

Высокомолекулярные соединения (серия Б), 2022, T. 64, № 2, стр. 107-115
МЕТАКРИЛАТСОДЕРЖАЩИЕ ФОСФАЗЕНОВЫЕ ОЛИГОМЕРЫ
И. С. Сиротин a, Ву Суан Шон a, Ю. В. Биличенко a, Р. С. Борисов a, b, c, Е. А. Горбунова a, В. В. Киреев a, *
a Российский химико-технологический университет им. Д.И. Менделеева
125047 Москва, Миусская пл., 9, Россия
b Институт нефтехимического синтеза им. А.В. Топчиева Российской академии наук
119991 Москва, Ленинский пр., 29, Россия
c Российский университет дружбы народов
117198 Москва, ул. Миклухо-Маклая, 6, Россия
* E-mail: kireev.v.v@muctr.ru
Поступила в редакцию 29.12.2021
После доработки 31.01.2022
Принята к публикации 07.02.2022
- EDN: XASZNI
- DOI: 10.31857/S2308113922020127
Аннотация
Реакцией фосфазенсодержащих эпоксидных соединений с метакриловой кислотой синтезированы метакрилатсодержащие фосфазеновые олигомеры, которые охарактеризованы спектрами ЯМР 1Н и ЯМР 31Р, MALDI-TOF-масс-спектрометрией и функциональным анализом. Метакрилатсодержащие фосфазеновые олигомеры с добавкой 0.5% гидрохинона стабильны в процессе хранения при обычной температуре в течение двух–трех недель, но легко сополимеризуются с метилметакрилатом в присутствии перекиси бензоила с образованием сополимеров с количественным содержанием гель-фракции.
Олигомерные фосфазены представляют все возрастающий интерес в связи с широкими возможностями их применения для получения термо- и огнестойких полимеров и композиционных материалов на их основе [1–5]. Для этого чаще используют олигофосфазены, содержащие в связанных с атомами фосфора органических радикалах функциональные группы – гидроксильные [6–9], альдегидные [10–12], карбоксильные, аминные [13–15], эпоксидные [16–21], эвгенольные [18, 22–25] и другие.
Особенно перспективными представляются фосфазенсодержащие метакриловые олигомеры (ФМО), которые получены взаимодействием гидроксиарилоксициклотрифосфазенов с метакрилоилхлоридом [12]. Однако последний является малодоступным, дорогим и неудобным в работе вследствие гидролитической нестабильности.
Более перспективной для синтеза ФМО представляется реакция метакриловой кислоты с фосфазенсодержащими эпоксидными олигомерами (ФЭО), удобные и эффективные методы синтеза которых разработаны в последние годы [16–21].
Цель настоящей работы – установление оптимальных условий реакции метакриловой кислоты с ФЭО, а также состава, строения и некоторых свойств образующихся ФМО.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Исходные ФЭО были синтезированы двухстадийным методом: сначала взаимодействием гексахлорциклотрифосфазена с избытком бисфенола А получали гидроксиарилоксициклотрифосфазены в смеси с бисфенолом, а затем обработкой этой смеси избытком эпихлоргидрина синтезировали ФЭО (табл. 1), содержащие 32–59% эпоксифосфазена и диглицидиловый эфир бисфенола А. Некоторые характеристики исходных ФЭО представлены в табл. 1. Эти смеси двух эпоксидов обработкой метакриловой кислотой переводили в ФМО, также состоящие из двух фракций – фосфазеновой и, преимущественно, бис-(4.4'-метакрилокси-2-гидроксипропоксифенил)-2.2-пропана:
(I – эпоксифосфазен, II – фосфазеновая фракция, III – диглицидиловый эфир бисфенола А, IV – бис-(4.4'-метакрилокси-2-гидроксипропоксифенил)-2.2-пропан; здесь и ниже
Таблица 1.
Характеристики исходных фосфазенсодержащих эпоксидных олигомеров
| Мольное соотношение гексахлорциклотрифосфазен : дифенилолпропан при синтезе гидроксиарилоксифосфазенов | Олигомер | Содержание фосфора, % | Эпоксидное число*, % | Содержание фосфазеновой фракции в ФЭО, %** |
|---|---|---|---|---|
| 1:12 | ФЭО-12 | 2.40 | 18.6/20.0 | 59/47 |
| 1:16 | ФЭО-16 | 1.79 | 20.1/21.3 | 46/35 |
| 1:24 | ФЭО-24 | 1.17 | 21.6/22.7 | 32/23 |
Строение исходных ФЭО было подтверждено спектрами ЯМР 1Н и ЯМР 31Р (рис. 1); последние характеризуются синглетным сигналом δр = 8.5 м.д. свидетельствующим о сохранении неизменным трифосфазенового цикла в процессе их образования.

Присоединение метакриловой кислоты к смеси эпоксидов контролировали по спектрам ЯМР 1Н, а также по данным функционального анализа (табл. 2). Как следует из этой таблицы, содержание фосфазеновой фракции и бис-(4.4'-метакрилокси-2-гидроксипропоксифенил)-2.2-пропана в образующихся ФМО соответствует содержанию их предшественников в исходном ФЭО, хотя содержание метакриловых групп (бромное число) несколько ниже расчетного для смесей P3N3[OArOCH2CH(OH)CH2OC(O)–C(CH3) = CH2]6 и бис-(4.4'-метакрилокси-2-гидроксипропоксифенил)-2.2-пропана различного состава, особенно для олигомера ФМО-12.
Таблица 2.
Характеристики фосфазенсодержащих метакриловых олигомеров
| Олигомер | Бромное число*, г Br/100 г | Отношение интенсивностей $\delta _{{\text{H}}}^{{{\text{C = C}}{{{\text{H}}}_{2}}}}$/$\delta _{{\text{H}}}^{{{\text{C}}{{{\text{H}}}_{3}}{\text{ - ип}}}}$ | Содержание фосфазеновой фракции в ФМО, мас. % | ||
|---|---|---|---|---|---|
| вычислено** | найдено | ||||
| по содержанию фосфора | по соотношению $\delta _{{\text{H}}}^{{{\text{C = C}}{{{\text{H}}}_{2}}}}$/$\delta _{{\text{H}}}^{{{\text{C}}{{{\text{H}}}_{3}}{\text{ - ип}}}}$ | ||||
| ФМО-12 | 56.1/58.1 | 0.54 | 43.4 | 32.4 | 38 |
| ФМО-16 | 53.5/55.7 | 0.57 | 31.5 | 26.3 | 29 |
| ФМО-24 | 51.0/53.1 | 0.59 | 20.3 | 19.7 | 23 |
Сопоставление спектров ЯМР 31Р трех полученных ФМО (рис. 1) позволяет заключить, что реакция (1) протекает полностью в случае олигомеров ФМО-16 и ФМО-24, спектры которых одинаковы и характеризуются наличием синглетного сигнала δр = 8.4 м.д. В то же время спектр ЯМР 31Р олигомера ФМО-12 (рис. 1а) содержит незначительные по интенсивности сигналы в области δр = 8.0 м.д., которые предположительно соответствуют соединениям с одной или двумя неконвертированными эпоксидными группами. Это согласуется с отмеченными выше пониженными значениями бромного числа данного олигомера.
Рис. 2.
Спектры ЯМР 1Н продуктов реакции метакриловой кислоты с ФЭО при мольном соотношении эпоксидные группы : метакриловая кислота = 1.0 : 1.3. а – расчетные значения δH по программе Chemdraw Ultra версии 12.0.2.1076; б – ФМО-12; в – ФМО-16; г – ФМО -24. Пояснения в тексте.

Рис. 2.
Окончание

Строение образующих ФМО подтверждают их спектры ЯМР 1Н (рис. 2), которые содержат сигналы протонов двух типов метильных групп, связанных с углеродом метакрилового ($\delta _{{\text{H}}}^{{{\text{C}}{{{\text{H}}}_{3}}{\text{ - мет}}}}$ = 2.0 м.д.) и изопропильного фрагментов ($\delta _{{\text{H}}}^{{{\text{C}}{{{\text{H}}}_{3}}{\text{ - ип}}}}$ = 1.6 м.д.).

Соотношение интегральных интенсивностей сигналов $\delta _{{\text{H}}}^{{{\text{C}}{{{\text{H}}}_{3}}{\text{ - ип}}}}$ : $\delta _{{\text{H}}}^{{{\text{C}}{{{\text{H}}}_{3}}{\text{ - мет}}}}$ равно единице для бис-(4.4'-метакрилокси-2-гидроксипропоксифенил)-2.2-пропана и 0.5 для фосфазеновой фракции. Аналогичная ситуация наблюдается и для соотношения $\delta _{{\text{H}}}^{{{\text{C = C}}{{{\text{H}}}_{2}}}}$ : $\delta _{{\text{H}}}^{{{\text{C}}{{{\text{H}}}_{3}}{\text{ - ип}}}}$; именно по значению последнего соотношения в табл. 2 приведено рассчитанное содержание в ФМО фосфазенового компонента.
Состав фосфазеновой фракции ФМО подтверждают их лазерные масс-спектры (рис. 3): основной продукт – искомый P3N3[OArOCH2CH(OH)CH2OC(O)-C(CH3)=CH2]6 c m/z = = 2372 (+Na). Вторым по интенсивности является пик соответствующий соединению с m/z = 2283 (+Na), содержащему пять метакрилокси-групп.
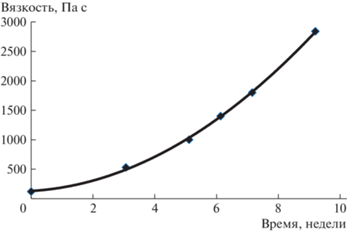
Рис. 4.
Изменение динамической вязкости ФМО-24 в процессе выдержки при 25°С в присутствии 0.5 мас. % гидрохинона.
Следует отметить плохую воспроизводимость MALDI-TOF-масс-спектров, обусловленную, по-видимому, нестабильностью ФМО в условиях эксперимента: под действием высокоэнергетического лазерного воздействия в процессе десорбции и ионизации фосфазеновых молекул возможны их побочные превращения и даже разрушение, о чем свидетельствуют малоинтенсивные пики с m/z = 2137, 2320 и 2394.
Тем не менее представленные на рис. 3 MALDI-TOF масс-спектры подтверждают факт образования метакрилатсодержащих фосфазеновых олигомеров, формула которых приведена на схеме (1) .
Как известно, метакриловые мономеры и олигомеры, как и многие соединения с кратными связями, могут подвергаться самопроизвольной полимеризации, поэтому для оценки возможности практического применения ФМО была исследована их стабильность при хранении в обычных условиях. Как следует из рис. 4, нарастание динамической вязкости ФМО-24 при 25°С от 100 до 3000 Па с происходит в течение 8 недель.
Рис. 5.
Влияние продолжительности блочной сополимеризации ММА с ФМО-24 на выход гель-фракции при содержании фосфазенового олигомера 25 (1), 10 (2), 5 (3) и 1 мас. % (4).

Высокая функциональность ФМО, обусловленная наличием в этих олигомерах фосфазеновой фракции, позволяет использовать их для сополимеризации при синтезе трехмерных полимеров с регулируемыми параметрами сетки. На рис. 5 видно резкое возрастание выхода гель-фракции при увеличении содержания олигомера ФМО-24 в процессе его сополимеризации с метилметакрилатом даже при минимальном количестве фосфазеновой фракции в исходной смеси.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Исходные вещества
Гексахлорциклотрифосфазен – коммерческий продукт очищали перекристаллизацией из гексана с последующей возгонкой в вакууме, Тпл = 113°C.
Дифенилолпропан – промышленный продукт очищали перекристаллизацией из хлорбензола, Тпл = 155°C.
Эпихлоргидрин (“Solvay”, Tavaux, Франция) применяли без дополнительной очистки, Ткип = = 117–118°С.
Гидроксид калия (Общество с ограниченной ответственностью “Химкомплект”), гидрохинон (Тпл = 174°C), триэтилбензиламонний хлористый (Тпл = 185°С), использовали без дополнительной очистки.
Толуол (Ткип = 110°C), метакриловую кислоту (Ткип = 162°C) применяли в свежеперегнанном виде.
Константы использованных растворителей соответствовали приведенным в литературе [26 ] .
Синтез фосфазенсодержащих метакриловых олигомеров
Стадия 1. В трехгорлую колбу с механической мешалкой, обратным холодильником и насадкой Дина-Старка загружали 3.48 г гексахлорциклотрифосфазена (0.01 моля), 54.72 г дифенилолпропана (0.24 моля), 40 г свежевысушенного К2СО3 и 500 мл ацетонитрила. Реакционную смесь перемешивали при 80°С в течение 5 ч и после охлаждения фильтрования и удаления растворителя на роторном испарителе получили 55.0 г смеси гексагидроксициклотрифосфазена, и избыточного дифенилолпропана. По данным спектроскопии ЯМР 31Р образовавшийся гексагидроксициклотрифосфазен содержит преимущественно гексагидроксиарилоксициклотрифосфазен P3N3(ОArOH)6.
Стадия 2. 15.3 г указанной смеси растворяли в 250 мл эпихлоргидрина и после образования гомогенного раствора постепенно при 60°С добавляли 8.1 г твердых пластинок КОН, содержащего 12% воды. Скорость введения КОН определяли по температуре реакционной смеси, которая не должна превышать 70°С. После перемешивания в течение 6 ч при указанной температуре охлажденную смесь отфильтровывали от солей и удаляли эпихлоргидрин отгонкой под вакуумом. Остаток растворяли в ацетоне, повторно отфильтровывали, отгоняли ацетон и высушивали продукт в вакууме.
Получили 16.7 г вязкой смолы с эпоксидным числом 21.6%, содержащей 28% эпоксифосфазена, который по данным ЯМР 31Р и MALDI-TOF содержал преимущественно гексаарилоксициклотрифосфазен с небольшой примесью соответствующего пентаэпоксида.
Стадия 3. Синтезированный фосфазенсодержащий эпоксидный олигомер растворяли в 250 мл толуола, вводили 3 мл метакриловой кислоты, 0.1 мас. % триметилбензиламмоний хлорида (катализатор) и 0.5% гидрохинона (стабилизатор). Процесс осуществляли при 100°С, контролируя его по содержанию в реакционной смеси метакриловой кислоты. По достижении ее постоянного содержания охлажденный раствор промывали 3%-ным водным раствором карбоната калия затем несколько раз водой. Раствор олигомера в толуоле сушили прокаленным сульфатом магния и после добавления 0.3% гидрохинона отгоняли толуол в вакууме. Получили 17 г ФМО-24 с содержанием фосфазеновой фракции около 20%.
Аналогично синтезировали ФМО-16 и ФМО-12.
Методы анализа
Спектры ЯМР 31P и ЯМР 1H снимали на приборе “Bruker CXP-200” при рабочей частоте 145 и 200 МГц соответственно, растворитель – дейтерохлороформ, Т = 25°С.
Масс-спектрометрический анализ MALDI-TOF выполняли на приборе “Brukers AutoFlex II”.
Содержание гель-фракции полимеризуемых образцов определяли методом равновесного набухания в ацетоне с помощью аппарата Сокслета.
Вязкость измеряли на реометре “Netzsch” с использованием плоского шпинделя при зазоре 0.1 мм и скорости сдвига 1 с–1.
Эпоксидное число определяли методом обратного кислотно-основного титрования (ГОСТ 12497-78).
Бромное число находили по методу [27 ] .
Список литературы
Allen C.W., Hernandez-Rubio D. Applicative Aspects of Poly(organophosphazenes). New York: Nova Sci. Publ., 2004. P. 119.
Allcock H.R. // Phosphorus, Sulfur, Silicon Related Elements. 2004. V. 179. № 4–5. P. 661.
Liu H., Wang X., Wu D. // Polym. Degrad. Stab. 2014. V. 103. № 1. P. 96.
Liu J., He Z., Wu G., Zhang X., Zhao C., Lei C. // Chem. Eng. J. 2020. V. 390. P. 124620.
You G., Cai Z., Peng H., Tan X., He H. // Phosphorus, Sulfur, Silicon Related Elements. 2014. V. 189. N 4. P. 541.
Chistyakov E.M., Kireev V.V., Filatov S.N., Terekhov I.V., Buzin M.I., Komarova Z.I. // Polymer Science B, 2012. V. 54. № 7–8. P. 407.
Чистяков Е.М., Филатов С.Н., Киреев В.В., Лысенко К.А., Бузин М.И., Чуев В.П. // Журн. общ. химии. 2012. Т. 81. № 6. С. 906.
Sirotin I.S., Bilichenko Yu.V., Brigadnov K.A., Kireev V.V., Suraeva O.V., Borisov R.S. // Russ. J. Appl. Chem. 2013. V. 86. № 12. P. 1903.
Kireev V.V., Chistyakov E.M., Filatov S.N., Borisov R.S., Prudskov B.M. // Polymer Science B. 2011. V. 53. № 7–8. P. 412.
Chistyakov E.M., Panfilova D.V., Kireev V.V. // Russ. J. General Chem. 2017. V. 87. № 5. P. 997.
Chistyakov E.M., Panfilova D.V., Kireev V.V., Volkov V.V., Bobrov M.F. // J. Molec. Struct. 2017. V. 1148. P. 1.
Филатов С.Н. Дис. … д-ра хим. наук. РХТУ им. Д.И. Менделеева, 2015.
Terekhov I.V., Filatov S.N., Chistyakov E.M., Borisov R.S., Kireev V.V. // Phosphorus, Sulfur Silicon Related Elements. 2017. V. 192. № 5. P. 544.
Sarychev I.A., Sirotin I.S., Borisov R.S., Mu J., Sokolskaya I.B., Bilichenko J.V., Filatov S.N., Kireev V.V. // Polymers. 2019. V. 11. № 4. P. 614.
Brigadnov K.A., Bilichenko Yu.V., Polyakov V.A., Borisov R.S., Gusev K.I., Rudakova T.A., Filatov S.N., Kireev V.V. // Polymer Science B. 2016. V. 58. № 5. P. 549.
Sirotin I.S., Bilichenko Yu.V., Brigadnov K.A., Kireev V.V., Prudskov B.M., Borisov R.S. Polymer Science B. 2014. V. 56. № 4. P. 471.
Kireev V.V., Bredov N.S., Bilichenko Yu.V., Lysenko K.A., Borisov R.S., Chuev V.P. // Polymer Science A. 2008. V. 50. № 6. P. 609.
Биличенко Ю.В., Филатов С.Н., Киреев В.В., Онучин Д.В., Горбунова И.Ю., Сиротин И.С. // I Коршаковская всерос. конф. “Поликонденсационные процессы и полимеры”. М., 2019. С. 44.
Sirotin I.S., Sarychev I.A., Vorobyeva V.V., Kuzmich A.A., Bornosuz N.V., Onuchin D.V., Gorbunova I.Yu., Kireev V.V. // Polymers. 2020. V. 12. № 6. P. 1225.
Weissberger A., Proskauer E.S., Riddick J.A., Toops E.E. Organic Solvents: Physical Properties and Methods of Purification. New York: Interscience, 1955.
Торопцева A.M., Белогородская К.В., Бондаренко В.М. Лабораторный практикум по химии и технологии высокомолекулярных соединений. Л.: Химия, 1972.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)