

Высокомолекулярные соединения (серия С), 2019, T. 61, № 1, стр. 96-109
ЦИКЛОДОДЕЦЕН В МЕТАТЕЗИСЕ ОЛЕФИНОВ: ПОЛИМЕРИЗАЦИЯ И МАКРОМОЛЕКУЛЯРНЫЙ КРОСС-МЕТАТЕЗИС С ПОЛИНОРБОРНЕНОМ
Ю. И. Денисова a, *, В. А. Жигарев a, М. Л. Грингольц a, Г. А. Шандрюк a, А. С. Перегудов b, Е. Ш. Финкельштейн a, Я. В. Кудрявцев a
a Институт нефтехимического синтеза им. А.В. Топчиева Российской академии наук
119991 Москва, Ленинский пр., 29, Россия
b Институт элементоорганических соединений им. А.Н. Несмеянова Российской академии наук
119334 Москва, ул. Вавилова, 28, Россия
* E-mail: denisova@ips.ac.ru
Поступила в редакцию 08.05.2019
После доработки 24.05.2019
Принята к публикации 31.05.2019
Аннотация
Впервые изучена реакция макромолекулярного кросс-метатезиса полинорборнена и полидодеценамера с использованием Ru-карбенового комплекса Граббса первого поколения. Полидодеценамер с высокими молекулярно-массовыми характеристиками (Mw = 406 × 103, Ð = 2.5) и выходом 96% синтезирован метатезисной полимеризацией циклододецена с раскрытием цикла в присутствии катализатора Граббса второго поколения. Полимер характеризуется температурой плавления 62–83°C, зависящей от содержания транс-двойных связей, и плохой растворимостью в органических растворителях при комнатной температуре. В результате его кросс-метатезиса с полинорборненом при 40°С синтезированы новые статистические мультиблок-сополимеры норборнена и циклододецена с различной степенью блочности. Методами ЯМР, ГПХ и ДСК изучено влияние условий реакции на строение и термические свойства сополимеров. На начальном этапе реакции на хроматограммах ГПХ наряду с высокомолекулярным пиком наблюдается пик низкомолекулярной фракции (M = (1–2) × 103), свидетельствующей об образовании циклоолигомеров. По мере увеличения глубины межцепного обмена доля олигомерной фракции значительно снижается. Методами мониторинга in situ ЯМР 1Н и ex situ ЯМР 13С изучена кинетика кросс-метатезиса полинорборнена с полидодеценамером и полиоктенамером при 40°С. Скорость элементарных реакций в этих смесях различается незначительно за исключением стадии превращения исходных карбенов в полимерные. Карбен [Ru]=полиоктенамер образуется в 3 раза быстрее, чем [Ru]=полидодеценамер. В обоих случаях доля Ru-карбенов, присоединенных к звеньям норборнена, крайне мала на протяжении всего процесса.
Циклододецен (ЦДД) является одним из доступных нефтехимических циклоолефинов [1]. Продукт его метатезисной полимеризации полидодеценамер (ПЦДД) – эффективная добавка к эластомерам, прежде всего для повышения твердости бутадиен-стирольных каучуков [2] и других вулканизатов.
Большой цикл работ посвящен реакции циклораскрывающей метатезисной полимеризации ЦДД. Для этого использовали классические каталитические системы на основе W с алюмоорганическими соединениями и промоторами (эпихлоргидрин, хлоранил) [3, 4], системы на основе WCl6 с тетрафенилпорфирином [5], электрохимически восстановленные системы на основе W и Mo [6], а также катализаторы Шрока W(=CHtBu)-(OtBu)2(NAr), Mo(=CH-t-Bu)(OC(CH3)3)2(NAr), Mo(=CH-t-Bu) (OC(CH3)2CF3)2(NAr), Mo(=CH-t-Bu) (OC(CH3)(CF3)2)2(NAr) [7, 8]. Подробный анализ микроструктуры полимера, получаемого на катализаторах Шрока [8], показал, что высокое содержание транс-звеньев (80–95%), отвечающих за кристалличность полимера, достигается, если вести реакцию в 3М-растворе хлороформа в течение 20 ч при комнатной температуре. Стоит отметить, что активность транс-изомера ЦДД гораздо выше, чем цис-изомера, что было продемонстрировано в кросс-метатезисе ЦДД с гексеном-1 [9].
В условиях поверхностно-инициируемой метатезисной полимеризации ЦДД в газовой фазе с использованием Ru-катализатора Граббса второго поколения [10] снижается вклад реакции переноса цепи, но полностью избежать ее не удается из-за близости расположения молекул на привитой поверхности. ПЦДД с фторалкильными концевыми группами получали полимеризацией ЦДД под действием WCl6/Bu4Sn в присутствии агента переноса цепи C10F21(CH2)6CH=CH(CH2)6C10F21; такие полимеры обладают низкой поверхностной энергией в сочетании с химической стойкостью и хорошими механическими свойствами [11]. Использование в качестве агента передачи цепи цис-1,4-диацетокси-2-бутена позволило синтезировать ПЦДД с концевыми ацетокси-группами, что придало полимеру повышенную устойчивость к окислению на воздухе [12]. Постмодификацией ПЦДД путем исчерпывающего гидроборирования двойных связей и последующего окисления гидроборатов получены строго линейные сополимеры этилена и винилового спирта с заданным распределением спиртовых звеньев [7]. Используя ЦДД в реакции метатезиса с расширением цикла в присутствии катализатора Граббса второго поколения удалось выделить ряд макроциклов [14].
Синтез сополимеров на основе ЦДД ограничен двумя примерами. Сополимеризацией ЦДД с 1,5-циклооктадиеном в присутствии 1,4-диацетокси-2-бутена в качестве агента передачи цепи и катализатора Граббса первого поколения синтезировали случайные сополимеры с концевыми ацетокси-группами, после удаления которых получили дигидрокси-телехелики [12]. Под действием катализатора Граббса третьего поколения проведена чередующаяся метатезисная сополимеризация ЦДД с мономером, содержащим замещенный циклобутеновый фрагмент – бицикло[4.2.0]окт-1(8)-ен-8-карбоксамид [14].
В данной работе впервые показана возможность синтеза статистических мультиблок-сополимеров ЦДД и норборнена (НБ), изучена кинетика процесса и основные факторы, определяющие строение продукта. Интерес к таким сополимерам обусловлен их способностью сочетать в одном материале свойства соответствующих гомополимеров без потери эксплуатационных свойств, характерной для полимерных смесей и даже для диблок-сополимеров, с простотой синтеза, присущей сополимерам с нерегулярным распределением звеньев в цепи [15].
Для синтеза сополимеров ЦДД–НБ использовали реакцию кросс-метатезиса между гомополимерами полинорборненом (ПНБ) и ПЦДД. Макромолекулярный кросс-метатезис – малоизученный процесс, представленный в литературе исследованиями реакции между полидиенами и полиэфирами/полиуретанами, синтезированными по различным механизмам [16–18], между полициклооктенами с различными заместителями [19], между термодинамически несовместимыми полимерами с целью реакционной компатибилизации [20], а также исследованиями кросс-метатезиса в системе ПНБ–полиоктенамер (ПЦО) [21–24]. Нами получен ряд сополимеров, которые трудно синтезировать из мономеров – НБ и циклооктена (ЦО), а также изучены их термические и кристаллические свойства.
Активность ЦДД в метатезисной полимеризации ниже, чем у ЦО, а сометатезис ЦДД и НБ затруднен из-за значительной разницы в энергии напряжения цикла (27.2 ккал/моль для НБ и 11.1 ккал/моль для ЦДД) [10, 25, 26]. Насколько нам известно, по этой причине сополимеры НБ с ЦДД до сих пор не были получены. ПЦДД отличается более высокой степенью кристалличности и температурой плавления по сравнению с другими полиалкенамерами [27–30], а его сополимеры и продукты их дальнейшей модификации по двойным связям могут найти применение в материалах с памятью формы, обладающими температурными переходами плавления и стеклования в различных диапазонах [31], композитах широкого назначения и материалах с повышенной газопроницаемостью [32].
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реагенты
Все опыты с соединениями, чувствительными к воздуху и влаге, проводили на стандартной вакуумной установке и линии Шленка в атмосфере аргона с использованием абсолютированных растворителей. Мономер ЦДД (“Aldrich”) абсолютировали над CaH2. НБ (“Aldrich”) абсолютировали над натрием, использовали в виде 4.6 М раствора в абсолютированном толуоле, который хранили в атмосфере аргона. Катализаторы Граббса первого (G-1, Cl2(PCy3)2Ru=CHPh)
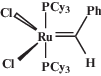
(Cy – циклогексил) и второго (G-2, RuCl2(=CHPh)(H2IMes)(PCy3)) поколения
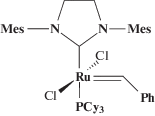 ,
,
где Mes –  (“Aldrich”) применяли без очистки в виде 0.002–0.022 M растворов в абсолютированном
хлороформе. Ингибитор окисления 2,2'-метилен-бис-(6-трет-бутил-4-метилфенол) (“Aldrich”) использовали без очистки. CDCl3 абсолютировали над СаН2. Другие реагенты и растворители (“Aldrich”) очищали и абсолютировали по стандартным
методикам.
(“Aldrich”) применяли без очистки в виде 0.002–0.022 M растворов в абсолютированном
хлороформе. Ингибитор окисления 2,2'-метилен-бис-(6-трет-бутил-4-метилфенол) (“Aldrich”) использовали без очистки. CDCl3 абсолютировали над СаН2. Другие реагенты и растворители (“Aldrich”) очищали и абсолютировали по стандартным
методикам.
Методы измерений
Спектры ЯМР 1Н и ЯМР 13С{1H} регистрировали на спектрометре “Avance 500” (“Bruker”) с рабочей частотой 500.13 и 125.77 МГц соответственно. Параметры съемки спектров ЯМР 13С{1H}: 30° импульс (3 мкс), время сбора данных 1.3 с, релаксационная задержка 1.4 с, размер фида 64 К, размер реального спектра 32 К. Химические сдвиги (м.д. от ТМС) определяли для спектров ПМР относительно остаточного сигнала растворителя (7.28 м.д.), для спектров ЯМР 13C – относительно CDCl3 (77.23 м.д.). По данным метода ЯМР 13С определяли состав сополимера НБ–ЦДД и долю диад различного типа в нем.
Молекулярно-массовые характеристики находили методом ГПХ на жидкостном хроматографе “Waters” с рефрактометрическим детектором и колонкой “Waters Styragel HR 5E” (1–5 мкм, 7.8 × × 300 мм) при 40°C в ТГФ с калибровкой по ПС-стандартам (“Polymer Labs”).
Термические свойства исследовали методом ДСК в калориметре “DSC823e” (“Mettler Toledo”). Нагревание и охлаждение образцов осуществляли со скоростью 10 град/мин в атмосфере аргона со скоростью потока 70 мл/мин в диапазоне температур –100 … +150°C. Результаты измерений обрабатывали с помощью сервисной программы “STARe”. Точность измерения температуры ±0.3К, энтальпии ±1 Дж/г.
Методы синтеза полимеров
Все операции проводили в инертной атмосфере (Ar) с использованием вакуумной техники Шленка.
Полидодеценамер. В вакуумированную и заполненную аргоном двугорлую колбу объемом 20 мл, снабженную магнитной мешалкой, вносили навеску катализатора G-2 массой 1.9 мг (0.0022 ммоля), добавляли 0.04 мл абсолютированного CHCl3 и 422 мг (2.54 ммоля) ЦДД. Спустя 25 мин реакционная смесь полностью теряла подвижность. Через 24 ч реакционную смесь нагревали до 40°C и добавляли 36 мл CHCl3 и винилэтиловый эфир (500 моль/моль G-2) в качестве стоп-агента, еще через 30 мин вводили ингибитор окисления. ПЦДД осаждали в метанол и сушили в вакууме при комнатной температуре (25°С) до постоянной массы. Выход полимера 406 мг, 96%. ЯМР-спектры соответствовали литературным данным [6, 8].
Полинорборнен. ПНБ синтезировали из бицикло[2.2.1]гепт-2-ена (норборнена) метатезисной полимеризацией с раскрытием цикла в присутствии катализатора G-1. В заполненную аргоном круглодонную двугорлую колбу объемом 50 мл, снабженную магнитной мешалкой, помещали навеску катализатора 25.0 мг (0.0304 ммоля) и добавляли 26.6 мл абсолютированного хлороформа. В охлажденный до 0°С раствор катализатора быстро приливали 4.63 мл (21.3 ммоля) раствора НБ в хлороформе и убирали охлаждающую баню. Спустя 1 ч реакционную смесь разбавляли хлороформом (23 мл), добавляли винилэтиловый эфир (500 моль/моль G-1) и через 30 мин вводили ингибитор окисления. Полимер осаждали в этанол и сушили в вакууме при 25°С до постоянной массы. Выход ПНБ составил 1.94 г, 97%. Отнесения сигналов на 1Н и 13С в ЯМР-спектрах ПНБ были сделаны согласно [33, 34].
Кросс-метатезис между ПНБ и ПЦДД
Непосредственно перед опытами по кросс-метатезису исходные гомополимеры очищали от остатков катализатора и ингибитора пропусканием их растворов в хлороформе через колонку с силикагелем (элюент – хлороформ), осаждая очищенный полимер в этанол. Полимеры тщательно сушили в вакууме до постоянной массы не менее 2 суток.
Методика кросс-метатезиса полимеров приведена для мольного соотношения [ПНБ] : [ПЦДД] : : [G-1] = 50 : 50 : 1. Гомополимеры ПНБ и ПЦДД (по 0.28 ммоля) помещали в двугорлую круглодонную колбу объемом 10 мл, оборудованную магнитной мешалкой, вакуумировали и заполняли аргоном, добавляли 0.7 мл абсолютного хлороформа и растворяли полимеры не менее 12 ч. Затем полученный раствор трижды дегазировали, используя цикл замораживание–вакуумирование–оттаивание, и добавляли к нему при постоянном перемешивании 0.35 мл (0.0061 ммоля) раствора катализатора в хлороформе концентрацией 17.4 ммоль/л. Концентрация гомополимеров в растворе составляла 5 мас. %. Реакцию вели при температуре 40°С. В качестве стоп-агента добавляли винилэтиловый эфир в мольном отношении к катализатору 500 : 1, затем через 30 мин вводили ингибитор окисления. Сополимеры выделяли осаждением в этанол с последующей сушкой в вакууме до постоянной массы. Для спектральных исследований реакционную массу концентрировали в вакууме и добавляли CDCl3.
Спектральные характеристики мультиблок-сополимеров:
ЯМР 1Н (500 МГц, CDCl3), δ м.д.: 5.39 (CH=CH, 1Н транс-ПНБ), 5.36 (CH=CH, 2Н, ПЦДД), 5.22 (СН=СН, 1Н, цис-ПНБ), 2.79 (уш. с, 2Н1,4 ПНБ), 2.44 (уш. с, 2Н1,4 ПНБ), 1.98 (4H, CH2), 1.86–1.76 (уш. м, 3Н5а,6а,7а ПНБ), 1.44–1.25 (м, 2Н5в,6в ПНБ, 16H, CH2, ПЦДД), 1.10–1.04 (м, 1Н7в ПНБ).
ЯМР 13C (126 МГц, CDCl3) δ, м.д.: 135.39, 135.36, 135.07, 134.91 (C=C, гетеродиады НБ–ЦДД); 134.12, 134.06. 133.89 (C=C, гомодиады цис-НБ–НБ); 133.29, 133.17, 133.01 (C=C, гомодиады транс-НБ–НБ); 130.51 (C=C, гомодиады транс-ЦДД–ЦДД), 130.04 (C=C, гомодиады цис-ЦДД–ЦДД); 128.8–128.56 (C=C, гетеродиады ЦДД–НБ); 43.56, 43.27 (С1,4 ПНБ); 42.89, 42.26, 41.54 (С7 ПНБ), 38.85, 38.60 (С1,4 ПНБ), 33.02 (С5,6, ПНБ), 33.11 (С5,6, ПНБ), 32.77 (транс-CH2 ПЦДД) 32.58 (С5,6, ПНБ), 32.42 (С7, ПНБ), 29.96, 29.85, 29.81, 29.74, 29.70, 29.50, 29.35 (8 CH2); 27.40 (цис-CH2 ПЦДД).
Мониторинг реакции кросс-метатезиса ПНБ и ПЦДД методом in situ ЯМР 1H
Гомополимеры ПНБ (20 мг, 0.213 ммоля), ПЦДД (35.3 мг, 0.213 ммоля) и 0.51 мл CDCl3 помещали в ЯМР-ампулу Янга (“Aldrich”) и растворяли при 40°С в течение 2 ч, затем оставляли на ночь. Полученную смесь трижды дегазировали и добавляли к замороженному раствору полимеров отдельно приготовленный 0.074 М раствор катализатора G-1 (17 мг, 0.021 ммоля) в CDCl3 (0.28 мл). Смесь размораживали, нагревали и перемешивали, сразу помещали в ЯМР-спектрометр и снимали спектры при температуре 40°С вначале через 15–30 мин, затем каждые 3 ч до 1 суток.
Мониторинг ЯМР 1H взаимодействия гомополимеров с G-1
Гомополимер ПЦДД (35.3 мг, 0.21 ммоля) и 0.53 мл CDCl3 помещали в ЯМР-ампулу Янга и растворяли в течение ночи, затем полученную смесь трижды дегазировали и добавляли к замороженному раствору полимера отдельно приготовленный 0.055 М раствор катализатора G-1 (17.9 мг, 0.022 ммоля) в CDCl3 (0.4 мл). Смесь размораживали, нагревали и перемешивали, сразу помещали в ЯМР-спектрометр и снимали спектры при температуре 40°С вначале через 15–30 мин, затем каждые 3 ч до 1 суток. Аналогичным образом исследовали взаимодействие ПНБ с G-1.
Исследование стабильности катализатора G-1
Катализатор G-1 (17 мг, 0.21 ммоля) помещали в ЯМР-ампулу Янга, вакуумировали, затем добавляли 0.85 мл CDCl3 и перемешивали. Ампулу сразу помещали в ЯМР-спектрометр и снимали спектры при температуре 40°С вначале через 15–30 мин, затем каждые 3 ч до 1 суток.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Синтез ПЦДД и ПНБ
Для проведения макромолекулярного кросс-метатезиса были приготовлены образцы ПЦДД и ПНБ. Исходный гомополимер ПЦДД получали метатезисной полимеризацией ЦДД с раскрытием цикла:
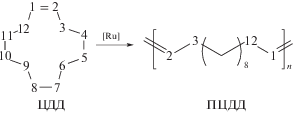
В качестве инициаторов полимеризации ЦДД применяли коммерчески доступные Ru-карбеновые комплексы G-1 и G-2. Была разработана методика и подобраны условия полимеризации ЦДД (табл. 1). В присутствии катализатора G-1 был получен полимер с низким выходом и небольшой ММ (опыт 1), плохо осаждавшийся спиртом. Катализатор G-2 оказался более эффективным. Варьированием времени реакции и мольного соотношения мономер/катализатор были подобраны условия получения ПЦДД с высокими выходом, ММ и содержанием транс-С=С звеньев (опыт 3), который далее использовали в кросс-метатезисе. В табл. 1 также приведены условия синтеза и характеристики ПЦО (опыт 6) и ПНБ (опыты 4, 5), полученных метатезисной полимеризацией ЦО и НБ в присутствии катализатора G-1:
Таблица 1.
Метатезисная полимеризация ЦДД, НБ, ЦО и характеристики полученных гомополимеров ([НБ] = = 0.8 моль/л, [ЦДД] = 4.5 моль/л, [ЦО] = 5 моль/л, Т = 25°С)
| Опыт, № | Катали- затор | Мольное отношение [кат] : [мономер] | Время, ч | Выход, % | Mw × 10–3 | Ð | Tc, °С | Tпл, °С | Цис/транс-С=С |
|---|---|---|---|---|---|---|---|---|---|
| ЦДД | |||||||||
| 1 | G-1 | 1 : 330 | 24 | 22 | 41.4 | 1.5 | – | – | – |
| 2 | G-2 | 1 : 4600 | 2 | 26 | 372 | 2.5 | –56 | 62 | 39/61 |
| 3 | G-2 | 1 : 1150 | 24 | 96 | 406 | 2.5 | –50 | 78 | 15/85 |
| НБ | |||||||||
| 4 | G-1 | 1 : 700 | 1 | 97 | 307 | 2.0 | 39 | отс | 17/83 |
| 5 [21] | G-1 | 1 : 500 | 1 | 99 | 310 | 3.1 | 39 | отс | 12/88 |
| ЦО | |||||||||
| 6 [21] | G-1 | 1 : 700 | 2 | 65 | 225 | 1.8 | –78 | 45 | 35/64 |
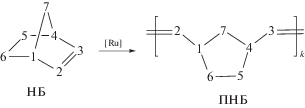
Активность мономеров в метатезисной полимеризации определяется напряженностью раскрываемого цикла, поэтому в присутствии катализатора G-1 выход и Мw полимера уменьшаются в ряду ПНБ–ПЦО–ПЦДД.
Кросс-метатезис в системе ПНБ–ПЦДД–G-1
Реакцию межцепного кросс-метатезиса между ПНБ и ПЦДД проводили в растворе хлороформа под действием катализатора G-1:
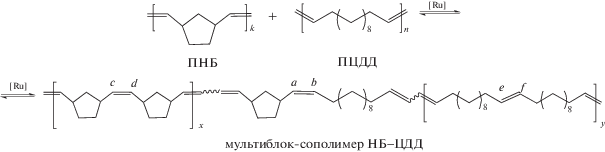
Непосредственно перед этим исходные гомополимеры очищали от остатков катализатора и ингибитора на хроматографической колонке. Одним из условий эффективного межцепного обмена между гомополимерами по схеме кросс-метатезиса является высокая концентрация полимеров в растворе. В противном случае конкурирующим становится процесс внутрицепного метатезиса, приводящий к образованию циклоолигомеров. Ранее для кросс-метатезиса ПНБ и ПЦО в качестве растворителя использовали хлороформ, позволявший готовить квазигомогенные растворы смеси гомополимеров с концентрацией 6–8 мас. % [21–24]. При большей концентрации по данным динамического светорассеяния происходила агрегация цепей ПЦО [22]. Концентрация гомополимеров [ПНБ + ПЦДД] в проводимых опытах была выбрана равной 0.5 моль/л для сравнения с ранее изученным кросс-метатезисом между ПНБ и ПЦО, проводившимся в аналогичных условиях. При попытке повысить суммарную концентрацию гомополимеров ПНБ и ПЦДД до 1 моль/л их смесь набухала, но не растворялась. Особенностью ПЦДД оказалась его более высокая Тпл в сравнении с ПЦО и ограниченная растворимость в хлороформе при комнатной температуре. Полная растворимость ПЦДД в хлороформе наблюдалась при нагревании до 40°С, поэтому приготовление реакционной смеси, проведение экспериментов и съемку ЯМР спектров в основном осуществляли при указанной температуре. После добавления в реакционную смесь раствора катализатора G-1 вязкость смеси снижалась, что обусловлено уменьшением средней ММ гомополимеров за счет взаимодействия катализатора с полимерными цепями. Спустя расчетное время реакцию останавливали добавлением винилэтилового эфира. Полученные новые сополимеры НБ–ЦДД были выделены осаждением в метанол и охарактеризованы методами ЯМР, ДСК и ГПХ (табл. 2). Об образовании сополимеров свидетельствует появление в области двойных связей спектров ЯМР сигналов углерода в гетеродиадах (рис. 1). Отнесения были сделаны на основании литературных данных [34–36] и наших предыдущих исследований [21–24]: сигнал углерода при двойной связи в звене НБ (Сa) гетеродиады НБ–ЦДД находится в более слабом поле относительно аналогичного сигнала в гомодиаде НБ–НБ (Сc,d), а сигнал углерода при двойной связи в звене ЦДД (Сb) гетеродиады ЦДД–НБ сдвинут в более сильное поле относительно этого сигнала в гомодиаде ЦДД–ЦДД (Сe,f).
Таблица 2.
Кросс-метатезис между ПНБ и ПЦДД или ПЦО под действием G-1
| Опыт, № | Мольное соотношение ПНБ : ПЦДД | Способ подготовки образца* | Время, ч | Доля А : В, % | Mw × 10–3, А/В | Ð, А/В | Тс, °С | Тпл, °С | Средняя длина блока | |
|---|---|---|---|---|---|---|---|---|---|---|
| LНБ | LЦДД | |||||||||
| ПНБ–ПЦДД | ||||||||||
| 1 | 1 : 1 | в | 0.67 | 70 : 30 | 26.1/0.8 | 1.5/1.1 | –32 | 59 | 8.5 | 8.5 |
| 2 | 1 : 1 | в | 1.5 | 50 : 50 | 30.0/0.92 | 1.5/1.2 | –40 | 51 | 4.7 | 5.2 |
| 3 | 1 : 1 | в | 5 | 52 : 48 | 31.2/1.20 | 1.5/1.2 | –46 | 42 | 2.7 | 2.6 |
| 4 | 1 : 1 | в | 9 | 70 : 30 | 30.2/0.91 | 1.6/1.1 | –43 | 33 | 2.3 | 2.4 |
| 5 | 1 : 1 | в | 24 | 83 : 17 | 25.6/0.86 | 1.6/1.1 | –41 | 27 | 2.1 | 2.0 |
| 7 | 1 : 1 | о | 1 | 96 : 3 | 92.4/0.74 | 2.2/1.0 | –23 | 66 | 14.5 | 11.6 |
| 8 | 1 : 1 | в | 5 | 84 : 15 | 88.2/1.18 | 1.8/1.2 | –34 | 58 | 7.8 | 7.6 |
| 9 | 1 : 1 | в | 24 | 60 : 39 | 106.5/2.37 | 1.5/1.9 | –47 | 63 | 6.5 | 6.3 |
| 10 | 1.8 : 1 | в | 5 | 33 : 67 | 74.1/1.09 | 1.9/1.2 | –27 | 58 | 13 | 7.5 |
| 12 | 1 : 1 | в | 5 | 86 : 14 | 170/1.34 | 2.1/1.3 | – | – | – | – |
| 13 | 1 : 1 | о | 5 | 99 : 1 | 168/2.35 | 2.3/1.0 | –23 | 73 | 49 | 50 |
| 14 | 1 : 1 | о | 24 | 78 : 22 | 168/0.84 | 2.1/1.2 | –21 | 72 | 31 | 34 |
| 15 | 1.8 : 1 | в | 5 | 89 : 11 | 114/0.74 | 2.1/1.1 | – | – | 29 | 17 |
| 1.8 : 1 | о | 5 | – | – | – | –14 | 66 | – | – | |
| 16 | 1 : 1.8 | в | 5 | 83 : 17 | 165/2.31 | 1.9/2.1 | – | – | – | – |
| 1 : 1.8 | о | 5 | 100 : 0 | 162/нет | 2.1/нет | –30 | 73 | 34 | 48 | |
| ПНБ–ПЦО | ||||||||||
| 17 | 1 : 1 | в | 5 | 47 : 53 | 45/1.13 | 1.6/1.5 | –45 | 38 | 6.5 | 5.4 |
| 18** [21] | 1 : 1 | в | 4.5 | – | 31/– | 3.4/– | – | – | 9.5 | 9.3 |
| 19** [23] | 1 : 1 | в | 24 | – | 45/– | 2.8/– | –35 | 36 | 6.8 | 7.3 |
| 20** [23] | 2 : 1 | в | 24 | – | 52/– | 1.8/– | –31 | отс | 6.2 | 3.4 |
| 21 | 1 : 1 | о | 5 | – | 75/– | 1.9/– | – | – | 18 | 20 |
| 22 [22] | 1 : 1 | о | 5 | 100 : 0 | 101/– | 2.6/– | –32 | 41 | 24 | 22 |
| 23** [21] | 1 : 1 | о | 24 | – | 107/– | 2.9/– | –35 | 45 | 14 | 15 |
Рис. 1.
Область связей С=С спектра ЯМР 13С эквимольной смеси гомополимеров ПНБ и ПЦДД (1) и сополимера НБ–ЦДД (2–4). Исходная концентрация G-1 равна 0.33 мол. % за 5 ч (2), 5 мол. % за 5 ч (3) и 24 ч (4). Т = 40°С.
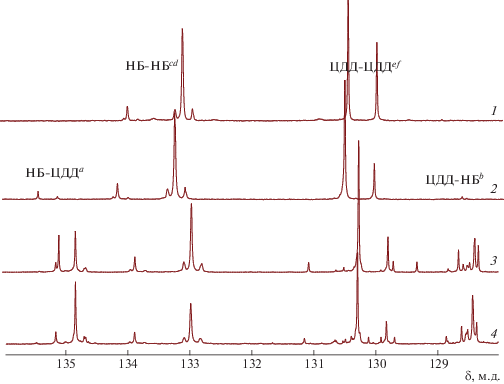
Проводя опыты с повышенным содержанием катализатора, в которых интегральные сигналы гетеродиад больше (рис. 1, спектры 3, 4), и используя специально подобранный режим съемки спектров ЯМР 13С{1H}, удалось выполнить более точные отнесения сигналов в области двойных связей, приведенные в Экспериментальной части. Как и в работах [21, 23], среднюю длину блоков в сополимерах находили из соотношения интегральных интенсивностей сигналов гомо- и гетеродиад:
Из данных ЯМР следует, что при 40°С в результате взаимодействия ПНБ с ПЦДД в присутствии 5 мол. % G-1 длина блоков уже через 5 ч достигает ≈2.5 звеньев, и далее медленно уменьшается, через 24 ч приближаясь к равновесному значению, равному двум, характерному для полностью случайного сополимера эквимольного состава (табл. 2, опыты 3–5; рис. 2).
Рис. 2.
Изменение со временем средней длины блоков ЦДД (1–3) и НБ (1 '–3 ') в ходе кросс-метатезиса при концентрации катализатора G-1 = 0.33 (1, 1 '), 1 (2, 2 ') и 5 мол. % (3, 3 ').

При уменьшении концентрации катализатора за заданное время образуется мультиблок-сополимер с более длинными блоками (табл. 2, опыты 9, 14 и рис. 2). Соотношение средней длины блоков НБ и ЦДД в сополимере можно регулировать, варьируя состав исходной смеси (опыты 15, 16). При увеличении доли ПНБ в исходной смеси при фиксированных концентрациях гомополимеров и катализатора кросс-метатезис идет интенсивнее (опыт 15): степень блочности возрастает, а средняя длина блоков и ММ сополимера, напротив, уменьшаются по сравнению с реакцией в эквимольной исходной смеси (опыты 12, 13). Аналогичное поведение ранее было отмечено для смеси ПНБ и ПЦО. Это согласуется с кинетическими данными, полученными при изучении эволюции концентраций активных центров реакции – Ru-карбеновых комплексов [22]. На начальной стадии кросс-метатезиса комплексы Ru=ПЦО образуются в результате взаимодействия G-1 с ПЦО. В дальнейшем лимитирующей стадией является взаимодействие Ru=ПЦО с макромолекулами ПНБ, поэтому увеличение концентрации ПНБ повышает общую скорость процесса.
Интересно, что на хроматограммах ГПХ сополимеров НБ–ЦДД, выделенных концентрированием реакционной смеси в вакууме, наблюдаются два пика, соответствующих высоко- и низкомолекулярному продуктам реакции (табл. 2, рис. 3). После осаждения в спирт доля низкомолекулярного продукта значительно уменьшается (табл. 2, опыт 7).
Рис. 3.
Кривые ГПХ продуктов кросс-метатезиса в системе ПНБ–ПЦДД–G-1 через 9 (1) и 24 ч (2). [G-1] = 5 мол. %, [ПНБ] : [ПЦДД] = 1 : 1, Т = 40°С.
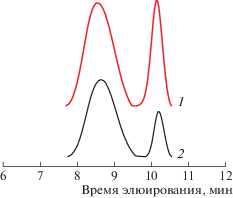
Известно, что при метатезисной полимеризации ЦДД склонен к циклизации в ходе внутримолекулярного метатезиса: на классических W-содержащих катализаторах образуются циклоолигомеры из 2–7 звеньев [34, 37–39], причем методом хроматомасс-спектрометрии зафиксировано даже образование катенанов, сцепленных между собой химически несвязанных циклических молекул [40]. Для наблюдаемого нами низкомолекулярного пика Мw ≈ (1–2) × 103, а ММ мономерного звена ЦДД равна 166, откуда следует, что низкомолекулярную фракцию могут составлять молекулы из 4–6 звеньев ЦДД. Это согласуется с приведенными выше литературными данными. Доля низкомолекулярной фракции зависит от концентрации катализатора и времени реакции (рис. 3, 4).
Рис. 4.
Временная зависимость доли низкомолекулярной фракции В в сополимерах НБ–ЦДД, полученных при исходной концентрации катализатора G-1, равной 5 (1), 1 (2) и 0.33 мол. % (3).
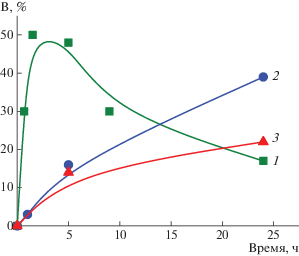
При высокой концентрации катализатора (рис. 4, кривая 1) низкомолекулярные продукты образуются очень быстро, и уже через 1.5 ч их доля достигает 50% от общего количества продуктов реакции. В дальнейшем доля олигомерной фракции уменьшается до 17% (табл. 2, опыт 5). Снижение особенно заметно на последних стадиях реакции, когда средняя длина блоков составляет ≈2 звена. Можно предположить, что вначале происходит образование циклоолигомеров путем внутримолекулярного метатезиса с участием гибких цепей ПЦДД. По мере протекания кросс-метатезиса в структуру олигомеров включаются более жесткие звенья НБ, которые, как известно, препятствуют формированию циклов с малым количеством звеньев [34]. В результате доля низкомолекулярной фракции со временем снижается. При уменьшении количества вносимого катализатора концентрация активных центров падает, а средняя ММ полимера возрастает, из-за чего как скорость образования, так и доля олигомеров в продуктах реакции оказываются заметно ниже (рис. 4, кривые 2, 3).
Ранее в исследованиях кросс-метатезиса между ПНБ и ПЦО при комнатной температуре на хроматограммах ГПХ низкомолекулярных продуктов не наблюдали (табл. 2, опыт 22) [22]. Однако в данной работе при 40°С мы обнаружили появление продуктов c молекулярной массой близкой к ММ олигомеров ЦДД, соответствующей ≈10 звеньям ЦО (опыт 17). Согласно литературным данным, ЦО способен образовывать циклоолигомеры с числом звеньев от 2 до 12 [39, 41–45]. По-видимому, рост температуры увеличивает гибкость цепи ПЦО, что повышает вклад внутримолекулярной реакции на начальной стадии кросс-метатезиса и приводит к более интенсивному образованию циклоолигомеров. Степень блочности сополимеров ЦО–НБ, полученных за заданное время, также возрастает с температурой (ср. опыты 17 и 18, 21 и 22 в табл. 2).
Сравнивая среднюю длину блоков в сополимерах, образующихся в смесях ПНБ с ПЦО и ПЦДД при 40°С, можно заключить, что в первом случае кросс-метатезис протекает быстрее. Разница в активности между ПЦО и ПЦДД в кросс-метатезисе с ПНБ более заметна при низкой концентрации катализатора. По данным табл. 2, при [G-1] = 0.33 мол. % средняя длина блоков равна 18–20 в сополимере НБ–ЦО (опыт 21) и 49–50 в сополимере НБ–ЦДД (опыт 13). На наш взгляд, это различие связано с большей длиной метиленовой последовательности в звене ЦДД, что может снижать полярность полимера и ухудшать его сродство к полярному растворителю (хлороформу). В результате число межцепных контактов в смеси ПЦДД–ПНБ оказывается меньше, чем в смеси ПЦО–ПНБ, о чем свидетельствует более высокая температура плавления ПЦДД по сравнению с ПЦО, и скорость реакции падает. С увеличением концентрации катализатора формируется значительное количество олигомеров, раствор становится более гомогенным и в результате средняя длина блоков в сополимерах НБ–ЦО и НБ–ЦДД принимает близкие значения.
Интересно, что и при комнатной температуре, несмотря на ограниченную растворимость в хлороформе, ПЦДД вовлекается в кросс-метатезис с ПНБ под действием G-1 (табл. 3), о чем свидетельствует появление сигналов гетеродиад в спектрах ЯМР 13С. Вместе с тем широкое ММР и термические свойства, близкие к свойствам ПЦДД, могут указывать на присутствие в продуктах реакции значительного количества непрореагировавших гомополимеров.
Таблица 3.
Кросс-метатезис в системах ПНБ–ПЦДД–G-1 и -ПНБ–ПЦО–G-1 при 25 °С
Сравнение поведения ПЦО и ПЦДД в реакции с ПНБ показывает, что растворимый при комнатной температуре ПЦО реагирует c ПНБ без нагревания более активно, чем ПЦДД, образуя мультиблок-сополимеры НБ–ЦО с более короткими блоками (табл. 3, опыты 2 и 3).
Кинетические исследования
Достоинством Ru-содержащих каталитических систем, помимо стабильности на воздухе и толерантности по отношению ко многим функциональным группам, является возможность наблюдать в спектре ЯМР 1Н сигнал карбенового протона Сl2(PCy3)2Ru=СН-Ph (20 м.д.), что позволяет следить за активными центрами реакции метатезиса. Нами была разработана методика изучения превращений Ru-карбеновых комплексов в ходе кросс-метатезиса полимеров методом мониторинга ЯМР 1Н [22]. На основе экспериментальных данных была предложена кинетическая модель, количественно описывающая кросс-метатезис в системе ПНБ–ПЦО–G-1 при комнатной температуре как совокупность элементарных реакций превращения исходного карбена в полимерный путем разрыва цепи и присоединения активного центра к концевой группе, гибели карбенов и межцепного обмена с участием концевых групп полимера, несущих карбены [22].
В данной работе этот подход применяется к исследованию кросс-метатезиса в системе ПНБ–ПЦДД–G-1. Ввиду ограниченной растворимости ПЦДД в хлороформе при комнатной температуре кинетические исследования с этим полимером проводили при 40°С. Для сравнения аналогичные измерения при указанной температуре выполнены и для системы ПНБ–ПЦО–G-1.
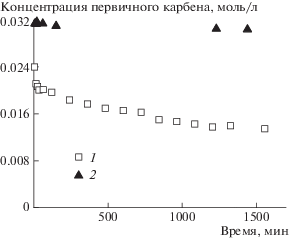
Первоначально была изучена стабильность раствора G-1 в СDCl3 (рис. 5). Катализатор стабилен при 25°С в течение 1 суток, однако при 40°С за первые 2 ч количество активных центров снижается на 15%, а через сутки ≈50% их погибает. Считая распад катализатора реакцией первого порядка, находим из данных рис. 5 ее константу скорости kd = 5.4 × 10–6 с–1.
Далее было исследовано взаимодействие катализатора G-1 с гомополимерами ПЦДД и ПНБ по отдельности:
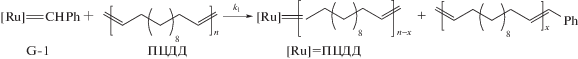
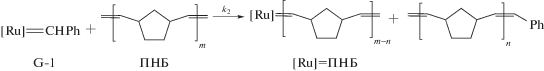
В системе ПЦДД–G-1 на начальной стадии первичный карбен катализатора практически полностью трансформируется в [Ru]=ПЦДД, концентрация которого затем постепенно снижается и спустя 20 ч составляет 50% от максимальной (рис. 6а). Взаимодействие ПНБ с G-1 (40°С) протекает заметно медленнее, так что стадии образования полимерного карбена [Ru]=ПНБ и гибели карбенов перекрываются во времени (рис. 6б). При повышении температуры от 25 до 40°С, скорость всех элементарных реакций возрастает. Сравнивая рис. 6а и рис. 6б, видно, что при 40°С карбен [Ru]=ПНБ гибнет быстрее, чем [Ru]=ПЦДД. Поскольку полимеры находятся как минимум в 20-кратном избытке по отношению к катализатору, можно считать превращение исходного карбена [Ru]=CHPh в полимерный карбен необратимой реакцией первого порядка, равно как и гибель карбена любого типа. Анализ кинетических данных позволяет рассчитать константы скорости образования полимерных карбенов k1 и k2 и их гибели k1d, k2d (табл. 4).
Рис. 6.
Зависимость от времени концентрации исходного карбена [Ru]=CHPh (ромбы) и полимерных карбенов [Ru]=ПЦДД (а) и [Ru]=ПНБ (б) при 40 (темные точки) и 25°С (светлые точки).
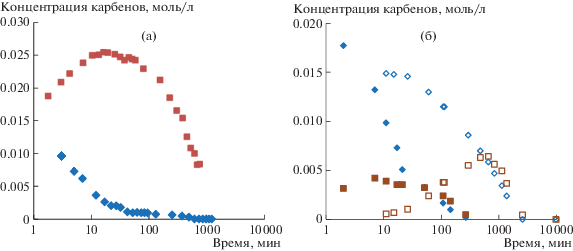
Таблица 4.
Константы скорости элементарных реакций в кросс-метатезисе ПНБ с ПЦДД и ПЦО
| Полимеры | k1, л/(моль с) | k2, л/(моль с) | k1d, c–1 | k2d, c–1 | k211, л/(моль с) | k122, л/(моль с) |
|---|---|---|---|---|---|---|
| ПНБ–ПЦДД | 9.5 × 10–3 | 2.2 × 10–3 | 2.0 × 10–5 | 1.7 × 10–4 | 2.8 × 10–1 | 1.9 × 10–3 |
| ПНБ–ПЦО | 3.1 × 10–2 | 2.2 × 10–3 | 8.2 × 10–5 | 7.5 × 10–4 | 1.7 × 10–1 | 1.7 × 10–3 |
| ПНБ–ПЦО* [22] | 3.1 × 10–3 | 5.4 × 10–5 | 2.6 × 10–6 | 2.4 × 10–5 | 2.2 × 10–2 | 2.2 × 10–4 |
Уравнение для концентрации с0 исходных карбенов имеет вид dc0/dt = –k1cpc0 – kdc0, где первое слагаемое в правой части описывает превращение исходного карбена [Ru]=CHPh в полимерный, второе – его гибель, а cp – концентрация полимера. Поскольку в опытах с ПЦДД kd = 5.4 × 10–6 с–1$ \ll $ k1cp ≈ ≈ 9.5 × 10–3 л/(моль с) × 0.5 моль/л ≈ 5 × 10–3 с–1, самопроизвольный распад катализатора G-1 можно не принимать во внимание.
В растворе смеси полимеров ПНБ–ПЦДД–G-1 полимерные карбены могут вступать в обменные реакции со связями С=С в звеньях другого типа, приводящие вначале к образованию сополимеров, а затем к увеличению степени их блочности:
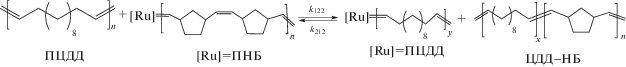
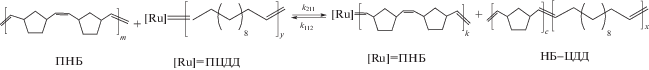
Эти обратимые элементарные реакции характеризуются константами скорости k122, k212, k211 и k112. Система кинетических уравнений, описывающих эволюцию системы в терминах концентраций карбенов и доли гетеродиад в сополимере, приведена в работе [22].
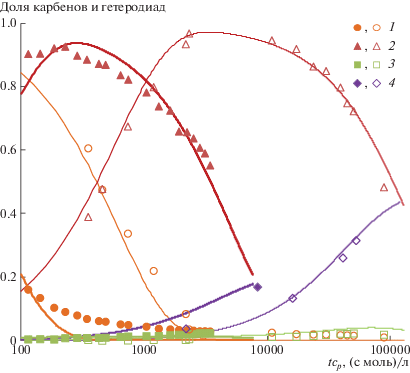
По данным мониторинга ЯМР 1Н (рис. 7), за исключением начальной стадии реакции, в системе ПНБ–ПЦДД–G-1 присутствуют главным образом карбены [Ru]=ПЦДД, в значительно меньшей концентрации карбены [Ru]=ПНБ и исходные карбены [Ru]=СНРh (рис. 8). Несмотря на это, по данным ЯМР 13С, в смеси образуется сополимер НБ–ЦДД, доля гетеродиад в котором за сутки приближается к 0.5. Как уже отмечалось в работе [22], такое поведение возможно, если скорость элементарной реакции k211 между карбеном [Ru]=ПНБ и связью С=С в звене ЦДД существенно (на два порядка) превышает скорость реакции k122 между карбеном [Ru]=ЦДД и связью в звене НБ. Причина этого может быть связана со стерическими факторами и требует дальнейших исследований.
Рис. 7.
Область Ru-карбеновых комплексов спектра ЯМР 1Н в системе ПНБ–ПЦДД–G-1 через 94 мин после начала реакции. [Ru]=ПНБ –18.88, [Ru]=ПЦДД – 19.33, [Ru]=СНРh – 20.05 м.д.
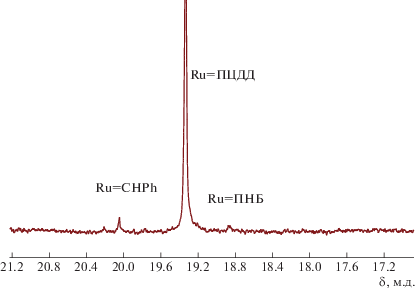
Рис. 8.
Кинетика долей карбенов [Ru]=СНРh (1), [Ru]=ПЦДД (2), [Ru]=ПНБ (3) и гетеродиад НБ–ЦДД (4) в ходе кросс-метатезиса между ПНБ и ПЦДД. Мольное соотношение [ПНБ] : [ПЦДД] : [G-1] = 10 : 10 : 1 , cp = 0.5 моль/л, Т = 40°С. Точки – экспериментальные данные, кривые – расчет по кинетической модели, предложенной в работе [22].

Значения констант скорости обменных и других элементарных реакций, приведенные в табл. 4, позволяют удовлетворительно описать экспериментальные данные по изменению долей карбенов различного типа и гетеродиад в сополимере НБ–ЦДД для системы ПНБ–ПЦДД–G-1 в рамках кинетической модели, разработанной ранее для системы ПНБ–ПЦО–G-1 (рис. 8).
Для корректного сравнения интенсивности кросс-метатезиса в смесях ПНБ с ПЦО и с ПЦДД была изучена кинетика превращений Ru-карбеновых комплексов в системе ПНБ–ПЦО–G-1 при 40°С и проведено ее сопоставление с данными, полученными ранее [21] при 25°С. По описанной выше методике сначала с помощью мониторинга ЯМР 1Н изучали взаимодействие G-1 с гомополимерами ПНБ и ПЦО по отдельности, а затем со смесью этих полимеров. Для смеси использовали также данные ЯМР 13С об изменении доли гетеродиад в сополимере НБ–ЦО, формирующемся в ходе кросс-метатезиса. Из рис. 9 следует, что наша кинетическая модель позволяет единым образом описывать экспериментальные данные для системы ПНБ–ПЦО–G-1 как при 25°С, так и при 40°С. Использованные для этого константы скорости элементарных реакций приведены в табл. 4.
Рис. 9.
Кинетика долей карбенов [Ru]=СНРh (1), [Ru]=ПЦО (2), [Ru]=ПНБ (3) и гетеродиад НБ–ЦО (4) в ходе кросс-метатезиса в системе ПЦО–ПНБ–G-1 при 25 (светлые точки ) и при 40°С (темные точки). Мольное соотношение [ПНБ] : [ПЦДД] : [G-1] = 10 : 10 : 1. Точки – экспериментальные данные, кривые – расчет по кинетической модели, предложенной в работе [22].
Видно, что повышение температуры реакции на 15°С увеличивает скорость образования полимерных карбенов [Ru]=ПЦО (k1) на порядок. Скорость гибели карбенов (k1d, k2d) возрастает в ≈ 30 раз, а скорость межцепного обмена (k211, k122) в ≈ 8 раз. Существенно, в ≈ 40 раз, повышается скорость образования полимерных карбенов [Ru]=ПНБ из [Ru]=CHPh (k2), однако она по-прежнему остается на порядок ниже, чем для карбенов [Ru]=ПЦО.
Отметим также, что при 40°С и исходной концентрации G-1, равной 5 мол. %, скорость всех элементарных реакций для смесей ПНБ с ПЦО и ПЦДД различается незначительно за исключением реакции превращения исходных карбенов в полимерные, когда Ru=[ПЦО] образуется в 3 раза быстрее, чем Ru=[ПЦДД], по-видимому, из-за более высокого термодинамического сродства между ПЦО и хлороформом.
В настоящей работе подробно изучена реакция макромолекулярного кросс-метатезиса в растворе смеси полинорборнен–полидодеценамер в хлороформе под действием катализатора Граббса первого поколения. Основные особенности этой системы по сравнению с парой полинорборнен–полициклооктен связаны с большей длиной метиленовой последовательности в звене ПЦДД, обусловливающей худшее качество растворителя для данного полимера. Впервые синтезированы мультиблок-сополимеры норборнена и циклододецена. Особенности термических и кристаллических свойств этих сополимеров будут представлены в отдельной публикации.
Строение полученных соединений изучено с использованием оборудования Центра коллективного пользования ИНХС РАН и Центра исследования строения молекул ИНЭОС РАН.
Работа выполнена при финансовой поддержке Российского научного фонда (проект 17-73-10450).
Список литературы
Fel’dblyum V.Sh. Synthesis and Application of Unsaturated Cyclic Hydrocarbons Moscow: Khimiya, 1982.
Pat. 4.048.262 USA. 1977.
Vardanyan L.M., Korshak Y.V., Peterina M.P., Dolgoplosk B.A. // Dokl. Akad. Nauk SSSR. 1972. V. 207. P. 345.
Ceausescu E., Cornilescu A., Nicolescu E., Popescu M., Coca S., Cuzmici M., Dragutan V. // J. Mol. Cat. 1988. V. 46. P. 415.
Coca S., Dimonie M., Dragutan V., Ion R., Popescu L., Teodorescu M., Moise F., Vasilescu A. // J. Mol. Cat. 1994. V. 90. P. 101.
Karabulut S., Çetinkaya S., Düz B., Imamoğlu Y. // Appl. Organometal. Chem. 2004. V. 18. P. 375.
Ramakrishnan S. // Macromolecules. 1991. V. 24. P. 3753.
Panagiotis Dounis W., Feast J., Kenwright A.M. // Polymer. 1995. V. 36. № 14. P. 2787.
Bykov V.I., Belyaev B.A., Butenko T.A., Finkel’shtein E.Sh. // Petroleum Chemistry. 2016. V. 56. № 1. P. 62.
Lerum M.F.Z., Chen W. // Langmuir. 2011. V. 27. P. 5403.
McLain S.J., Sauer B.B., Firment L.E. // Macromolecules. 1996. V. 29. P. 8211.
Gascon C., Lucas F., Carlotti S., Deffieux A. // J. Appl. Polym. Sci. 2010. V. 118. P. 1830.
Lee C.W., Choi T.-L., Grubbs R.H. // J. Am. Chem. Soc. 2002. V. 124. P. 3224.
Zhang J., Li G., Sampson N.S. // ACS Macro Lett. 2018. V. 7. P. 1068.
Gringolts M.L., Denisova Y.I., Finkelshtein E.Sh., Kudryavtsev Y.V. // Beilstein J. Org. Chem. 2019. V. 15. P. 218.
Otsuka H., Muta T., Sakada M., Maeda T., Takahara A. // Chem. Commun. 2009. № 9. P. 1073.
Maeda T., Kamimura S., Ohishi T., Takahara A., Otsuka H. // Polymer. 2014. V. 55. № 24. P. 6245.
Ohishi T., Suyama K., Kamimura S., Sakada M., Imato K., Kawahara S., Takahara A., Otsuka H. // Polymer. 2015. V. 78. P. 145.
Radlauer M.R., Matta M.E., Hillmyer M.A. // Polym. Chem. 2016. V. 7. № 40. P. 6269.
Descour C., Macko T., Schreur-Piet I., Pepels M.P.F., Duchateau R. // RSC Adv. 2015. V. 5. № 13. P. 9658.
Gringolts M.L., Denisova Yu.I., Shandryuk G.A., Krentsel L.B., Litmanovich A.D., Finkelshtein E.S., Kudryavtsev Y.V. // RSC Adv. 2015. V. 5. № 1. P. 316.
Denisova Yu.I., Gringolts M.L., Peregudov A.S., Krentsel L.B., Litmanovich E.A., Litmanovich A.D., Finkelshtein E.Sh., Kudryavtsev Y.V. // Beilstein J. Org. Chem. 2015. V. 11. P. 1796.
Denisova Yu.I., Gringolts M.L., Krentsel’ L.B., Shandryuk G.A., Litmanovich A.D., Finkelshtein E.Sh., Kudryavtsev Y.V. // Polymer. Science B. 2016. V. 58. № 3. P. 292.
Shandryuk G.A., Denisova Yu.I., Gringolts M.L., Krentsel L.B., Litmanovich A.D., Finkelshtein E.Sh., Kudryavtsev Y.V. // Eur. Polym. J. 2017. V. 86. P. 143.
Walker R., Conrad R.M., Grubbs R.H. // Macromolecules. 2009. V. 42. № 3. P. 599.
Schleyer P.v.R., Williams J.E., Blanchard K.R. // J. Am. Chem. Soc. 1970. V. 92. P. 2377.
Keller A., Martuscelli E. // Makromol. Chem. 1971. B. 141. S. 189.
Keller A., Martuscelli E. // Makromol. Chem. 1972. B. 151. S. 169.
Martuscelli E., Vittoria V. // Polymer. 1972. V. 13. P. 360.
Werden K.V., Holland-Moritz K. // Colloid Polym. Sci. 1981. V. 259. P. 731.
Liu C., Qin H., Mather P. T. // J. Mater. Chem. 2007. V. 17. P. 1543.
Finkelshtein E.S., Bermeshev M.V., Gringolts M.L., Starannikova L.E., Yampolskii Y.P. // Russ. Chem. Rev. 2011. V. 80. № 4. P. 341.
Floros G., Saragas N., Paraskevopoulou P., Psaroudakis N., Koinis S., Pitsikalis M., Hadjichristidis N., Mertis K. // Polymers. 2012. V. 4. P. 1657.
Ivin K.J., Mol J.C. Olefin Metathesis and Metathesis Polymerization. London: Acad. Press, 1997.
Lichtenheldt M., Wang D., Vehlow K., Reinhardt I., Kuhnel C., Decker U., Blechert S., Buchmeiser M.R. // Chem. Eur. J. 2009. V. 15. P. 9451.
Amir-Ebrahimi V., Rooney J.J. // J. of Molecular Catalysis, Chem. 2004. V. 208. P. 115.
Wolovsky R. // J. Am. Chem. Soc. 1970. V. 92. № 7. P. 2132.
Кутейников В.М., Коршак Ю.В., Долгоплоск Б.А. // Тр. Московского хим.-технол. ин-та, 1975. Т. 86. С. 117.
Höcker H., Reimann W., Riebel K., Szentivanyi Z. // Makromol. Chem. 1976. V. 177. P. 1707.
Ben-Efraim D.A., Batich C., Wassermaт E. // J. Am. Chem. Soc. 1970. V. 92. № 7. P. 2133.
Höcker H., Müsch R. // Macromol. Chem. 1972. B 157. S. 201.
Höcker H., Müsch R. // Macromol. Chem. 1974. B. 175. S. 1395.
Höcker H., Reif L., Reinmann W., Riebel K. // Rec. Trav. Chim. Pays-Bas. 1977. V. 96. P. M47.
Höcker H. // J. Mol. Cat. 1991. V. 65. P. 95.
Reif L., Höcker H. // Macromolecules. 1984. V. 17. P. 952.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия С)