1967. Ch. 20.
Николаев А.Ф., Ван Эр-Тень, Зырянова Г.А., Лебедева Э.В., Афанасьева А.С. // Пласт. массы. 1966. № 3. P. 17.
Николаев А.Ф., Ван Эр-Тень., Зырянова Г.А., Балаева Г.Л., Григорьева Г.М., Дрейман М.А. // Пласт. массы. 1967. № 9. P. 24.
Hayes R.F., Allen C.W. // Dalton Trans. 2016. V. 45. № 5. P. 2060.
El Gouri M., El Bachiri A., Hegazi S.E., Rafik M., El Harfi A. // Polym. Degrad. Stab. 2009. V. 94. № 11. P. 2101.
El Gouri M., Hegazi S.E., Rafik M., El Harfi A. // Ann. Chim., Science des Materiaux. 2010. V. 35. № 1. P. 27.
El Gouri M., Cherkaoui O., Ziraoui R., El Harfi A. // J. Mater. Environ. Sci. 2010. V. 1. № 3. P. 157.
El Gouri M., El Bachiri A., Hegazi S. E., Ziraoui R., Rafik M., El Harfi A. // J. Mater. Environ. Sci. 2011. V. 2. № 4. P. 319.
El Gouri M., El Mansouri A., El Gouri R., Hadik N., Outzourhit A., El Harfi A. // J. Mater. Environ. Sci. 2014. V. 5. № 2. P. 400.
Dagdag O., El Gouri M., El Mansouri A., Outzourhit, A., El Harfi A., Cherkaoui O., El Bachiri A., Hamed O., Jodeh S., Hanbali G., Khalaf B. // Polymers. 2020. V. 12. № 4. P. 921.
Liu J., Tang J., Wang X., Wu G. // Polym. Degrad. Stab. 2014. V. 103. № 1. P. 96.
Gleria M., Minto F., Tiso B., Bertani R., Tondello E., Pò R., Fiocca L., Lucchelli E., Giannotta G., Cardi N. // Designed Monomers Polymers. 2001. V. 4. № 3. P. 219.
Bertani R., Boscolo-Boscoletto A., Dintcheva N., Ghedini E., Gleria M., La Mantia F., Pace G., Pannocchia P., Sassi A., Scaffaro R., Venzo A. // Designed Monomers Polymers. 2003. V. 6. № 3. P. 245.
Kireev V.V., Bredov N.S., Bilichenko Yu.V., Lysenko K.A., Borisov R.S., Chuev V.P. // Polymer Science A. 2008. V. 50. № 6. P. 609.
Kireev V.V., Bilichenko Yu.V., Borisov R.S., Sirotin I.S., Filatov S.N. // Polymer Science B. 2018. V. 60. № 3. P. 243.
Liu J., He Z., Wu G., Zhang X., Zhao C., Lei C. //Chem. Eng. Jl. 2020. V. 390. P. 124620.
Sirotin I.S., Bilichenko Yu.V., Solodukhin A.N., Kireev V.V., Buzin M.I., Borisov R.S. // Polymer Science B. 2013. V. 55. № 5–6. P. 241.
Scaffaro R., Botta L., La Mantia F.P., Magagnini P., Acierno D., Gleria M., Bertani R. // Polym. Degrad. Stab. 2005. V. 90(2 SPEC. ISS.). P. 234.
Chen-Yang Y.W., Lee H.F., Yuan C.Y. // J. Polym. Sci., Polym. Chem. 2000. V. 38. № 6. P. 972.
Lakshmikandhan T., Sethuraman K., Chandramohan A., Alagar M. // Polym. Compos. 2017. V. 38. P. E24.
Xu G.-R., Xu M.-J., Li B. // Polym. Degrad. Stab. 2014. V. 109. P. 240.
You G., Cai Z., Peng H., Tan X., He H. // Phosphorus, Sulfur, Silicon and the Related Elements. 2014. V. 189. № 4. P. 541.
Chistyakov E.M., Kireev V.V., Filatov S.N., Terekhov I.V., Buzin M.I., Komarova L.I. // Polymer Science B. 2012. V. 54. № 7–8. P. 407.
Филатов С.Н. Дис. … д-ра хим. наук. М.: РХТУ им. Д.И. Менделеева, 2016.
Liu J., Tang J., Wang X., Wu D. // RSC Adv. 2012. V. 2. № 13. P. 5789.
Liu F., Wei H., Huang X., Zhang J., Zhou Y., Tang X. // J. Macromol. Sci., Phys. 2010. V. 49. № 5. P. 1002.
Bai Y., Wang X., Wu D. // Industr. Eng. Chem. Res. 2012. V. 51. № 46. P. 15064.
Terekhov I.V., Filatov S.N., Chistyakov E.M., Borisov R.S., Kireev V.V. // Russ. J. Appl. Chem. 2013. V. 86. № 10. P. 1600.
Терехов И.В. Дис. … канд. хим. наук. М.: РХТУ им. Д.И. Менделеева, 2014.
Jian S., Xiaodong W., Dezhen W. // ACS Appl. Mater. Interfaces. 2012. V. 4. № 8. P. 4047.
Hua F., Xiaodong W., Dezhen W. // Industr. Eng. Chem. Res. 2013. V. 52. № 30. P. 10160.
Бригаднов К.А. Дис. … канд. хим. наук. М.: РХТУ им. Д.И. Менделеева, 2018.
Liu H., Wang X., Wu D. // Polym. Degrad. Stab. 2015. V. 118. P. 45.
Kireev V.V., Chistyakov E.M., Filatov S.N., Borisov R.S., Prudskov B.M. // Polymer Science B. 2011. V. 53. № 7–8. P. 412.
Сиротин И.С. Дис. … канд. хим. наук. М.: РХТУ им. Д.И. Менделеева, 2013.
Sirotin I.S., Bilichenko Y.V., Brigadnov K.A., Kireev V.V., Prudskov B.M., Borisov R.S. // Polymer Science B. 2014. V. 56. № 4. P. 471.
Пат. 2537403 Россия // Б.И. 2015. № 1.
Sarychev I.A., Sirotin I.S., Borisov R.S., Mu Jianxin, Sokolskaya I.B., Bilichenko J.V., Filatov S.N., Kireev V.V. // Polymers. 2019. V. 11. № 4. P. 614.
Kireev V.V., Bilichenko Y.V., Borisov R.S., Jianxin Mu, Kuznetsov D.A., Eroshenko A.V., Filatov S.N., Sirotin I.S. // Polymers. 2019. V. 11. № 12. P. 1914.
Биличенко Ю.В., Зыонг Н.Т., Лось Н.С., Сиротин И.С., Киреев В.В., Филатов С.Н. // Хим. пром-сть сегодня. 2020. № 1. С. 18.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)