

Высокомолекулярные соединения (серия Б), 2023, T. 65, № 2, стр. 83-107
Особенности влияния хлорорганических соединений на полимеризацию олефинов и диенов в присутствии катализаторов Циглера–Натта
И. И. Салахов a, *, В. Г. Козлов b, Л. Б. Сосновская a, И. Э. Нифантьев c, П. В. Ивченко c
a Нижнекамский химико-технологический институт (филиал) ФГБОУ ВО
“Казанский национальный исследовательский технологический университет”
423578 Нижнекамск, пр. Строителей, 47, Россия
b Институт экологии Волжского бассейна Российской академии наук
445003 Самарская обл., Тольятти, ул. Комзина, 10, Россия
c Институт нефтехимического синтеза им. А.В. Топчиева Российской академии наук
19991 Москва, Ленинский пр., 29, Россия
* E-mail: i.i.salahov@gmail.com
Поступила в редакцию 02.11.2022
После доработки 16.01.2023
Принята к публикации 16.02.2023
- EDN: WZFSOL
- DOI: 10.31857/S2308113923700419
Аннотация
Проанализированы публикации отечественных и зарубежных авторов, посвященные исследованиям каталитических систем Циглера‒Натта на основе неодима, ванадия и титана, модифицированных хлорорганическими соединениями и используемых в синтезе полиолефинов и полидиенов. Проведен анализ патентной литературы и проанализированы основные тренды развития в области металлокомплексного катализа с использованием хлорорганических соединений. Рассмотрены преимущества и недостатки подобных каталитических систем по сравнению с классическими немодифицированными катализаторами, в которых в качестве источника хлора применяются алкил-алюминийхлориды. Авторами подробно проанализированы опубликованные данные о роли атома хлора как лиганда в активных центрах полимеризации олефинов и диенов. Выявлена и описана роль моно- и полихлорированных органических соединений в неодимовой, ванадиевой и титан-магниевой каталитических системах.
ВВЕДЕНИЕ
В последние десятилетия полиолефины прочно заняли лидирующую позицию среди продуктов нефтехимической индустрии [1, 2], в то время как полидиены по-прежнему незаменимы в производстве резины для шинной промышленности [3, 4]. Современные технологии производства основной массы полиолефинов и каучуков основаны на использовании координационных катализаторов полимеризации. Работы Карла Циглера и Джулио Натта в 50-х годах XX века открыли новый этап в развитии индустрии полимеров [5]. В 1953 г. Карл Циглер в ходе исследования “никель-эффекта” обнаружил, что активация соединений некоторых переходных металлов алюминийалкилами приводит к образованию частиц, способных катализировать олигомеризацию и полимеризацию этилена [6]. Вскоре после первых публикаций Циглера с сотрудниками о синтезе полиэтилена при низких температурах на катализаторах, полученных взаимодействием галогенидов титана и алюминийорганических соединений [7–9], появилась серия работ Натта с сотрудниками [10–15] по синтезу изотактического полипропилена и других полиолефинов. После публикации Циглером и Натта результатов исследований по эффективной координационной полимеризации этилена и пропилена в конце 1950-х годов ведущие нефтехимические компании активно занялись разработкой технологий, нашедших отражение в патентных заявках, описывающих каталитические процессы полимеризации изопрена, бутадиена-1,3, этилена и пропилена с использованием катализаторов Циглера–Натта [6].

В последующие десятилетия было создано несколько поколений катализаторов и технологических процессов, организовано производство разнообразных полимерных материалов – полиэтилена высокой плотности, линейного полиэтилена низкой плотности, изотактического полипропилена, этилен-пропиленового каучука (СКЭП), этилен-пропилен-диенового каучука (СКЭПТ), цис- и транс-поли(1,3-бутадиена) и поли(1,3-изопрена) [1, 2, 4, 16–21].
С конца 1950-х годов исследования в области координационной полимеризации α-олефинов и 1,3-диенов получили широкое развитие. В нашей стране фундаментальный вклад в исследования на катализаторах и разработку катализаторов Циглера–Натта полимеризации диенов внесли работы Б.А. Долгоплоска и Ю.Б. Монакова. Одним из важных направлений исследований Ю.Б. Монакова и его научной группы – изучение полимеризации диенов в присутствии лантаноидсодержащих катализаторов [22]. Результаты этих исследований имеют фундаментальное значение для теории одно- и многоцентрового катализа полимеризации.
Процессы полимеризации диенов и олефинов в присутствии различных каталитических систем на основе Циглера–Натта, вызывают закономерный интерес из-за высокой стереоспецифичности катализаторов и направлены преимущественно на повышение стереорегулярности эластомеров, в случае полипропилена – на увеличение степени изотактичности, кристалличности и ударопрочностных показателей полимера. Актуальные исследования в области полимеризации этилена нацелены на эффективное регулирование плотности, управление молекулярно-массовыми характеристиками и улучшение физико-механических свойств полимера.
Классические катализаторы Циглера–Натта на основе титана, ванадия и неодима для координационной полимеризации диенов и олефинов представляют собой комбинацию соединения переходного металла (MLn), триалкилалюминия (АlR3) и источника галогена, который добавляется отдельно или входит в состав MLn. В ходе взаимодействия MLn и АlR3 происходит образование активных центров, позволяющих синтезировать стереорегулярные полимеры [23, 24]. Однако для подобных катализаторов характерны реакции восстановления металла триалкилалюминием, что приводит к понижению активности каталитической системы [23–25]. Для поддержания активности в катализаторах Циглера–Натта дополнительно используют алкилалюминийхлориды.
Одним из путей дополнительной активации катализаторов Циглера–Натта служит введение в каталитический комплекс или непосредственно в процесс полимеризации хлорсодержащих углеводородных соединений, позволяющих реактивировать катализатор. Хлорсодержащие углеводородные соединения менее пирофорны и не чувствительны к влаге и кислороду: так, например, гексахлор-п-ксилол (ГХПК), является дешевым, малотоксичным и негорючим соединением [26]. В связи с этим с точки зрения дальнейшего совершенствования технологий получения пластмасс и каучуков модификация катализаторов Циглера–Натта является актуальной и важной задачей, так как позволяет решить вопросы улучшения качества полимеров за счет улучшения однородности, понижения их зольности (остатков каталитического комплекса), повышения безопасности производства путем использования более инертных компонентов и увеличения эффективности промышленных установок.
В настоящее время отсутствуют обзоры, в которых комплексно проанализирована роль хлорорганических соединений в различных типах каталитических систем. Роль хлорсодержащих соединений в каталитической полимеризации и олигомеризации олефинов и диенов до сих пор не ясна, обзор, посвященный участию M–(μ-Cl)–Al комплексов в каталитических превращениях α-олефинов [27], в основном касается проблем механизма координационной полимеризации и структуры каталитических частиц, фактически рассматривая алкилалюминийхлориды в качестве единственного источника хлора. Мы полагали, что детальный анализ научной периодики и патентной литературы за последние годы и систематизация данных по полимеризации олефинов и диенов на неодимовых, ванадиевых и титан-магниевых катализаторах (ТМК) смогут выявить основные особенности влияния хлорсодержащих углеводородных соединений на каталитическое поведение Циглер-Наттовских систем. Актуальность обзора также обусловлена тем, что в России по-прежнему широко используют титан-магниевые катализаторы и другие “классические” катализаторы Циглера–Натта, поэтому изучение путей их модификации хлорсодержащих углеводородных соединений способно существенно повысить эффективность процессов получения полимеров, что важно как с научной, так и прикладной точек зрения.
Цель настоящего обзора – рассмотрение основных результатов экспериментальных исследований полимеризации олефинов и диенов под действием катализаторов Циглера–Натта, модифицированных моно- и полихлорированными органическими соединениями.
ПОЛИМЕРИЗАЦИЯ ДИЕНОВ НА ЛАНТАНОИДНЫХ (НЕОДИМОВЫХ) КАТАЛИЗАТОРАХ
Согласно классификации, предложенной Ю.Б. Монаковым с сотрудниками [22], лантаноидсодержащие катализаторы подразделяются на три основные группы.
I. Двухкомпонентные системы. В этих катализаторах тригалогенид лантаноида (Ln) представлен в виде комплексного соединения LnHal3 ∙ nL, где L – различные органические лиганды с n ≤ 3, вторым компонентом является алюминийорганическое соединение R3Al.
II. Трехкомпонентные системы. Лантаноидная компонента в них не содержит галоген, представляя собой комплекс Ln c карбоксилат-, фосфонат-, алкоголят-анионами, например Nd(OR)3–${\text{R}}_{3}^{'}$Al–R'AlСl2.
III. Двухкомпонентные системы, в которых лантаноидная компонента представляет собой смешанно-лигандный комплекс, причем по меньшей мере один из лигандов является галогенид-анионом (например, R'LnHal2 ∙ nТГФ–R3Al). Следует отметить, что к группе III относятся каталитические системы на основе (η5-циклопентадиенил)LnCl2 [28–30] или (η5-инденил)LnCl2 [31, 32] и AlR3, катализирующие 1,4-цис-полимеризацию диенов [32–35]. Показано, что металлоорганические производные лантаноидов, формирующие каталитический комплекс, должны содержать не менее двух атомов галогена. Системы с одним атомом галогена, например R2LnHal–AlR3, не катализируют полимеризацию диенов.
В полимеризации диенов наибольшую активность демонстрируют неодимсодержащие катализаторы первых двух групп.
Влияние природы галогена на эффективность катализаторов группы I изучено достаточно хорошо в полимеризации бутадиена-1,3. Фторсодержащие катализаторы дают лишь следовые количества полимера (табл. 1), конверсия мономера для катализаторов, содержащих другие галогены, при прочих равных условиях понижается в ряду С1–Вr–I, т.е. хлорсодержащие каталитические системы являются наиболее активными. Микроструктура 1,4-цис-полибутадиена не зависит от природы галогена в составе лантаноидного катализатора.
Таблица 1.
Влияние природы галогена в составе катализаторов NdHal3/EtOH/Et3Al на их выход и молекулярно-структурные характеристики 1,4-цис-полибутадиена [36, 37]
| Галогенид | Выход, % | [η], дл/г | Массовая доля звеньев, % | ||
|---|---|---|---|---|---|
| цис-1,4- | транс-1,4- | 1,2- | |||
| F | 2 | – | 95.7 | 2.5 | 1.8 |
| Cl | 94 | 8.3 | 96.2 | 3.5 | 0.3 |
| Br | 80 | 11.0 | 96.8 | 2.0 | 1.2 |
| I | 24 | 14.8 | 96.7 | 2.2 | 1.1 |
Установлено [37–40], что при варьировании природы органического лиганда изменяется степень ковалентности связей Ln–Наl, что влияет на процесс алкилирования лантаноидной компоненты и уровень активности катализатора [36, 41, 42].
Катализаторы первой и второй групп обладают одинаковой стереоспецифичностью [43]. Активность двухкомпонентных и трехкомпонентных каталитических систем зависит от природы галогена [36, 37, 43]. В каталитических системах группы II помимо лантаноидсодержащего соединения LnX3 и триалкилалюминия (R3Al) присутствует третий компонент, содержащий галоген. В качестве третьего компонента используют алкилалюминийгалогениды [36, 44–48], алкилтитангалогениды [47, 48], галогениды некоторых металлов III–VI и VIII групп [44, 47, 48], галогенводородные кислоты [44–48], галогены в элементарном виде [45], алкилхлорсиланы [49–53]. Роль третьего компонента, очевидно, сводится к галоидированию лантаноида с образованием соединений, содержащих от одного до трех атомов галогена. Реакции галоидирования протекают при взаимодействии галоидирующего агента с карбоксилатами, алкоголятами и другими подобными соединениями общей формулы LnX3:
В трехкомпонентных каталитических системах формируются активные центры на основе и моно- и дигалогенсодержащих производных (LnX2Hal, LnXHal2), что подтверждают результаты, полученные на катализаторах группы III [43]. Активность каталитических систем группы II зависит от глубины галоидирования лантаноидной компоненты. Если идет галоидирование до стадии LnHal3, то должны образовываться активные центры, идентичные катализаторам группы I. Действительно, в работе [54] установлено близкое значение констант скорости реакции роста для этих двух видов каталитических систем.
Был исследован широкий круг галоидирующих агентов, начиная от ионогенных галогенидов до ковалентно связанных атомов галогена [55]. Наиболее подробно изучены галогениды, включающие связи Al–Hal, C–Hal, Si–Hal и другие. Алкилалюминийгалогениды наиболее предпочтительны из-за их хорошей растворимости в углеводородных растворителях и достаточно высокой сокаталитической активности [56–59]. Примерами галоидов со связью С–Cl являются такие хлорсодержащие углеводородные соединения как трет-BuCl [60, 61], CCl4 [62, 63], CHCl3 [64, 65] и ГХПК [66, 67]. Из кремнийсодержащих галогенидов можно выделить SiCl4 [49, 62] и RSiCl3 [62, 68, 69].
Компания JSR заявила об использовании хлоридов олова, метилхлорсиланов, галогенидов бериллия, галогенидов серебра, золота, магния, кальция, бария, цинка, марганца в качестве галоидирующих агентов [70–75].
Для каталитических систем группы II установлены следующие ряды активности в полимеризации бутадиена-1,3 в зависимости от источника галогена:
Et3Al2Сl3 > Et2AlСl > трет-BuCl > SiCl4 [49]
Me2АlСl > MeАlСl2 ≈ Me3Al2Сl3 > трет-BuCl [76]
Et2AlСl > EtAlСl2 ≈ Et3Al2Сl3 > трет-BuCl [76]
Общие для этих рядов – высокая активность систем, содержащих алкилалюминийхлориды (табл. 2). Выявлено, что значения ММ и Mw/Mn для алкилалюминийхлоридов ниже, для хлорсодержащих углеводородных соединений (трет-BuCl) выше, а для кремнийсодержащего галоида еще выше. Содержание 1,4-цис-звеньев для образцов полибутадиена находится на одном уровне.
Таблица 2.
Влияние доноров хлора на полимеризацию 1,3-бутадиена, катализируемую системами Nd(Versatate)3/галоидирующий агент/i-Bu2AlH [49]
| Образец, № | Источник хлора | Мольное соотношение Cl : Nd | Конверсия, % | Mw ×10–3 | Mw/Mn | Доля 1,4-цис-звеньев, % |
|---|---|---|---|---|---|---|
| 1 | Et3Al2Сl3 | 3.0 | 84 | 240 | 4.3 | 97 |
| 2 | Et2AlСl | 3.0 | 83 | 220 | 5.0 | 96 |
| 3 | трет-BuCl | 3.0 | 77 | 380 | 5.9 | 97 |
| 4 | SiCl4 | 2.0 | 19 | 550 | 11.1 | 97 |
Значительная разница полимеризационной активности систем при замене галоидирующего агента позволяет заключить, что он участвует не только в процессе галоидирования атомов лантаноида, но и в формировании лигандного окружения центрального атома. Это также подтверждает характер ММР полимеров (рис. 1), полученных на лантаноидных каталитических системах, содержащих в качестве галоидирующих агентов соединения различной природы [49].
Рис. 1.
Кривые ММР 1,4-цис-полибутадиена, полученные с использованием разных источников галогена. Каталитические системы: 1 – Nd(Versatate)3/SiCl4/i-Bu2AlH, 2 – Nd(Versatate)3/Et2AlCl/iBu2AlH (Et2AlCl, модифицированный трибутилфосфат) [49].

Если в качестве галоидирующего агента использовать диалкилалюминийгалогениды, то область соотношений Hal : Ln, обеспечивающих максимальную активность, практически не зависит от природы галогена (за исключением фтора) и составляет 2.0–2.5 [45, 77]. Положение максимума активности при варьировании соотношения Hal : Ln и активность лантаноидных систем зависят от природы заместителя X в LnX3. Это связано с полнотой галоидирования соединения лантаноида, характером связи Ln–X, возможным присутствием X в качестве одного из лигандов в активном центре. Показано, что для систем на основе карбоксилатов максимальная активность достигается при соотношениях Сl : Ln = 2.0–2.5, а для алкоголятов лантаноидов – при Сl : Ln = 3 [40, 44–46, 78, 79].
В работе [80] исследована природа хрорирующего агента (хлорсилана, хлорсодержащих углеводородных соединений и алкилалюминийхлоридов) на полимеризацию бутадиена-1,3 в присутствии каталитической системы Nd(OiPr)3/Al(i-Bu)2H/хлорирующий агент (табл. 3). Показано, что значение ММ образцов ПБ изменяется в ряду Me2SiCl2 > > хлорсодержащие углеводородные соединения (CCl4, трет-BuCl) > алкилалюминийхлориды для Cl : Nd = 2.0.
Таблица 3.
Влияние доноров хлора на полимеризацию 1,3-бутадиена, катализируемую системами Nd(OiPr)3/Al(i-Bu)2H/Cl [80]
| Образец, № | Источник хлора | Мольное соотношение Cl : Nd | Выход, % | Mn ×10–3 | Mw/Mn |
|---|---|---|---|---|---|
| Хлорсилан | |||||
| 1 | Me2SiCl2 | 2.0 | 99.3 | 7.49 | 1.36 |
| 3.0 | 100 | 7.37 | 1.54** | ||
| 3.0* | 68.2 | 114 | 3.82 | ||
| Хлорорганические соединения | |||||
| 2 | CCl4 | 2.0 | 95.0 | 6.68 | 3.67 |
| 3.0 | 99.6 | 7.27 | 4.39 | ||
| 3 | трет-BuCl | 2.0 | 97.6 | 6.44 | 3.24 |
| 3.0 | 97.8 | 6.39 | 5.54 | ||
| Алкилалюминийхлориды | |||||
| 4 | Al(i-Bu)2Cl | 2.0 | 95.8 | 5.99 | 3.04 |
| 3.0 | 96.4 | 5.69 | 3.59 | ||
| 5 | AlEt2Cl | 2.0 | 94.9 | 5.82 | 3.36 |
| 3.0 | 97.4 | 5.62 | 3.70 | ||
| 6 | AlEtCl2 | 2.0 | 94.8 | 5.63 | 2.56 |
| 3.0 | 100 | 6.45 | 4.25 | ||
| 7 | Al2Et3Cl3 | 2.0 | 95.3 | 5.65 | 1.74*** |
| 3.0 | 96.6 | 4.87 | 2.43 | ||
| 2.0* | 66.6 | 59.0 | 3.82 | ||
При этом для каталитических систем Nd(OiPr)3/Al(i-Bu)2H/Me2SiCl2 и Nd(OiPr)3/ Al(i-Bu)2H/Al2Et3Cl3 значения полидисперности при прочих равных условиях ниже (1.36–1.74), чем для систем с остальными хлорирующими агентами. В то же время при увеличении мольного соотношения бутадиен : Nd с 1000 до 6000 значения Mw/Mn для указанных двух систем возрастают до уровня 3.8. Следует отметить, что с этилалюминийсесквихлоридом (ЭАСХ) доля 1,4-цис-звеньев в обазцах ПБ оказывется выше, чем чем с хлорсиланами.
Большое влияние на активность трехкомпонентных каталитических систем и молекулярные характеристики полидиенов оказывает концентрация алкилалюминийгалогенида. Снижение активности катализатора при дозировках диалкилалюминийгалогенида, превышающих оптимальные, обусловлено, вероятно, образованием прочных мостиковых комплексов типа
 ,
,
в которых отсутствует связь L–С, обеспечивающая полимеризацию. Таким образом, R2AlHal может в определенных условиях проявлять себя как ингибитор [43].
Учитывая представления, сформулированные при изучении реакций галоидирующего агента в трехкомпонентных каталитических системах группы II [43, 56, 81–83], с увеличением соотношения хлорсодержащего соединения и металлокомплекса группы II можно предположить образование комплексов Nd–Al
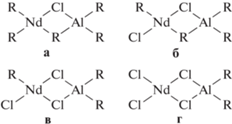
В трехкомпонентных каталитических системах для эффективного ведения полимеризации необходимо достижение определенного донорно-акцепторного равновесия, которое регулируется введением атомов хлора в систему [84]. По данным работ [85, 86] комплексы типа R2NdCl–R3Al (а) менее активны по сравнению с комплексами типа RNdCl2-R3Al (б). В первом случае, по всей видимости, равновесие не достигается по причине недостатка анионов хлора, что приводит к понижению реакционной способности центров. Кроме того, на центрах первого комплекса возможен одновременный рост двух полимерных цепочек (по двум связям R–Nd) и можно предположить, что их пониженная реакционная способность по сравнению с центрами типа RNdCl2–R3Al (б) связана со стерическими затруднениями [86].
Согласно литературным данным [22, 56, 58, 87–89] обладают более высокой реакционной способностью комплексы RNdCl2–R3Al (б) и RNdCl2–R2AlCl (в). Это подтверждают положения максимума активности катализатора при мольных соотношениях Cl : Nd = 2–3 (табл. 4) [90].
Таблица 4.
Кинетические параметры полимеризации бутадиена-1,3 при разном мольном соотношении Cl : Nd, в присутствии каталитической системы Nd(OCOС9Н19)3/i-Bu2AlH/ГХПК [90]
| Мольное соотношение Cl : Nd | Скорость полимеризации wp, моль/л мин | Константа скорости роста kр, л/моль мин | Доля активных центров γа, % |
|---|---|---|---|
| 1.5 | 0.13 | 2100 | 32 |
| 2.0 | 0.18 | 2900 | 32 |
| 2.4 | 0.25 | 3500 | 36 |
| 3.0 | 0.25 | 4800 | 27 |
| 4.0 | 0.16 | 4800 | 17 |
Примечание. Здесь и в табл. 6 условия полимеризации: 60°С; i-Bu2AlH : Nd = 20; см = 1.4 моль/л; скат = 1.4 × 10–4 моль/л; н-гексан.
Комплекс г, по-видимому, неактивен в полимеризации, или, по меньшей мере, имеет существенно более низкую реакционную способность по сравнению с комплексами а–в, что обусловлено отсутствием связи Nd–R обеспечивающей процесс полимеризации в мостиковом комплексе г [43]. Подтверждением может служить уменьшение скорости процесса и доли активных центров при Cl : Nd = 4.0 (табл. 4).
Необходимо отметить, что при варьировании соотношения Cl : Nd в каталитической системе Nd(OCOС9Н19)3–i-Bu2AlH–ГХПК изменение скорости процесса связано не только с изменяющейся долей активных центров при различной дозировке ГХКП, но и с различающимися константами скорости реакции роста цепи (табл. 4). Изменение kр свидетельствует о формировании активных центров различной реакционной способности, что, вполне возможно, связано с разным лигандным окружением атома неодима в активном центре полимеризации. Вероятно, всегда происходит формирование нескольких типов активных центров, показанных выше, и при изменении Cl : Nd в каталитической системе меняется соотношение концентрации различных типов активных центров. Это выражается изменением кажущейся константы скорости реакции роста цепи.
Использование RAlСl2 вместо R2AlСl не приводит к изменению зависимости активности системы от соотношения Сl : Ln [78]. Галоидирущий агент оказывает заметное влияние на ММР полидиенов. Увеличение соотношения кислота Льюиса : соединение лантанода от 0.8 до 1.4 приводит к уширению ММР и переходу от унимодального распределения к бимодальному [57]. Формирование бимодального ММР для 1,4-цис-ПБ с ростом соотношения Cl : Nd с 1.5 до 4.0 также подтверждают результаты, полученные в работах [90, 91].
Исследование каталитической системы Nd(OCOС9Н19)3–i-Bu2AlH–галоидирующий агент в полимеризации бутадиена-1,3 позволило установить, что при замене хлорсодержащего компонента Et3Al2Cl3 на ГХПК изменений в характере конверсионных зависимостей нет (табл. 5), но при Cl : Nd = 2.0–2.4 использование Et3Al2Cl3 вызывает некоторое повышение скорости полимеризации (рис. 2). При увеличении соотношения Cl : Nd до 3.0 для системы на основе Et3Al2Cl3 наблюдается снижение скорости полимеризации, в то время как для системы Nd(OCOС9Н19)3 – i-Bu2AlH–ГХПК скорость остается стабильной и высокой в интервале Cl : Nd от 2.4 до 3.0.
Таблица 5.
Значения конверсии, средних молекулярных масс и полидисперсности образцов 1,4-цис-полибутадиена, полученных в присутствии разных хлорирующих агентов [91]
| Образец, № | Время, мин | Конверсия, % | Mw × 10–3 | Mw/Mn |
|---|---|---|---|---|
| Nd(OCOС9Н19)3|i-Bu2AlH|Et3Al2Cl3 | ||||
| 1 | 0.25 | 5 | 244 | 5.7 |
| 2 | 0.50 | 9 | 281 | 4.8 |
| 3 | 0.75 | 15 | 300 | 4.0 |
| 4 | 5 | 43 | 326 | 3.8 |
| 5 | 10 | 55 | 329 | 3.7 |
| 6 | 30 | 79 | 385 | 3.5 |
| 7 | 60 | 92 | 410 | 3.4 |
| Nd(OCOС9Н19)3|i-Bu2AlH|ГХПК | ||||
| 8 | 0.25 | 4 | 230 | 5.9 |
| 9 | 0.50 | 9 | 265 | 4.9 |
| 10 | 0.75 | 14 | 271 | 3.9 |
| 11 | 5 | 37 | 283 | 3.4 |
| 12 | 10 | 48 | 314 | 3.2 |
| 13 | 30 | 72 | 398 | 2.8 |
| 14 | 60 | 85 | 422 | 2.8 |
Рис. 2.
Зависимость скорости полимеризации wp от мольного соотношения Cl : Nd в интервале 1.5–4.0 при полимеризации на каталитической системе Nd(OCOС9Н19)3/i-Bu2AlH − хлорирующий агент ГХПК (1) и Et3Al2Cl3 (2) [91]. Условия полимеризации: Т = 60°С, i-Bu2AlH : Nd = 20, cм = 1.4 моль/л, cкат = 1.4 × 10-4 моль/л, н-гексан.

На основании полученных кинетических параметров процесса полимеризации бутадиена-1,3 (табл. 6) можно сделать вывод, что при формировании каталитических систем Nd(OCOС9Н19)3–i-Bu2AlH–ГХПК и Nd(OCOС9Н19)3– i-Bu2AlH – Et3Al2Cl3 образуются активные центры с различной реакционной способностью, что подтверждает изменение величины kp. Кроме того, изменяется и вероятность ограничения роста цепи. Для систем с ГХПК вероятность передачи цепи как на алюминийорганическое соединение ($k_{o}^{{{\text{Al}}}}$/kp), так и на мономер ($k_{o}^{{\text{м}}}$/kp) ниже, чем для систем с Et3Al2Cl3. Для лантаноидных каталитических систем передача цепи на мономер и алюминийорганическое соединение являются основными реакциями ограничения роста цепи [22, 43], следовательно, системы, содержащие ГХПК позволяют получать полимер с более высокой ММ. Этот факт позволяет сделать заключение, что ГХПК не полностью удаляется из координационной сферы Nd3+ после реакции хлорирования и входит в состав активного центра полимеризации и, таким образом, влияет на реакционную способность каталитической системы.
Таблица 6.
Кинетические параметры полимеризации бутадиена-1,3 на каталитической системе Nd(OCOС9Н19)3/i-Bu2AlH/галоидирующий агент [91].
| Галоидирующий агент | Скорость полимеризации wp, моль/л ⋅ мин | Константа скорости роста kp, л/моль мин | Константа передачи цепи, моль/л ⋅ мин | Доля активных центров, % | |
|---|---|---|---|---|---|
| на алюминий $k_{o}^{{{\text{Al}}}}$ | на мономер $k_{o}^{{\text{м}}}$ | ||||
| Et3Al2Cl3 | 0.27 | 4200 | 2.0 | 0.017 | 33 |
| ГХПК | 0.25 | 3500 | 1.2 | 0.012 | 36 |
Установлено, что с повышением концентрации хлорсодержащих углеводородных соединений возрастают средние ММ (рис. 3). При этом наиболее интенсивный рост отмечен на начальном этапе полимеризации (до 10%), далее с увеличением конверсии средние ММ повышаются монотонно. В то же время наиболее широкое ММР полибутадиена характерно на начальных участках полимеризации, а увеличение степени превращения мономера вызывает существенное сужение полидисперности.
Рис. 3.
Зависимость Mw (1–3) и Mw/Mn (4–6) от конверсии мономера х при мольном соотношении Cl : Nd = 1.5 (1, 4), 2.4 (2, 5) и 4.0 (3, 6) в каталитическом комплексе Nd(OCOС9Н19)3/i-Bu2AlH/ГХПК. Условия указаны в табл. 4.

Увеличение концентрации ГХПК в реакционной системе приводит к росту стереоспецифичности катализатора (рис. 4) – подобные закономерности наблюдаются и для других хлорорганических соединений [79, 92]. Так, содержание цис-1,4-звеньев в полибутадиене возрастает с 96 до 98.1%, а доля транс-1,4-звеньев соответственно уменьшается.
Рис. 4.
Зависимость микроструктуры полибутадиена от мольного соотношения Cl : Nd в каталитическом комплексе Nd(OCOС9Н19)3/i-Bu2AlH/ГХПК. 1 – цис-1,4-звенья, 2 – транс-1,4-звенья, 3 – 1,2 – звенья. Условия указаны в табл. 4.

Одной из важных молекулярных характеристик полимера, определяющей его физико-механические показатели, является наличие разветвленности в макромолекулярных цепях. Фактор разветвленности g – это отношение гидродинамического параметра разветвленного и неразветвленного образцов полимера при одинаковом значении их молекулярной массы. Отклонение g-фактора от единицы свидетельствует о наличии разветвленных макромолекул. Известно, что для неодимовых каталитических систем не характерно наличие разветвленной структуры в ПБ, как это присуще для кобальтовых и никелевых катализаторов. Однако для исследуемой каталитической системы Nd(OCOС9Н19)3–i-Bu2AlH–ГХПК было обнаружено присутствие разветвленных макромолекул [26, 90].
На рис. 5 приведены значения фактора ветвленности g для ПБ, полученного при различном содержании ГХПК в системе в начальный период полимеризации и при глубоких степенях превращения мономера. Как видно, имеется явная тенденция к появлению разветвленных макромолекул в ПБ при малых значениях конверсий мономера. Эта тенденция усиливается с увеличением мольного соотношения Cl : Nd. Полимеры, полученные при глубоких конверсиях, практически не содержат разветвленных макромолекул и только при соотношении Cl : Nd = 4.0 можно отметить некоторую разветвленность макромолекул в области высокомолекулярных фракций (рис. 6). Если сопоставить данные по разветвленности с кривыми ММР для соответствующих образцов, можно видеть, что разветвленность характерна для образцов, включающих достаточно высокое количество высокомолекулярной фракции (рис. 6).
Рис. 5.
Зависимость фактора разветвленности g от мольного соотношения Cl : Nd в каталитическом комплексе Nd(OCOС9Н19)3/i-Bu2AlH/ГХПК. 1 – начальный этап полимеризации (х = 4–9%), 2 – х > 60%. Условия указаны в табл. 4.

Рис. 6.
Логарифмические зависимости характеристической вязкости [η] (1,2) и фактора разветвленности g (3) от молекулярной массы; 4 – кривая ММР полибутадиена, полученная при Сl : Nd = 2.0 (а, б) и 4.0 (в, г). Конверсия мономера 5 (а, в) и 74% (б, г). 1 – расчет, 2 – эксперимент. Условия указаны в табл. 4.

По мере роста конверсии доля высокомолекулярной фракции уменьшается и соответственно понижается доля разветвленных молекул. Вероятно, с этим связано отсутствие ветвленности на конечных этапах полимеризации. И только при соотношении Cl : Nd = 4.0, где доля высокомолекулярной фракции достаточно большая даже при высоких конверсиях, можно наблюдать ветвленность и при конверсияи 60–70%. Появление разветвленности на исследованной каталитической системе связано с наличием реакций передачи на полимерную цепь. Появление разветвленных макромолекул на начальных стадиях полимеризации, когда содержание макромолекул в системе еще сравнительно небольшое, явление весьма необычное. Вероятность передачи на полимер как правило возрастает при глубоких конверсиях мономера. Если принять возможность участия в полимеризации на рассматриваемых каталитических системах активных центров различной реакционной способности, а следовательно, и с различной вероятностью ограничения роста цепи, то исходя из вида ММР (рис. 6), на начальном этапе процесса формируется значительная доля центров с низкой вероятностью ограничения роста материальной цепи. Именно они ответственны за формирование наиболее высокомолекулярных фракций при малых конверсиях и, как показывает значение фактора ветвленности (g ≤ 1, рис. 6), эти центры имеют склонность к реакциям передачи цепи на полимерную цепь. В ходе процесса полимеризации изменяется состав полимеризационной системы и, как показывает вид ММР, меняется и распределение по активности центров полимеризации или распределение активных центров по вероятности ограничения роста цепи (это взаимосвязанные понятия). Доля высокомолекулярной фракции, полученной в начале процесса, становится мала, и присутствие разветвленных макромолекул не определяется гидродинамическими методами. Предполагаемые изменения в составе активных центров наиболее вероятно связать с изменением лигандного окружения у центрального атома или с процессами сольватации [93].
Таким образом, для всех рассмотренных лантаноидных каталитических систем роль галоидирующего агента сводится не только к функции образования связи Ln–Hal, необходимой для проявления координирующей и каталитической активности связью Ln–С в каталитическом комплексе; наличие галоидирующего агента определяет параметры этих связей за счет его присутствия в координационной сфере каталитического комплекса. Алкилалюминийхлориды являются эффективными галоидирующими агентами в трехкомпонентных неодимовых системах и позволяют синтезировать ПБ с высоким выходом и содержанием 1,4-цис-звеньев, но пониженной ММ, чем с хлорсодержащими соединениями. Последние также демонстрируют высокие каталитические свойства (активность и стереоспецифичность) при полимеризации 1,3-бутадиена, а образующийся ПБ характеризуется повышенной ММ и, в случае ГХПК, появляется разветвленность в полимере. При использовании ГХПК взамен ЭАСХ на неодимовых каталитических системах при синтезе ПБ наблюдаются сужение ММР, что обычно положительно отражается на физико-механических и эксплуатационных характеристиках полимеров и вулканизатов на их основе [94].
СОПОЛИМЕРИЗАЦИЯ ЭТИЛЕНА С ПРОПИЛЕНОМ НА ВАНАДИЕВЫХ КАТАЛИЗАТОРАХ
Полимеризация олефинов с использованием катализаторов Циглера–Натта (комплексных соединений ванадия) ранее рассматривали в обзорах [95–101], где основное внимание уделено различным вариантам соединений на основе ванадия, в том числе, металлоценам. Показано, что в отличие от катализаторов на основе титана и циркония многие ванадийсодержащие катализаторы активируются R2AlCl или RAlCl2; использование R3Al и метилалюмоксана менее эффективно. Важное значение связи V–(µ-Cl)–Al в каталитических процессах обсуждено в обзоре [27]. Катализаторы на основе ванадия зачастую уступают титан-магниевым катализаторам и комплексам металлов VI группы по активности, однако они обладают рядом преимуществ. Так, в случае сополимеризации этилена с пропиленом они обеспечивают более высокое включение сомономера; они эффективны в тройной сополимеризации этилен-пропилен-диен [99].
Особенностью каталитических систем на основе ванадия является восстановление ванадия алкилами алюминия в ходе процесса до степени окисления +2, которая неактивна в полимеризации. В результате происходит падение каталитической активности вплоть до полной остановки процесса [102–106]. Один из способов решения этой проблемы – модификация ванадиевых катализаторов окислителями, способствующими реактивации активных центров в полимеризационой системе [103, 105–107]. В том случае, когда окислитель вводят в систему с компонентами катализатора, с мономером или углеводородным растворителем, скорость полимеризации в отличие от процесса в отсутствие окислителя продолжительное время сохраняется постоянной. Помимо этого полимеризация на промотированных окислителями системах вызывает повышение эффективности катализатора. По мнению многих авторов [102, 103, 105, 106, 108], данные эффекты обусловлены регенерацией активных центров катализатора в результате окисления неактивных или малоактивных при (со)полимеризации низковалентного ванадия или понижением скорости восстановительных процессов в реакционной системе. Например, добавка гексахлорциклопентадиена окисляет двухвалентный ванадий в трехвалентное состояние (рис. 7) [109].
Рис. 7.
Зависимость валентности ванадия от времени в системах VOCl3–Et3Al2Cl2 (1) и VOCl3–Et3Al2Cl2–гексахлорциклопентадиен (2) [109].

С целью совершенствования ванадиевых катализаторов (со)полимеризации олефинов опробованы различные модификаторы, в том числе хлорсодержащие соединения. Например, в работе [110] исследованы гексахлорциклопентадиен, гексахлорацетон, гексахлорпропилен и сложные эфиры трихлоруксусной кислоты при полимеризации этилена в присутствии ванадиевых прекатализаторов и алкилалюминийхлоридов. Влияние различных галоидных углеводородов (CCl4, CHCl3, CHBr3, CH2Cl2, CH2Br2, CH3CCl3, CF3CCl3) на активацию в полимеризацию этилена при высоких температурах и соотношениях галоген : V = = 30–300 в присутствии ванадиевых катализаторов исследовали в работе [105]. Было показано, что на каталитической системе VCl3/0.3AlCl3/ триэтилалюминий или тригексилалюминий активность существенно возрастает с увеличением числа атомов галогена при одинаковом количестве атомов углерода: СH2Cl2 < CHCl3 < CCl4 (табл. 7).
Таблица 7.
Влияние добавок хлорорганических соединений к ванадиевым катализаторам на их активность при полимеризации этилена и на изменения степени оксиления VCl2 (при 80°С) [105]
| Образец, № | Хлорорганическое соединение | Активность для VCl3/0.3AlCl3/ТЭА, кг ПЭ/г V | Активность для VCl3/ТГА, кг ПЭ/г V | Доля V III, % |
|---|---|---|---|---|
| 1 | Без промотора | 1.4 | 0.9 | 0 |
| 2 | CH2Cl2 | 4.6 | 1.8 | 31 |
| 3 | CHCl3 | 11.7 | 2.3 | 77 |
| 4 | CCl4 | 14.8 | 4.5 | 100 |
При этом количество VIII, образующегося в присутствии различных хлорорганических соединений, увеличивается в том же порядке, что и активность полимеризации; это подтверждает предположение, что два указанных процесса связаны. Уровень ММ и ММР образцов ПЭ существенно не изменяются, стоит лишь отметить некоторое изменение Mn через оптимум и расширение полидисперности при высоком содержании CCl4 (табл. 8).
Таблица 8.
Влияние мольного соотношения CCl4 : V на молекулярные характеристики образцов ПЭ [105] (катализатор VCl3/0.3AlCl3/ТГА; давление 5 бар, продолжительность полимеризации 1 мин)
| Мольное соотношение CCl4 : V | Mw × 10–3 | Mn × 10–3 | Mw/Mn |
|---|---|---|---|
| 0 | 423 | 143 | 3.0 |
| 50 | 460 | 170 | 2.7 |
| 100 | 394 | 125 | 3.2 |
| 250 | 415 | 102 | 4.0 |
Авторы [104] изучали влияние сомономеров на сополимеризацию этилена с α-олефинами, при этом для поддержания активности катализатора на основе VCl4(ТГФ)/SiO2 в качестве промотора использовали CHCl3.
Ниже представлены наиболее изученные хлорорганические соединения при полимеризации на VOCl3–Et3Al2Cl3 [104–106, 109, 112–114].

В работе [109] показано, что введение в систему гексахлорциклопентадиена (соединение 2а) после потери активности каталитической системой VOCl3–Et3Al2Cl3 при сополимеризации этилена с пропиленом вызывает возобновление процесса. Молекулярная масса сополимеров непрерывно понижается с увеличением соотношения гексахлорциклопентадиен : V, что свидетельствует об участии окислителя в процессах обрыва цепи. ММР сополимера, полученного на модифицированной гексахлорциклопентадиеном системе VOCl3–Et3Al2Cl3 остается таким же, как и у сополимера, синтезированного на неактивированных катализаторах [102]. Схожие закономерности для молекулярных характеристик наблюдаются и для систем, содержащих ди(хлорсульфонил)-2,4-тиофен и дихлорфенилфосфин, но активирующее действие этих добавок почти в два раза превышает активирующее действие гексахлорциклопентадиена. Модифицирование систем, включающих VOCl3, VCl4, упомянутыми окислителями позволяет в 4–5 раз повысить интегральный выход продуктов полимеризации [111].
G.H. Zohuri и соавторами [112] изучали сополимеризацию этилена с пропиленом и терполимеризацию этилена, пропилена и 5-этилиден-2-норборнена с использованием каталитической системы на основе VCl4–i-Bu3Al и в присутствии промоторов CHCl3 и CCl4. Продемонстрировано, что CHCl3 как промотор увеличивает активность сополимеризации при синтезе СКЭПТ в 2.5 раза, а CCl4 оказывает незначительное влияние на процесс. Авторы работы связывают эти различия с тем, что радикал CH${\text{Cl}}_{2}^{ \bullet }$ более стабилен по сравнению с CCl3, и в данном случае хлороформ, обладая более подвижным атомом хлора, легче освобождается от него, чем CCl4.
Авторы работы [108] указали, что галогенированные сложные эфиры высокоэффективны в производстве СКЭПТ. Ранее G. Evens [113] запатентовал целый класс подобных соединений. В работе [106] показано, что использование в системе VOCl3–Et3Al2Cl3 такого хлорорганического соединения, как этилдихлорфенилацетат (соединение 4б), позволяет в несколько раз повысить активность каталитической системы. При этом отмечено, что каталитическая система без промотора и без водорода формирует сополимер с бимодальными ММР, предполагая двухцентровое поведение катализатора [106, 108, 114]. Добавление промотора катализатора вызывает повышение производительности катализатора на порядок и понижает долю высокомолекулярной фракции в ММР сополимера. Введение водорода также предотвращает формирование бимодального ММР, независимо от присутствия промотора. Точно так же G. Evens с соавторами [115] при описании эффектов влияния галоидоуглеводородных промоторов на сополимеризацию этилена и пропилена наблюдали сильную зависимость ММ от промотора и бимодальное ММР в его отсутствие.
В работе [116] приведены данные по сополимеризации этилена с пропиленом на каталитической системе VOCl3/Et3Al2Cl3 с промотором этилтрихлорацетатом (табл. 9). Самая высокая активность была получена при соотношении этилтрихлорацетат : V = 10 во всех случаях, а дальнейшее увеличение содержания ЭТХА привело к незначительному снижению активности. Кроме того, молекулярный вес полученных полимеров уменьшился с увеличением соотношения этилтрихлорацетат : V. Такой результат может быть связан с более высокой концентрацией активных частиц, образованных в присутствии реактиватора. Более того, система с VОCl3 позволяет получить сополимеры с более широким молекулярно-массовым распределением (5.46–7.69). Между тем, присутствие этилтрихлорацетата также влияло на включение звеньев пропилена в образующиеся сополимеры. Увеличение соотношения этилтрихлорацетат : V привело к росту содержания звеньев пропилена.
Таблица 9.
Данные по сополимеризации этилена и пропилена на каталитической системе VOCl3/Et3Al2Cl3, модифицированной с промотором ЭТХА [116]
| Мольное соотношение ЭТХА : V | Активность, кг сополимера/моль V ч бар | Mw × 10–4 | Mw/Mn | Содержание пропиленовых звеньев, мол. % |
|---|---|---|---|---|
| 0 | 18.6 | 19.5 | 5.69 | 25 |
| 5 | 38.4 | 10.3 | 7.25 | 29 |
| 10 | 64.0 | 6.4 | 7.69 | 33 |
| 20 | 61.2 | 2.2 | 5.46 | 40 |
Результаты сравнительных исследований сополимеризации этилена с пропиленом на каталитических системах типа VOCl3/Et3Al2Cl3 с хлорорганическими соединениями имеет единичный характер, и обобщенных сравнительных зависимостей по активности для указанных выше промоторов в литературе приведено мало. В связи с этим в работе [117] нами проведено сравнительное изучение сополимеризации этилена и пропилена в присутствии каталитической системы VOCl3/Et3Al2Cl3, модифицированной хлорсодержащими соединениями с различными типами углеводородного каркаса и количеством атомов хлора в молекуле от 1 до 6 (соединения 1a, 2a, 3a, 3б, 4a): алифатический трет-BuCl, циклический гексахлорциклопентадиен, ароматические – 1,2,4-трихлорбензол и ГХПК (соединение 3б), а также галогенированный сложный эфир – этилтрихлорацетат, при этом мольное соотношение Cl : V варьировали от 4 до 10. Показано, что выход этилен-пропиленового сополимера определяется химической природой хлорорганического соединения (рис. 8).
Рис. 8.
Уровни активности каталитической системы VOCl3–Et3Al2Cl3, модифицированной различными хлорсодержащими соединениями. Условия сополимеризации: 30°С, давление 0.6 МПа, время 60 мин; содержание Н2 в газовой фазе 0.005 об. %, мольное соотношение Al : V = 50. Мольное соотношение Cl : V = 4 (темные столбцы) и 10 (светлые). ТХБ ‒ трихлорбензол, ГХЦПД ‒ гексахлорциклопентадиен.

В целом для каталитической системы VOCl3/Et3Al2Cl3/хлорорганическое соединение установлены следующие ряды активности в зависимости от источника хлора:
Этилтрихлорацетат ≥ ГХПК > 1,2,4-трихлорбензол > гексахлорциклопентадиен > трет-BuCl > > без промотора [117].
Видно, что наличие в промоторе (этилтрихлорацетат и ГХПК) трех атомов хлора при атоме углерода (–ССl3) может быть благоприятным фактором для активации ванадиевого катализатора. С другой стороны, ГХПК содержит две группы –ССl3, а этилтрихлорацетат – одну. В данном случае более важно присутствие сложноэфирной группы (–СООR), которая в большей степени способствует окислению двухвалентного ванадия (или активации катализатора), чем присутствие ароматического кольца в ГХПК. Трихлорбензол имеет значительно меньшую активность как промотор. Наличие трех атомов хлора, связанных непосредственно с ароматическим кольцом – явно менее эффективная комбинация, чем две трихлоруглеродных группы в пара-положении у ГХПК. Следует отметить, что заявляемый van Doremaele с сотрудниками этилдихлорфенилацетат, содержащий в своей структуре комбинацию ароматического кольца, группы –ССl2 и сложноэфирной группы –СООR, которая сочетает в себе элементы как ГХПК, так и этилтрихлорацетат, также является эффективным промотором.
Присутствие в молекуле гексахлорциклопентадиена шести атомов хлора несколько ускоряет процесс (соединение 2а), но в значительно меньшей степени, чем содержащий также шесть атомов хлора ГХПК (рис. 8). В работе [16] показано, что оптимум активности при сополимеризации этилена с пропиленом для системы VOCl3–Et3Al2Cl3–гексахлорциклопентадиен наблюдался при мольном соотношении гексахлорциклопентадиена к ванадию в интервале 60–100. Это соотношение значительно выше, чем при использовании ГХПК. Трихлорбензол практически не оказывает активирующего действия на процесс сополимеризации этилена и пропилена на VOCl3–Et3Al2Cl3. Таким образом, ГХПК проявляет себя эффективно как промотор и по сравнению с другими исследованными хлорорганическими соединениями ближе по силе активации к этилтрихлорацетату.
Учитывая тот факт, что этилтрихлорацетат как активатор достаточно хорошо изучен [103, 116, 118], а ГХПК нет, были исследованы его некоторые особенности и преимущества. С увеличением мольного соотношения C8H4Cl6 : VOCl3 выше пяти окраска образующегося сополимера меняется от белой до светло-фиолетовой. Изменение цвета сополимера, наиболее вероятно, связано с окислением ванадия под действием ГХПК. Известно, что ион V+4 имеет темно-голубую окраску, а V+5 красно-желтую. Смесь двух указанных ионов вполне может давать фиолетовый оттенок. При мольном соотношении C8H4Cl6 : VOCl3 = 4 сополимер не окрашивается, поэтому дальнейшие исследования проводили при соотношении, равном четырем, оптимальным с точки зрения, как активности катализатора, так и цветности сополимера [117].
Расчет кинетических параметров процесса сополимеризации этилена и пропилена на каталитических системах VOCl3/Al2(C2H5)3Cl3 и VOCl3/Al2(C2H5)3Cl3/ГХПК показал (табл. 10), что добавление этого промотора в реакционную систему приводит к явному увеличению kи, констант скоростей роста и понижению константы реакции дезактивации центров полимеризации.
Таблица 10.
Кинетические константы элементарных реакций процесса сополимеризации этилена и пропилена (при мольном соотношении ГХПК/VOCl3 = 4), рассчитанных по методу Файнмана–Росса [119] (условия указаны в подписи к рис. 12 )
| Каталитическая система | Константа скорости инициирования с участием этилена kи (± 0.04), л/моль с | Константа скорости реакции дезактивации центров сополимеризации kо(±0.0006), с–1 | Константы скоростей реакций роста цепи*, л/моль с | |||
|---|---|---|---|---|---|---|
| k11 ± 450 | k12 ± 11 | k22 ± 1 | k21 ± 12 | |||
| VOCl3/Al2(C2H5)3Cl3 | 0.24 | 0.0243 | 2150 | 76 | 7 | 91 |
| VOCl3/Al2(C2H5)3Cl3/ГХПК | 0.54 | 0.0100 | 4550 | 231 | 26 | 410 |
Изменение констант скоростей роста цепи свидетельствует о том, что повышение активности системы связано не просто с изменением числа активных центров, а свидетельствует о различной химической природе активных центров в присутствии и отсутствии промотора (ГХПК) при использовании системы VOCl3–Al2(C2H5)3Cl3. Следует отметить, что при этом разница в величинах констант реакций инициирования и роста цепи практически сохраняется. Введение ГХПК в систему увеличивает соотношение (k22 + k12)/ (k11 + k21) с 0.037 до 0.052, т.е. вероятность присоединения пропилена к растущему концу полимерной цепи возрастает практически в полтора раза, что должно приводить к обогащению сополимера вторым мономером. Это подтверждено при анализе полимера, где доля пропиленовых звеньев в сополимере увеличилась с 29 до 33 и 40% (при соотношении ГХПК : VOCl3 = 0, 4 и 10 соответственно). Полученные в работе результаты по кинетическим константам близки к значениям кинетических данных, установленных ранее на подобной системе [120, 121].
Данные в табл. 11 показывают, что к повышению выхода полимера при прочих равных условиях приводит увеличение мольного соотношения ГХПК : VOCl3 от 0 до 6. В отсутствие ГХПК выход сополимера составляет 0.8 кг СКЭП/г V, а при мольном соотношении ГХПК : VOCl3 = 6 наблюдается максимальный выход – 3.3 кг СКЭП/г V.
Таблица 11.
Влияние мольного соотношения ГХПК : VOCl3 на выход и характеристики сополимера этилена с пропиленом
| Образец, № | Мольное соотношение ГХПК : VOCl3 | Выход сополимера, кг/г V | Mw | Mw/Mn | Содержание пропиленовых звеньев, мас.% | Степень кристал-личности, % | Тс, °С | Цвет полимера |
|---|---|---|---|---|---|---|---|---|
| 1 | 0 | 0.8 | 172 | 1.8 | 29 | 13.2 | –48 | Белый |
| 2 | 0.5 | 0.9 | – | – | 29 | – | – | « |
| 3 | 1 | 1.0 | 198 | 2.8 | 29 | – | – | « |
| 4 | 4 | 2.7 | 217 | 3.0 | 33 | 11.8 | –51 | « |
| 5 | 4* | 1.9 | – | – | 30 | – | – | « |
| 5 | 3.0 | 250 | 3.0 | 34 | – | – | Белый с сиреневым оттенком | |
| 6 | 6 | 3.3 | 264 | 2.9 | 36 | – | – | Белый с фиолетовым оттенком |
| 7 | 10 | 3.1 | – | – | 40 | 9.2 | –57 | Желтый с фиолетовым оттенком |
| 8 | 15 | 2.9 | – | – | – | – | – | То же |
Дальнейшее увеличение соотношения ГХПК : VOCl3 до 10 и 15 не вызывает существенного изменения выхода полимера (табл. 11). Введение третьего сомономера этилиденнорборнена понижает активность полимеризационной системы – выход полимера уменьшается с 2.7 до 1.9 кг на грамм ванадия; это коррелирует с результатами работы [112], где показано, что ввод этилиденнорборнена при тройной сополимеризации также сопровождается понижением активности. Однако если провести сравнение с опытом 1 (табл. 11), то выход тройного сополимера увеличился более чем в два раза. Следовательно, ГХПК можно с полным основанием считать эффективным промотором для каталитической системы VOCl3–ЭАСХ при сополимеризации этилена и пропилена, а также при тройной сополимеризации этилена, пропилена и этилиденнорборнена.
Присутствие в системе ГХПК независимо от его количества в пределах исследованных концентраций приводит к расширению ММР сополимера приблизительно с двух до трех (табл. 11), что, видимо, можно связать с изменением соотношения скоростей реакций инициирования и гибели активных центров (табл. 10). Увеличение концентрации ГХПК дает некоторое возрастание среднемассовой молекулярной массы Mw (табл. 2), но если рассмотреть изменение среднечисленной молекулярой массы Mn, то введение ГХПК приводит к снижению Mn, но при соотношениях ГХПК : VOCl3 на уровне 6 наблюдается некоторая тенденция к повышению до значения, характерного для систем без ГХПК.
При увеличении содержания ГХПК в каталитической системе VOCl3–ЭАСХ–ГХПК при сополимеризации этилена с пропиленом доля пропиленовых звеньев в сополимере растет. Из приведенных в табл. 10 значений констант сополимеризации это логичный результат, поскольку введение ГХПК повышает вероятность вхождения пропилена в полимерную цепь. Изменение указанных констант свидетельствует о различной химической природе активных центров в присутствии и отсутствии промотора (ГХПК) при использовании системы VOCl3–ЭАСХ. Обнаруженный эффект позволяет оптимизировать начальное соотношение мономеров, так как одной из особенностей синтеза СКЭП(Т) является необходимость повышенной дозировки пропилена в исходной смеси мономеров для обеспечения заданного количества его звеньев в сополимере. С увеличением доли пропилена в сополимере степень кристалличности и температура стеклования сополимеров ожидаемо понижаются (табл. 11), что для статистической сополимеризации (см. величины r1 и r2, табл. 12) обусловлено более короткими последовательностями, способными к кристаллизации этиленовых звеньев.
Таблица 12.
Значения констант сополимеризации этилена (M1) и пропилена (M2) при разной температуре (уравнение Майо–Льюиса [122])
| Каталитическая система | Температура сополимеризации, С | r1 (±1.25) | r2 (±0.002) | r1r2 |
|---|---|---|---|---|
| VOCl3/ЭАСХ | 20 | 13.6 | 0.099 | 1.35 |
| 30 | 16.5 | 0.082 | 1.35 | |
| 40 | 21.3 | 0.063 | 1.34 | |
| 50 | 21.0 | 0.064 | 1.34 | |
| VOCl3/ЭАСХ/ГХПК | 20 | 8.4 | 0.049 | 0.41 |
| 30 | 12.5 | 0.047 | 0.59 | |
| 40 | 17.5 | 0.056 | 0.98 | |
| 50 | 22.5 | 0.060 | 1.35 |
Влияние ГХПК на структуру образуемого сополимера изучали с использованием ЯМР и поляризованного инфракрасного излучения. Было установлено [123], что модификация каталитической системы VOCl3–ЭАСХ гексахлор-п-ксилолом позволяет получать однородный по составу сополимер.
Процесс сополимеризации этилена и пропилена на VOCl3–Et3Al2Cl3 можно представить в соответствии с работами [105, 108] в виде следующих упрощенных реакций:

В отсутствие ГХПК процесс протекает по направлениям 1 – образование активных центров (+EASC), 2 – сополимеризация (Monomers) и 3 – побочный процесс восстановления ванадия (+EASC). При введении ГХПК добавляется новый процесс 4 – окисления V+2 и возврат какой-то доли ванадия к образованию новых активных центров (+HCPX). Именно этот процесс, видимо, и приводит к уширению ММР сополимера. Кроме того, к направлению 1 необходимо добавить новый компонент – ГХПК, так как повышение скорости процесса связано не только с изменением числа активных центров, но и с величиной констант относительной активности (табл. 10), что свидетельствует о бóльшей вероятности участия ГХПК в формировании активных центров.
Таким образом, модификация ванадиевых катализаторов с помощью хлорорганических соединений при сополимеризации этилена и пропилена является эффективным решением проблем низкой активности подобных систем, позволяющим повысить в несколько раз выход сополимера и улучшить его некоторые характеристики. Понимание процессов восстановления катализатора, когда может происходить постепенное понижение валентности ванадия вплоть до 0 [V(V) → V (IV) → V (III) → V (II) → V(I) → V(0)], является важным фактором для оптимизации процесса сополимеризации. В свою очередь подбор хлорорганических соединений определенного типа, содержащих повышенное количество атомов хлора, позволит сохранять баланс валентности ванадия и его окружения, необходимого для проведения высокоактивной полимеризации в заданном направлении. Что касается молекулярных характеристик, то выбор того или иного промотора может позволить получить продукт как с пониженной, так и с повышенной ММ, а также синтезировать СКЭП(СКЭПТ) с высокой композиционной однородностью.
ПОЛИМЕРИЗАЦИЯ ЭТИЛЕНА НА НАНЕСЕННЫХ ТИТАН-МАГНИЕВЫХ КАТАЛИЗАТОРАХ
К титан-магниевым катализаторам синтеза ПЭ предъявляется ряд таких требований, как высокая активность и способность к сополимеризации, повышенная чувствительность к водороду, удовлетворительные молекулярные/вязкостные характеристики и улучшенная морфология образующегося порошка полимера. В настоящее время при совершенствовании ТМК используют два основных подхода: оптимизацию методов подготовки катализаторов и модификацию каталитических систем введением различных добавок – промоторов [102, 124–127].
Анализ литературы показал, что до 2000-х годов активацию хлорорганическими соединениями преимущественно использовали для “ванадиевых” катализаторов [104–106, 113, 128–130]. В последние годы появилось достаточно большое число публикаций, посвященных промотированию ТМК хлорорганическими соединениями [126, 127, 131–138]. Промоторами полимеризации этилена, подходящими для катализаторов на основе ванадия, являются такие соединения как CHCl3, CFCl3, C3H2Cl6 и C3Cl8 [131, 139–142]; а компания “Union Carbide” (сейчас “Dow Chemical”) успешно использовала промоторы активности в катализаторах на основе ванадия при получении линейного полиэтилена низкой плотности: например, CHCl3 и CFCl3 [120–139], CH2Cl2 и CF2ClCFCl2 [121], C3H2Cl6 и C3Cl8 [142]. Однако, изучая ванадиевые катализаторы, K.J. Cann с сотрудниками [143, 144] обнаружили, что CFCl3, CHCl3 и CCl4 дезактивируют катализатор на основе титана, а CH2Cl2 может повысить активность этого же катализатора примерно на 50%. Для каждого промотора имеется оптимальное мольное соотношение галоген(Cl) : Ti, при котором активность катализатора достигает максимума. Влияние хлорорганических добавок на катализаторы Циглера–Натта ранее отмечено в обзорах, посвященных титан-магниевым катализаторам [126, 131].
H.K. Luo и сотрудниками изучено влияние галоидсодержащих промоторов (преимущественно, хлорсодержащих) на активность гомополимеризации этилена в присутствии ТМК [131]. Было исследовано 16 галоидоуглеводородов различной природы – алициклических, алифатических и ароматических в присутствии каталитической системы на основе TiCl3–MgCl2–ТГФ/SiO2 и Et3Al (табл. 13). Как видно из этой таблицы, при мольном соотношении промотор : Ti = 40 (за исключением Cl(CH2)3Cl, CHCl2CHCl2, CCl2FCCH3, CHCl3, CCl3CH3, которые отравляют катализатор), двенадцать галогенуглеродов повышают каталитическую активность, а C6H11Cl, C6H5Cl, Cl–(CH2)5–Cl, CF2ClCFCl2 обладают очень высокой эффективностью промотирования.
Таблица 13.
Сравнительные данные по активации катализатора TiCl3–MgCl2–ТГФ/SiO2 с использованием различных хлорорганических соединений [131]
| Вид хлорорганического соединения | Хлорорганическое соединение | P(H2/C2H4), МПа | Время, ч | Активность (×104), г ПЭ /г Ti ч |
|---|---|---|---|---|
| Без промотора | – | 0.15/0.58 | 2 | 7.3 |
| – | 0.25/0.48 | 2 | 5.6 | |
| Алициклический | Хлорциклогексан | 0.25/0.48 | 2 | 24.0 |
| Хлорциклогексан | 0.15/0.58 | 0.5 | 36.6 | |
| Ароматический | Хлорбензол | 0.25/0.48 | 2 | 12.0 |
| о-Дихлорбензол | 0.15/0.58 | 2 | 12.45 | |
| м-Дихлорбензол | 0.15/0.58 | 2 | 9.75 | |
| р-Дихлорбензол | 0.15/0.58 | 2 | 10.2 | |
| Алифатический | Cl–(CH2)–Cl | 0.15/0.58 | 1 | 15.6 |
| Cl–(CH2)2–Cl | 0.15/0.58 | 1 | 24.2 | |
| Cl–(CH2)3–Cl | 0.15/0.58 | 2 | 6.25 | |
| Cl–(CH2)4–Cl | 0.15/0.58 | 1 | 20.6 | |
| Cl–(CH2)5–Cl | 0.15/0.58 | 1 | 26.2 | |
| CHCl2CHCl2 | 0.15/0.58 | 2 | 5.0 | |
| CHCl3 | 0.15/0.58 | 2 | 0 | |
| CCl3CH3 | 0.15/0.58 | 2 | 0 | |
| н-C4H9Cl | 0.15/0.58 | 2 | 13.85 |
В целом активность каталитической системы при полимеризации этилена для TiCl3–MgCl2–ТГФ/SiO2 изменяется в зависимости от природы хлорорганического соединения в указанном ряду промоторов следующим образом:
для алифатических
CF2ClCFCl2 > Cl-(CH2)5-Cl > Cl-(CH2)2-Cl > > Cl-(CH2)4-Cl > CH2Cl2 > н-C4H9Cl;
для ароматических
C6H5Cl > (1,2-, орто)C6H4Cl2 > (1,3-, мета)C6H4Cl2 ≈ ≈ (1,4-, пара)C6H4Cl2;
для монохлоридов
C6H11Cl > C6H5Cl > н-C4H9Cl.
Хлорциклогексан (C6H11Cl) обладает наиболее высокой активностью – при его использовании для модификации TiCl3–MgCl2–ТГФ/SiO2 наблюдается увеличение выхода ПЭ в 3 раза. Однако этими же авторами выявлено [131], что модификация катализатора TiCl4–MgCl2–ТГФ/SiO2 тем же хлорциклогексаном не приводит к столь заметному изменению скорости процесса (повышение только на 27%), т.е. для разных катализаторов рост активности для одного и того же промотора может отличаться.
N. Bahri-Laleh с сотрудниками [133] исследовали серию хлорорганических соединений – хлорциклогексан, хлорциклопентан, н-C4H9Cl (хлорбутил), 1,4-дихлорбутан и CHCl3 (хлороформ) при модификации ТМК, где оптимальные соотношения хлорорганическое соединение : Ti составляли 128, 110, 20, 0,54 и 0,033 для хлорциклогексана, хлорциклопентана, хлорбутила, дихлорбутана и хлороформа (табл. 14).
Таблица 14.
Активность ТМК, модифицированного различными хлорорганическими соединениями и некоторые характеристики порошка ПЭ [133]
| Хлорорганическое соединение | Активность, кг ПЭ/г кат ч | Показатель текучести расплава* | Средний размер частиц порошка, мкм | Тпл, °С | Кристал-личность, % |
|---|---|---|---|---|---|
| Без промотора | 16.88 | 0.40 | 123 | 135 | 64 |
| Хлорциклогексан | 31.18 | 0.48 | 197 | 137 | 70 |
| Хлорциклопентан | 30.54 | 0.45 | 192 | 136 | 68 |
| Хлорбутил | 27.86 | 0.45 | 188 | 136 | 65 |
| 1,4-Дихлорбутан | 28.70 | 0.47 | 154 | 135 | 65 |
| Хлороформ | 28.07 | 0.44 | 180 | 136 | 67 |
Как видно из представленных данных, наибольший рост активности достигается при использовании хлорциклогексана и хлорциклопентана, однако, независимо от содержания водорода, хлорциклогексан явно выделяется среди монохлоридов. Остальные соединения также позволяют значительно повысить активность, при этом уровень изменения для хлорбутила, дихлорбутана и хлороформа практически одинаков. Следует отметить, что в отличие от результатов [131], полученных H.K. Luo и сотрудниками с катализатором на основе TiCl3, группа N. Bahri-Laleh [133] выявила, что сочетание CHCl3 с катализатором на основе TiCl4 позволяет получить существенный рост активности (до ~70%). В то же время обе группы установили, что хлорциклогексан является высокоэффективным промотором полимеризации этилена.
В дополнение к предыдущим исследованиям N. Bahri-Laleh и сотрудниками [126] для наилучшего понимания природы промоторов и механизма реактивации каталитических центров была проведена вторая серия исследований полимеризации этилена на титан-магниевом катализаторе TiCl4–MgCl2/трихлорэтан, модифицированного хлорсодержащими соединениями различной природы – алициклического, алифатического и ароматического типа [126]. Было использовано квантово-химическое моделирование на основе теории функционала плотности (DFT) в попытке установить возможный механизм реактивации/восстановления каталитического центра галогенорганическими соединениями [145]. Исследовали хлорсодержащие соединения, включая хлорциклогексан и хлорциклопентан (алициклические), CH3Cl, CH2Cl2 и CHCl3 (алифатические), хлорбензол и 1,2,4-трихлорбензол (ароматические) и 3,3,3-трихлорпроп-1-ен (органогалогенид алкенового типа) при различных молярных соотношениях хлорорганическое соединение : Ti. Показано, что активность ТМК, модифицированного указанными соединениями, понижается в следующем ряду хлорсодержащих углеводородов: алициклические–алифатические–ароматические. Использование добавки алкенового типа (аллилхлорида) приводило к отравлению катализатора. Применение хлорциклогексана приводит к увеличению выхода полимера и показателя текучести расплава, а также улучшению морфологии полимерного порошка (средний размер частиц возрастает с 123 до 197 мкм) [133]. При этом повышается температура плавления и кристалличности гомополимеров этилена (табл. 14).
С помощью DFT-моделирования установлено, что восстановление активности Ti катализатора (в ряду CH3Cl, CH2Cl2, CHCl3, CCl4) происходит в основном за счет окислительного присоединения органогалогенидов к менее активному центру Ti II и превращения его в центр Ti IV, а не в центр Ti III. Кроме того, для промоторов с бóльшим содержанием атомов хлора окисление (реактивация) центра Ti II становится термодинамически более благоприятным.
В ряде работ было исследовано влияние монохлорированных органических соединений, таких как С2H5Cl [134] и н-C4H9Cl [136]. Было показано, что в процессе полимеризации этилена активность каталитической системы TiCl4/Mg(OEt)2/Et3Al, модифицированной EtCl и н-C4H9Cl, имеет оптимум в области мольного соотношения хлорэтил : Et3Al до 0.5, при которой выход полимера возрастает на ~30%. Уменьшение или увеличение этого соотношения приводило к понижению активности реакционной системы. Общее для этих модифицированных каталитических систем – улучшение морфологии образующегося порошка ПЭ и понижение средних ММ с проявлением тенденции на расширение ММР.
В работах [135, 137] также подтверждена высокая эффективность хлорциклогексана при модификации ТМК, содержащего в своем составе внутренние доноры на основе спиртов и эфиров. Кроме того, ТМК с хлорциклогексаном показали высокий отклик на водород, высокие значения показателя текучести расплава, низкие значения Mw и Mw/Mn.
В работе [146] проведен анализ 122 публикаций по катализаторам синтеза линейного ПЭНП. Так, при полимеризации этилена на титан-магниевом катализаторе на основе TiCl3 в суспензии при 85°С введение в реакционную среду 1,2-дихлорэтана, 2,2,4,4-тетрахлорэтана, 1,1,2-трихлорпропана, дибромметана или хлорэтана приводило к расширению ММР полимера с 5–9 до 18–20 при практически постоянной активности катализатора и среднечисленной молекулярной массы. При полимеризации этилена на TiCl4/MgCl2 в суспензии при 90°С в присутствии триэтилалюминия введение в реакционную среду н-, втор- и трет-бутилхлорида, бромистого этила и хлорбензола резко изменились реологические свойства образующегося полимера: текучесть расплава возрастала в ряду н-BuCl–трет-BuCl–EtBr– втор-BuCl [147]. В присутствии хлорбензола наблюдалось двукратное увеличение среднечисленной молекулярной массы ПЭ.
Следует отметить, что практически все представленные работы [131, 133, 135–137] посвящены исследованию хлорциклогексана в гомополимеризации этилена, и в них практически отсутствуют данные о влиянии хлорорганического соединения на сополимеризацию этилена с α-олефинами. В то же время большинство производимых марок ПЭ являются сополимерами этилена и α-олефинов [148–150]. В связи с этим исследование сополимеризации этилена с α-олефинами на ТМК, модифицированных хлорорганическими соединениями, имеет важное научное и практическое значение. В работах [151, 152] в качестве промоторов ТМК использовали хлорциклогексан и ГХПК.
По результатам исследования полимеризации этилена и сополимеризации этилена и гекс-1-ена на каталитической системе TiCl4 ∙ MgCl2/i-Bu3Al в присутствии моно- и мультихлорированных хлорорганических соединений установлено, что хлорциклогексан и ГХПК позволяют увеличить активность ТМК в 2–4 раза в гомополимеризации и в 1.2–2.0 раза в сополимеризации (табл. 15). Природа хлорорганических соединений оказывает влияние на показатель текучести расплава и молекулярные характеристики ПЭ, а при введении гекс-1-ена, различия в этих параметрах становятся более явными.
Таблица 15.
Активность катализатора ТМК/триизобутилалюминий без промотора и в присутствии хлорциклогексан/ГХПК, а также некоторые характеристики образцов ПЭ [151]
| Хлорорганическое соединение | Содержание гексен-1 в системе, моль/л | wG, кгПЭ/ г кат ч | Показатель текучести расплава * | Mw | Mw/Mn | Tпл, °C | Степень кристалл- личности, % |
|---|---|---|---|---|---|---|---|
| Без промотора | 0 | 1.8 | 1.1 | 170 | 4.3 | 137.9 | 62 |
| 0.16 | 4.3 | 2.0 | 145 | 3.9 | 130.3 | 55 | |
| 0.32 | 3.2 | 3.1 | 120 | 3.8 | 130.8 | 40 | |
| Хлорциклогексан | 0 | 6.7 | 1.0 | 180 | 3.9 | 137.5 | 64 |
| 0.16 | 7.0 | 2.7 | 140 | 3.8 | 133.7 | 54 | |
| 0.32 | 6.5 | 5.1 | 100 | 3.8 | 131.0 | 48 | |
| ГХПК | 0 | 3.8 | 0.6 | 200 | 3.9 | 138.6 | 61 |
| 0.16 | 5.2 | 1.1 | 165 | 3.8 | 134.5 | 54 | |
| 0.32 | 4.0 | 1.3 | 160 | 3.8 | 132.3 | 51 |
Как видно из табл. 15, для ПЭ наибольшее значение показателя текучести расплава характерно для полимера, полученного на ТМК в отсутствие хлорорганического соединения. При гомополимеризации этилена на системе без хлорорганического соединения и на системе ТМК−хлорциклогексан близкий уровень показателя текучести расплава согласуется с достаточно близкими величинами Мw. Показано, что при увеличении содержания гекс-1-ена как в случае сополимеризации на ТМК без промотора, так и ТМК с хлорциклогексаном наблюдается понижение Мn и еще большее возрастание показателя текучести расплава. Но наиболее существенное снижение ММ и соответственно повышение показателя текучести расплава происходит при использовании в качестве промотора хлорциклогексана. Для катализатора с ГХПК уровень показателя текучести расплава как гомополимера, так и сополимеров достаточно низкий и мало изменяется с повышением содержания гексена-1, а ММ имеет наиболее высокое значение. Полидисперсность как гомоПЭ, так и сополимеров практически не зависит от присутствия хлорорганического соединения в каталитической системе (табл. 15). Модификация ТМК исследуемыми промоторами не приводит к существенному изменению вида кривых ММР. Кривые являются мономодальными для всех образцов полимеров [151]. Использованные в работе промоторы не оказывают влияния на термические свойства образующегося на ТМК полиэтилена.
Общее для всех исследованных каталитических систем – снижение средних ММ при введении гексена-1 вследствие увеличения роли процессов ограничения роста цепи при сополимеризации по сравнению с гомополимеризацией этилена. Уменьшение Мn при увеличении содержания гексена-1 в случае сополимеризации на ТМК и ТМК с хлорциклогексаном связано с высокой вероятностью участия α-олефина в процессе ограничения роста цепи. В случае использования ГХПК такое заключение сделать нельзя, хотя вероятность ограничения роста цепи также выше в сополимеризации, чем при гомополимеризации этилена.
Механизм активирующего действия промоторов с подвижным атомом хлора на титан-магниевые катализаторы Циглера–Натта (TiCl4·MgCl2/R3Al) при полимеризации этилена является предметом дискуссий. В литературе существует несколько предположений.
1. Регенерация (реактивация) низкоактивных центров катализатора в результате окисления низковалентного титана [102, 126, 145].
2. Снижение восстановительной способности алюминийорганических соединений при замещении алкильных групп на атомы хлора в реакциях R3Al c RCl и уменьшение скорости восстановительных процессов в каталитической системе [102]; это может изменять соотношение Ti2+/Ti3+ в составе активных центров.
3. Образование при взаимодействии R3Al c RCl небольшого количества кислот Льюиса (RAlCl2, AlCl3) [102]; кислоты Льюиса могут связывать продукты, образующиеся в процессе полимеризации (R2AlH, R2AlCl и т.д.), которые блокируют активные центры, тем самым повышая концентрацию центров роста [153].
4. Галоидоуглеводороды могут слабо координироваться на титановом центре, тем самым изменять его реакционную способность [131].
По мнению авторов [151], введение хлорорганического соединения в каталитический комплекс влияет на соотношение Ti2+/Ti3+ в составе активного компонента катализатора, который формируется после взаимодействия ТМК с алюминийорганическим сокатализатором (i-Bu3Al). Исходя из этого, максимальная доля Ti2+ образуется в немодифицированном ТМК, доля Ti2+ уменьшается, а Ti3+ возрастает в ТМК/хлорциклогексан. Минимальная доля Ti2+ (максимальная – Ti3+), по-видимому, в катализаторе ТМК/ГХПК. Результаты DFT-моделирования позволили подтвердить [126, 145], что для богатых хлором промоторов окисление (реактивация) центра Ti2+ в Ti3+ становится термодинамически более выгодным.
На основании данных работы [153] ТМК, содержащие в активном центре Ti2+, имеют более высокую вероятность вхождения α-олефина в полимерную цепь при сополимеризации с этиленом, чем системы ТМК/хлорорганическое соединение. В отсутствие в каталитической системе хлорорганического соединения доля активных центров, содержащих Ti2+, выше, чем в системах с промотором. В работе [153] для катализаторов, содержащих Ti3+ получены значения r1 в диапазоне 100–114, что близко к данным для систем ТМК с хлорциклогексаном. В связи с этим значение r1 для немодифицированного ТМК, ниже, а содержание звеньев α-олефина выше, чем у систем ТМК/хлорорганическое соединение, в которых в активных центрах преобладают соединения Ti3+.
С точки зрения механистических аспектов действия хлорсодержащих промоторов, по мнению авторов [151], наблюдаемые изменения в структуре сополимера, по-видимому, обусловлены образованием более высококристаллического полимера при модификации катализатора. Очевидно, что при введении хлорорганического соединения в исходный ТМК происходит изменение концентрации менее активных центров с повышенной сополимеризующей способностью Ti2+ в более активные центры с меньшей сополимеризующей способностью Ti3+ и дальнейшим формированием полимера с высоким выходом и кристалличностью. Что касается чувствительности к водороду в процессе сополимеризации, то предполагается, что в ТМК с хлорциклогексаном активные центры характеризуются повышенной лабильностью (повышенной вероятностью обрыва цепи), в то время как для ГХПК образуются активные центры с повышенной стабильностью (меньшей вероятностью обрыва цепи). Эту особенность можно объяснить другой причиной, а именно, природой углеводорода в хлорорганическое соединение. Циклические кольца в хлорциклогексане и ароматические в ГХПК, по-видимому, могут за счет координации создавать стерические затруднения для активного центра и снижать внедрение гекс-1-ена (низкое r2) или в случае ГХПК – влиять на взаимодействие с водородом (низкий показатель текучести расплава, повышенное значение Mw).
Процесс сополимеризации этилена и гексена-1 на ТМК/триизобутилалюминий/хлорорганическое соединение представлен в работе [151] в виде следующих упрощенных реакций, представленных ниже.

(ХЦГ – хлорциклогексан, ТИБА – триизобутилалюминий).
В отсутствие хлорорганических соединений процесс протекает по следующим направлениям: 1 – образование активных центров (+i-Bu3Al), 2 – сополимеризация на Ti3+ (Monomers&Сomonomers). 3 – дальнейший процесс восстановления титана (+i-Bu3Al), 4 – сополимеризация на Ti2+ (Monomers&Сomonomers). При введении хлорорганических соединений добавляется новый процесс 5 – окисление Ti+2 и возврат какой-то доли титана к образованию новых активных центров в более высокой степени окисления Ti+3. Кроме того, к направлению 1 необходимо добавить новый хлорорганический компонент – хлорциклогексан или ГХПК, поскольку повышение скорости процесса связано не только с окислением Ti+2 → Ti+3, но и с различным изменением констант сополимеризации, а также чувствительности к водороду. Это свидетельствует о большой вероятности участия хлорциклогексан/ГХПК в формировании центров сополимеризации. Что касается чувствительности к водороду для ТМК с ГХПК, то нельзя исключать [146], что хлорорганическое соединение с бензольным кольцом в случае координации на активном центре может одновременно активировать за счет окисления хлором и создавать стерические затруднения при взаимодействии с гекс-1-еном и водородом.
Таким образом, литературные данные показывают, что среди исследованных хлорорганических соединений наибольшее повышение активности каталитических систем на основе ТМК вызывают циклические соединения с одним атомом хлора. Подобные различия с ванадиевыми системами обсуловлены тем, что глубина и скорость восстановления соединений титана и ванадия различаются, так как восстановление ванадия протекает гораздо быстрее. При этом сам TiCl4 нанесен на хлорид магния, который, как известно, также влияет на активность катализатора. В то же время для систем на основе TiCl4–MgCl2, в отличие от TiCl3, хлорорганические соединения с числом атомов хлора более одного также влияет на процесс полимеризации, вызывая существенную активацию. Характеристики ПЭ в зависимости от используемого хлорорганического соединения, претерпевают заметные изменения. Особенно это наблюдается при сополимеризации этилена с α-олефинами, когда встраиваемость сомономеров в структуру полимера несколько снижается.
ЗАКЛЮЧЕНИЕ
В настоящем обзоре рассмотрены литературные данные, описывающие влияние хлорорганических соединений на полимеризацию диенов и олефинов под действием широко применяемых в промышленности катализаторов Циглера–Натта на основе неодима, ванадия и титана. Многие исследователи подчеркивали промотирующую роль атома хлора, который входит в состав активных центров катализаторов Циглера–Натта и обеспечивает важную функцию активации каталитической системы.
Отмечено разное влияние хлорсодержащих соединений – использование мультихлорированных соединений в неодимовой и ванадиевой системах благоприятно сказывается на их каталитических свойствах. Напротив, для титан-магниевых катализаторов высокоэффективными являются монохлорированные соединения. Важную роль играют и углеводородные заместители в хлорсодержащих соединениях: так, например, наличие циклических и ароматических колец является ключевым фактором, влияющим на лигандное окружение активных центров катализаторов и скорость полимеризации.
Применение промоторов, разумеется, не решит всех вопросов, которые возникают при создании новых полиолефиновых технологий и продуктов, но могут существенно повысить производительность катализаторов, улучшить молекулярно-массовые характеристики, композиционную однородность и микроструктуру полимеров.
Работа выполнена при поддержке Гос.задания ИНХС РАН.
Список литературы
Sauter D.W., Taoufik M., Boisson C. // Polymers. 2017. V. 9. № 6. P. 185.
Jubinville D., Esmizadeh E., Saikrishnan S., Tzoganakis C., Mekonnen T. // Sust. Mater. Technol. 2020. V. 25. P. e00188.
Ricci G., Leone G. // Polyolefins J. 2014. V. 1. № 1. P. 43.
Ricci G., Pampaloni G., Sommazzi A., Masi F. // Macromolecules. 2021. V. 54. № 13. P. 5879.
Долгоплоск Б.А., Тинякова Е.И. // Высокомолек. соед. А. 1994. Т. 36. № 10. С. 1653.
Ziegler Catalysts: Recent Scientific Innovations and Technological Improvements / Ed. by G. Fink, R. Mülhaupt, H. H. Brintzinger. NewYork: Springer, 1995.
Ziegler K., Gellert H., Holzkamp E., Wilke G. // Brennstoffe. 1954. B. 35. S. 321.
Ziegler K. // J. Petr. Refiner. 1955. V. 34. № 8. P. 111.
Ziegler K., Holzkamp E., Breil H., Martin H. // Angew. Chem. 1955. B. 67. S. 426, 541.
Natta G., Corradini P. // Rend. Accad. Naz. Lincei VIII. 1955. V. 19. № 5. P. 239.
Natta G., Corradini P. // J. Polym. Sci. 1956. V. 20. P. 251.
Natta G. // J. Polym. Sci. 1957. V. 26. P. 120.
Natta G., Corradini P., Porri L. // Rend. Accad. Naz. Lincei VIII. 1956. V. 20. P. 728.
Natta G. // Rubber Plastics Age. 1957. V. 38. P. 495.
Natta G., Porri L., Mazzei A., Morero D. // Chim. e Ind. (Milan). 1959. V. 41. № 5. P. 398.
Baier M.C., Zuideveld M.A., Mecking S. // Angew. Chem. Int. Ed. 2014. V. 53. № 37. P. 9722.
Stürzel M., Mihan S., Mülhaupt R. // Chem. Rev. 2016. V. 116. № 3. P. 1398.
Stalzer M.M., Delferro M., Marks T.J. // Catal. Lett. 2015. V. 145. P. 3.
Mohite A.S., Rajpurkar Y.D., More A.P. // Polym. Bull. 2021. V. 79. P. 1309.
Zanchin G., Leone G. // Progr. Polym. Sci. 2021. V. 113. P. 101342.
Kumawat J., Gupta V.K. // Polym. Chem. 2020. V. 11. № 38. P. 6107.
Монаков Ю.Б., Марина Н.Г., Сабиров З.М. // Высокомолек. соед. А. 1994. Т. 36. № 10. С. 1680.
The Stereo Rubbers / Ed. by W. Saltman. New York: Wiley-Interscience, 1977.
Taniike T., Terano M. // Adv. Polym. Sci. 2013. № 257. P. 81.
Trischler H., Höchfurtner T., Ruff M., Paulik C. // Kinet. Catal. 2013. V. 54. № 5. P. 559.
Salakhov I., Akhmetov I., Kozlov V. // Polymer Science B. 2011. V. 53. № 7–8. P. 385.
Nifant’ev I.E., Salakhov I.I., Ivchenko P.V. // Molecules. 2022. V. 27. № 21. P. 7164.
Yu G., Chen W., Wang Y. // Kexue Tongbao. 1983. V. 28. № 7. P. 408.
Yu G., Chen W., Wang Y. // Kexue Tongbao. 1984. V. 29. № 3. P. 421.
Qian H., Yu G., Chen W. // Gaofenzi Tongxun. 1984. № 3. P. 226.
Chen W., Xiao S., Wang Y., Yu G. // Kexue Tongbao. 1984. V. 29. № 7. P. 892.
Маркевич И.Н., Шараев О.К., Тинякова Е.И., Долгоплоск Б.А. // Докл. АН СССР. 1983. Т. 268. № 4. С. 892.
Яковлев В.А., Воллерштейн Е.Л., Черезова Л.С., Тинякова Е.И., Долгоплоск Б.А. // Докл. АН СССР. 1983. Т. 268. № 6. С. 1422.
Гайлюнас Г.А., Биктимиров Р.Х., Хайруллина Р.М., Марина Н.Г., Монаков Ю.Б., Толстиков Г.А. // Докл. АН СССР. 1987. Т. 295. № 6. С. 1385.
Гайлюнас Г.А., Биктимиров Р.Х., Савельева И.Г. Химия и физикохимия высокомолекулярных соединений. Уфа: Ин-т химии Башкирского филиала АН СССР, 1987.
Shen Z., Ouyang J., Wang F., Hu F., Yu F., Qian B. // Polym. Sci., Polym. Chem. Ed. 1980. V. 18. № 12. P. 3345.
Shen Z., Ouyang J. // Handbook on the Physics and Chemistry of Rare Earth. / Ed. by K.A. Gschneidner, Jr., L. Eyring. Elsevier Sci. Publ. 1987. V. 9. P. 395.
Yang J., Hu J., Feng S., Pan E., Xie D., Zhong C., Ouyang J. // Sci. Sin. 1980. V. 23. № 6. P. 734.
Gallazzi M.C., Bianchi F., Depero L., Zocchi M. // Polymer. 1988. V. 29. № 8. P. 1516.
Mazzei A. // Macromol. Chem. 1981. B. 182. № 4. S. 61.
Ceausescu E., Dimonie M., Fieroiu V., Hubca G., Gruber V., Badea E., Vladulescu V., Verestoi A., Iovu H., Vasile I. // Rev. Roum. Chim. 1989. V. 34. № 1. P. 5.
Санягин А.А., Кормер В.А. // Докл. АН СССР. 1985. Т. 283. № 5. С. 1209.
Монаков Ю.Б., Толстиков Г.А. Каталитическая полимеризация 1,3-диенов. М.: Наука, 1990.
Пат. 3794604 США // РЖХим. 1975. C279П.
Throckmorton M.C. // Kautsch. Gummi Kunstst. 1969. B. 22. № 6. S. 293.
Пат. 2011543 ФРГ // Chem. Abstrs. 1971. 13981y.
Пат. 2399447 Франция // РЖХим. 1980. С379П.
Пат. 2833721 ФРГ // Chem. Abstrs. 1979. 169316b.
Quirk R.P., Kells A.M. // Polym. Int. 2000. V. 49. № 7. P. 751.
Liao Y., Lin X., Zhang S. Пат. 85101899 Китай // Chem. Abstrs. 1986. V. 105. № 26. P. 227542.
Yang C., Zheng Y., Wang F. // Hecheng Xiangjiao Gongue. 1987. V. 10. № 6. P. 409.
Liao Y., Zhang S., Liu X. // Yingyong Huaxue. 1987. V. 4. № 1. P. 13.
Liao Y., Zhang S., Liu X. // Yingyong Huaxue. 1988. V. 5. № 2. P. 34.
Pan E., Zhong C., Xie D., Ouyang J. // Huaxue Xuebao. 1982. V. 40. № 4. P. 301.
Friebe L., Nuyken O., Obrecht W. // Neodymium Based Ziegler Catalysts – Fundamental Chemistry / Ed. by O. Nuyken Munich: Springer, 2006.
Friebe L., Nuyken O., Windisch H., Obrecht W. // Macromol. Chem. Phys. 2002. № 203. P. 1055.
Witte J. // Angew. Makromol. Chem. 1981. № 94. P. 119.
Friebe L., Windisch H., Nuyken O., Obrecht W. // Macromol. Sci., Pure Appl. Chem. 2004. 41. P. 245.
Friebe L., Muller J.M., Nuyken O., Obrecht W. // J. Macromol. Sci., Pure Appl. Chem. 2006. 43. P. 11.
Черненко Г.М., Яковлев В.А., Тинякова Е.И., Долгоплоск Б.А. // Высокомолек. соед. Б. 1989. Т. 31. № 8. С. 637.
Dong W., Endo K., Masuda T. // Macromol. Chem. Phys. 2003. № 204. P. 104.
Jenkins D.K. // Polymer. 1985. V. 26. № 1. P. 152.
Пат. 2206577 Россия. 2001 // Б.И. 2003. С08F136/06.
Zhang Q., Ni X., Shen Z. // Macromol. Sci., Pure Appl. Chem. 2004. № 41. P. 39.
Zhang Q., Ni X., Shen Z. // Polym. Int. 2002. № 51. P. 208.
Пат. 2268894 Россия 2004 // Б.И. 2006. С08F4/14.
Ахметов И.Г., Махиянов Н.К., Салахов И.И., Хачатуров А.С. // Каучук и резина. 2007. № 5. С. 2.
Knauf T., Braubach W. Pat. 1095952 Eur. 2001.
Knauf T., Braubach W. // Chem. Abstr. 2001. V. 134. № 312297.
Jang Y.C., Kwag K.H., Kim P.S. Pat. 2000032230 Korean. 2000.
Jang Y.C., Kwag K.H., Kim P.S. // Chem. Abstrs. 2002. V. 136. № 341170.
Sone T., Hattori I., Yamazaki D., Nonaka K. Eur. pat. 1099710 2001.
Sone T., Hattori I., Yamazaki D., Nonaka K. // Chem. Abstrs. 2001. V. 134. № 354405.
Ishino Y., Nakayama S., Mori Y., Hattori I. Pat. 6391990 USA. 2003.
Ishino Y., Nakayama S., Mori Y., Hattori I. // Chem. Abstrs. 2000. V. 133. № 151837.
Wilson D.J. // J. Polym. Sci. A. 1995. V. 33. P. 2505.
Монаков Ю.Б., Биешев Я.Х., Берг А.А., Рафиков С.Р. // Докл. АН СССР. 1977. Т. 234. № 5. С. 1125.
Рафиков С.Р., Монаков Ю.Б., Биешев Я.Х., Валитова И.Ф., Муринов Ю.И., Толстиков Г.А., Никитин Ю.И. // Докл. АН СССР. 1976. Т. 229. № 5. С. 1174.
Shokri A., Talebi S., Salami-Kalajahi M. // Polym. Bull. 2020. V. 77. P. 5245.
Wang F., Liu H., Zheng W., Guo J., Zhang C., Zhao L., Zhang H., Hu Y., Bai C., Zhang X. // Polymer. 2013. V. 54. P. 6716.
Quirk R.P., Kells R.P., Yunlu K., Cuif J.P. // Polymer. 2000. № 41. P. 5903.
Mello I.L., Coutinho F.M.B. // J. Appl. Polym. Sci. 2009. V. 112. P. 1496.
Manuiko G.V., Salakhov I.I., Aminova G.A., Akhmetov I.G., Dyakonov G.S., Bronskaya V.V., Demidova E.V. // Theor. Found. Chem. Eng. 2010. V. 44. № 2. P. 139.
Taube R. // Metallorganic Catalysts for Synthesis and Polymerization / Ed. by W. Kaminsky. Berlin; Heidelberg; New York: Spring-Verlag, 1999.
Авдеева О.Г., Маркевич И.Н., Шараев О.К., Бондаренко Г.Н., Тинякова Е.И., Долгоплоск Б.А. // Докл. АН СССР. 1986. Т. 286. № 3. С. 641.
Monakov Yu.B., Sabirov Z.M., Duvakina N.V., Glukhov E.A., Ponomareva O.A., Spirikhin L.V. // Polymer Science A. 2001. V. 43. № 2. P. 91.
Монаков Ю.Б., Марина Н.Г., Сабиров З.М. // Высокомолек. соед. A. 1994. Т. 36. № 3. С. 378.
Гольштейн С.Б., Яковлев В.А., Бондаренко Г.Н., Ямпольский Ю.П., Долгоплоск Б.А. // Докл. АН СССР. 1986. Т. 289. № 3. С. 657.
Воллерштейн Е.Л., Глебова Н.Н., Гольштейн С.Б., Завадовская Э.Н., Шараев О.К., Яковлев В.А., Тинякова Е.И., Долгоплоск Б.А. // Докл. АН СССР. 1979. Т. 249. № 4. С. 860.
Akhmetov I.G., Kozlov V.G., Salakhov I.I., Sakhabutdinov A.G., D’yakonov G.S. // Int. Polym. Sci. And Tech. 2010. V. 37.
Салахов И.И. Дис. … канд. тех. наук. Казань: Казанский национальный исслед. технол. ун-т, 2009.
De León R.D., Corral F.S., Enríquez-Medrano F.J., Ibarra G.B., Martínez P.L., Gámez F.H., López-González H.R., De Valle L.F.R. // Int. J. Polym. Sci. 2016. P. 1.
Zolotarev V.L., Markov B.A., Yartseva T.A. // Int. Polym. Sci. Tech. 2013. V. 40. № 11. P. 17.
Аксенов В.И., Галибеев С.С., Аширов Р.В., Тихомирова И.Н., Казаков Ю.М., Максимов Д.А., Каблов В.Ф. Координационная полимеризация бутадиена-1,3 на различных каталитических системах. Томск: Изд-во Томского политехн. ун-та, 2011.
Hagen H., Boersma J., van Koten G. // Chem. Soc. Rev. 2002. V. 31. № 6. P. 357.
Wu J.-Q., Li Y.-S. // Coord. Chem. Rev. 2011. V. 255. № 19–20. P. 2303.
Redshaw C. // Dalton Trans. 2010. V. 39. № 24. P. 5595.
Nomura K., Zhang S. // Chem. Rev. 2011. V. 111. № 3. P. 2342.
Zambelli A., Sessa I., Grisi F., Fusco R., Accomazzi P. // Macromol. Rapid Commun. 2001. V. 22. № 5. P. 297.
Soshnikov I.E., Semikolenova N.V., Shubin A.A., Bryliakov K.P., Zakharov V.A., Redshaw C., Talsi E.P. // Organometallics. 2009. V. 28. № 23. P. 6714.
Bravaya N.M., Faingol’d E.E., Badamshina E.R., Sanginov E.A. // Polymer Science C. 2020. V. 62. № 1. P. 1.
Чирков Н.М., Матковский П.Е., Дьячковский Ф.С. Полимеризация на комплексных металлорганических катализаторах. М.: Химия, 1976.
Zhang H.X., Hu Y.M., Zhang C.Y., Lee D.H., Yoon K.B., Zhang X.Q. // Catal. Commun. 2016. V. 83. P. 39.
Karol F.J., Kao S.C., Cann K.J. // Polym. Sci., Polym. Chem. 1993. V. 31. P. 2541.
Adisson E., Deffieux A., Fontanille M., Bujadoux K. // J. Polym. Sci., Polym. Chem. 1994. V. 32. P. 1033.
D’Agnillo L., Soares J.B.P., van Doremaele G.H.J. // Macromol. Mater. Eng. 2005. V. 290. P. 256.
Reinking M.K., Bauer P.D., Seyler J.W. // J. Appl. Catal. A. 1999. V. 189. P. 23.
van Duin M., van Doremaele G., van der Aar N. // Rubber World. 2018. 258. P. 20.
Gumboldt A., Helberg J., Schleitzer G. // Makromol. Chem. 1967. V. 101. P. 229.
Christman D.L. // J. Polym. Sci. A. 1972. V. 10. P. 471.
Maric G.J., Mornet Ph.J. // Rev. Gen. Caoutch. Plast. 1971. V. 48. № 10. P. 1016.
Zohuri G.H., Vakili M., Jamjah R., Ahmadjo S., Nekomanesh M. // Rubber Chem. Technol. 2005. V. 78. P. 682.
Evens G., Pijpers E., Seevens R. Eur. pat. 0044119B1. 1985.
van Doremaele G., Hazen J. NASCRE1, First North American Symposium on Chemical Reaction Engineering. Houston, USA. 2001.
Evens G.G., Pijpers E.M.J., Seevens R.H.M. // Transition Metal Catalyzed Polymerizations / Ed. by R. Quirk. Cambridge: Cambridge Univ. Press, 1988.
Hao X., Zhang C., Li L., Zhang H., Hu Y., Hao D., Zhang X. // Polymers. 2017. V. 9. № 8. P. 325.
Salakhov I., Tkacheva E., Kozlov V., Galanina D., Sakhabutdinov A. // RSC Adv. 2021. V. 11. P. 20916.
Cozewith C., Ver Strate G. // Macromolecules. 1971. V. 4. № 4. P. 482.
Fineman M., Ross S.D. // J. Polym. Sci. 1950. V. 5. P. 259.
Karasev V.N., Minsker K.S. // Polym. Sci. USSR. 1971. V. 13. P. 1652.
Подольный Ю.Б., Соловьева Г.В., Каменев Ю.Г. // Высокомолек. соед. А. 1974. Т. 16. № 12. С. 2767.
Mayo F.R., Lewis F.M. // J. Am. Chem. Soc. 1944. V. 66. P. 1594.
Bhargava R., Wang S.Q., Koenig J. // Liquid Chromatography / FTIR Microspectroscopy / Microwave Assisted Synthesis. Advances in Polymer Science. Springer, Berlin, Heidelberg, 2003. V. 163.
Mikenas T.B., Zakharov V.A., Nikitin V.E., Echev-skaya L.G., Matsko M.A. // Russ. J. Appl. Chem. 2010. V. 83. P. 2210.
Mikenas T.B., Koshevoy E.I., Cherepanova S.V., Zakharov V.A. // J. Polym. Sci., Polym. Chem. 2016. V. 54. P. 2558.
Bahri-Laleh N., Arabi H., Mehdipor-Ataei S., Nekoomanesh-Haghighi M., Zohuri G., Seifali M., Akbari Z. // J. Appl. Polym. Sci. 2012. V. 123. P. 2526.
Abedi S., Majdabadi-Farahani N., Daftari-Besheli M., Ghasempour H., Azadi F. // Polym. Bull. 2015. V. 72. P. 2377.
Rośario Ribeiro M., Deffieux A., Fontanille M., Portela M.F. // Macromol. Chem. Phys. 1995. V. 196. P. 3833.
Spitz R., Pasquet V., Patin M., Guyot A. // Ziegler Catalysts / Ed. by G. Fink, R. Mülhaupt, H.H. Brintzinger. Berlin; Heidelberg: Springer, 1995.
Linda N. Winslow. Pat. 5534472 USA. 1996.
Luo H.K., Tang R.G., Yang H., Zhao Q.F., An J.Y. // Appl. Catal. A. Gen. 2000. V. 203(2). P. 269.
Farrer D.K. Pat. 6646073 USA. 2003.
Bahri-Laleh N., Abbas-Abadi M.S., Haghighi M.N., Akbari Z., Mohammad R.T., Mirjahanmardi S.H. // J. Appl. Polym. Sci. 2010. V. 117(3). P. 1780.
Abedi S., Azadi F., Daftari-Besheli M., Majdabadi-Farahani N., Safinejad A. // J. Appl. Polym. Sci. 2014. V. 131. P. 40189.
Abbas-Abadi M.S. // Designed Monomers and Polymers. 2017. V. 20. P. 524.
Gholami Y., Abdouss M., Abedi S., Azadi F., Baniani P., Arsalanfar M. // Bull. Chem. React. Eng. Catal. 2018. V. 13. P. 412.
Abbas-Abadi M.S., Rashedi R., Sepahi A., Farhadi A., Biglarkhani M. // Iran Polym. J. 2020. V. 29. P. 659.
Bazgir H., Abbas-Abadi M.S., Haghighi M.N., Darounkola M.R.R., Issaabadi Z. // J. Polym. Res. 2021. V. 28. P. 301.
Beran D.L., Cann K.J., Jorgensen R.J. Eur. pat. 120501. 1984.
Karol F.J. WO 88 02377. 1988.
Nicoletti J.W., Cann K.J., Karol F.J. Eur. pat. 286001. 1988.
Cann K.J., Karol F.J., Lee K.H. Pat. 4892853 USA. 1990.
Cann K.J. // Polym. Mater. Sci. Eng. 1991. V. 64. P. 106.
Cann K.J., Nicoletti J.W., Karol F.J. Pat. 5442018 USA. 1995.
Bahri-Laleh N., Correa A., Mehdipour-Ataei Sh., Arabi H., Haghighi M.N., Zohuri G., Cavallo L. // Macromolecules. 2011. V. 44. № 4. P. 778.
Крыжановский А.В., Иванчев С.С. // Высокомолек. соед. А. 1990. Т. 32. № 7. С. 1383.
Крыжановский А.В., Захаров П.С., Богданов-Катьков Н.В., Поляков А.А., Иванчев С.С. // Пласт. массы. 1988. № 4. С. 20.
Geyer R., Jambeck J.R., Law K.L. // Sci. Adv. 2017. V. 3.
Gupta P., Wilkes G.L., Sukhadia A.M., Krishnaswamy R.K., Lamborn M.J., Wharry S.M., Tso C.C., DesLauriers P.J., Mansfield T., Beyer F.L. // Polymer. 2005. V. 46.
Meng W., Li H., Li J., Chen B. // J. Korean Chem. Soc. 2011. V. 55. № 4. P. 673.
Salakhov I.I., Mikenas T.B., Zakharov V.A., Kozlov V.G., Matsko M.A., Suslova T.N. // Int. J. Mol. Sci. 2022. V. 23. P. 10335.
Салахов И.И, Сахабутдинов А.Г, Батыршин А.З., Шайдуллин Н.М., Бородин Р.Г., Михайлов В.Н., Назаров В.В., Хакимова Т.М., Волков В.Л., Суслова Т.Н. Пат. 2759723 Россия. 2021.
Mikenas T.B., Tregubov A.A., Zakharov V.A., Echev-skaya L.G., Matsko M.A. // Polimery. 2008. V. 53. P. 353.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)