

Высокомолекулярные соединения (серия С), 2019, T. 61, № 1, стр. 110-121
КРОСС-МЕТАТЕЗИС МЕЖДУ ПОЛИНОРБОРНЕНОМ И ПОЛИ(5,6-ЭПОКСИ-1-ОКТЕНАМЕРОМ)
А. В. Роенко a, *, Ю. И. Денисова a, М. Л. Грингольц a, А. С. Перегудов b, Г. А. Шандрюк a, Е. Ш. Финкельштейн a, Я. В. Кудрявцев a
a Институт нефтехимического синтеза им. А.В. Топчиева Российской академии наук
119991 Москва, Ленинский пр., 29, Россия
b Институт элементоорганических соединений им. А.Н. Несмеянова Российской академии наук
119991 Москва, ул. Вавилова, 28, Россия
* E-mail: royenko@ips.ac.ru
Поступила в редакцию 14.03.2019
После доработки 02.04.2019
Принята к публикации 10.04.2019
Аннотация
Изучена макромолекулярная реакция межцепного кросс-метатезиса между полинорборненом и поли(5,6-эпокси-1-октенамером) в присутствии катализатора Граббса первого поколения. Химически стабильный поли(5,6-эпокси-1-октенамер) получен метатезисной полимеризацией 5,6-эпоксициклоокт-1-ена на рутениевом катализаторе Граббса второго поколения. Синтезированы и охарактеризованы методами ЯМР, ГПХ, ДСК новые статистические мультиблок-сополимеры норборнена и 5,6-эпоксициклоокт-1-ена с различной средней длиной блоков, определяемой временем реакции, типом растворителя, концентрацией катализатора и исходных гомополимеров. Несмотря на удаленность эпокси-заместителя от реакционного центра – двойной связи, поли(5,6-эпокси-1-октенамер) демонстрирует меньшую активность в реакции кросс-метатезиса с полинорборненом по сравнению с незамещенным полиоктенамером. Изучено влияние эпокси-групп в основной цепи гомополимеров и мультиблок-сополимеров норборнена и циклооктена на их термические характеристики. Показано, что температуры стеклования и плавления увеличиваются с повышением концентрации эпокси-групп при введении оксиранового фрагмента как в гомо-, так и в сополимеры. Кристалличность в поли(5,6-эпокси-1-октенамере) возрастает при увеличении содержания транс-связей С=С в нем.
ВВЕДЕНИЕ
Реакция метатезиса олефинов, открытая лишь в середине ХХ века, в настоящее время превратилась в эффективный инструмент органического и полимерного синтеза. Общепринятая классификация разновидностей реакции метатезиса в зависимости от типа субстрата и особенностей протекания реакции включает в себя гомометатезис, сометатезис, циклообразующий метатезис диенов (RCM), полимеризацию циклоолефинов с раскрытием цикла (ROMP), полимеризацию ациклических диенов (ADMET) и т.д. [1]. В последнее десятилетие получило развитие новое направление – реакция кросс-метатезиса между полимерными цепями, или макромолекулярный кросс-метатезис (МКМ) [2–14]. Это новая реакция межцепного обмена, приводящая к образованию статистических мультиблок-сополимеров – перспективной основы для создания наноматериалов, сочетающих разнообразные функциональные свойства с долговечностью и высокой механической прочностью [15]. Показано, что МКМ эффективен в синтезе сополимеров из гомополимеров, полученных по различным механизмам, например из полибутадиена или натурального полиизопрена и ненасыщенных полиэфиров или полиуретанов, синтезированных ступенчатой полимеризацией [2–4]. Данную реакцию успешно применяли для компатибилизации несовместимых полимеров [16] и улучшения податливости сшитого полибутадиена [17]. Наши недавние исследования на примере норборнена (НБ) и циклооктена (ЦО) продемонстрировали, что МКМ позволяет синтезировать сополимеры блочного строения, которые трудно получить из мономеров, обладающих значительной разницей в полимеризационной активности [9–14]. В цикле работ, посвященном изучению закономерностей кросс-метатезиса между полинорборненом (ПНБ) и полициклооктеном (ПЦО), показано, что строение и свойства мультиблок-сополимеров можно регулировать, варьируя условия реакции (время, концентрация катализатора, соотношение гомополимеров) [9, 10]. Использование в качестве катализатора Ru-карбеновых комплексов Граббса дало возможность с помощью in situ мониторинга методом ЯМР 1H изучить превращения активных центров реакции – металлокарбенов и на основе полученных данных разработать кинетическую модель МКМ в смеси ПНБ с ПЦО [8]. Изменяя строение мультиблок-сополимеров НБ–ЦО, можно регулировать их термические и кристаллические свойства [11]. Введение функциональных заместителей в сополимеры расширяет возможности их практического использования. Нами начаты исследования по синтезу функциональных мульти-блок-сополимеров путем модификации мономеров, из которых получают исходные гомополимеры для кросс-метатезиса [12–14]. Таким способом были введены боковые триметилсилильные группы в блок норборнена, способные улучшать газоразделительные свойства [12], и гидроксильные группы в блок циклооктена, придающие полимерам гидрофильность [13, 14]. Стоит отметить, что введение заместителей в циклооктен приводит к еще большему снижению напряженности цикла [1, 18, 19], что делает практически невозможным сометатезис замещенных циклооктенов с норборненом в присутствии коммерчески доступных катализаторов.
В продолжение исследований по синтезу функциональных мультиблок-сополимеров НБ–ЦО из замещенных циклооктенов по реакции МКМ в настоящей работе получены мультиблок-сополимеры из 5,6-эпоксициклоокт-1-ена. Известно, что введение оксиранового кольца в цепь полимера улучшает его адгезионные и термические свойства, придает метатезисным полимерам стабильность при хранении [20–22]. Будучи перспективными для синтеза так называемых “умных” материалов, эпоксидированные полимеры используются для получения самозаживляемых материалов и обладающих памятью формы [23, 24], находят применение в покрытиях [25]. В литературе описано введение оксирановых групп в ПНБ и ПЦО эпоксидированием полимеров [26–30]. Эпоксидирование ПНБ проводили для изучения влияния модификации на газоразделительные свойства, в связи с чем была разработана методика, позволяющая достичь количественного выхода [26, 28]. В ПЦО удавалось заменить на оксирановые фрагменты не более 30–40% двойных связей, что связано, скорее всего, с тенденцией эпоксидированного ПЦО к сшиванию [30].
Недавно мы провели эпоксидирование мультиблок-сополимеров НБ–ЦО различного строения, полученных с помощью МКМ [31]. Оказалось, что эпоксидирование повышает температуру стеклования и плавления сополимеров, причем кристаллические свойства сополимера определяют только блоки циклооктена. Процессу исчерпывающего эпоксидирования сополимера сопутствует его деструкция, которая особенно выражена для блоков норборнена.
В настоящей работе применен другой способ синтеза мультиблок-сополимеров НБ–ЦО, содержащих оксирановые циклы в цепи, а именно, введение эпокси-группы в мономерное звено ЦО на стадии синтеза мономера, полимеризация 5,6-эпоксициклоокт-1-ена (ЭЦО) и кросс-метатезис полученного поли(5,6-эпокси-1-октенамера) (ПЭЦО) с ПНБ. Такой подход позволяет направленно вводить эпоксидные фрагменты только в блок циклооктена при сохранении двойных связей в блоке норборнена. Получаемые сополимеры предоставляют широкие возможности их дальнейшей модификации как по двойным связям (сшивка), так и по эпокси-группам.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реагенты
Все операции с соединениями, чувствительными к воздуху и влаге, проводили на стандартной вакуумной установке и линии Шленка в атмосфере аргона с использованием абсолютированных растворителей.
Мономеры, норборнен (“AcrosOrganics”) и 1,5-циклооктадиен (ЦОД) (“Aldrich”), сушили над натрием, перегоняли и хранили в атмосфере аргона. Катализаторы Граббса первого (Гр-1) [Cl2(PCy3)2Ru=CHPh]
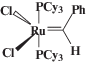
(Cy – циклогексил) и второго поколения (Гр-2) [Cl2(H2IMes)(PCy3)Ru=CHPh]
 ,
,
(Mes –  ) использовали без дальнейшей очистки в виде 0.002–0.022 M растворов в хлороформе.
Мета-хлорпербензойную кислоту (ХПБК) (“Aldrich”), ингибитор окисления 2,2´-метилен-бис-(6-трет-бутил-4-метилфенол) (“Aldrich”) применяли также без очистки. Винилэтиловый эфир абсолютировали
кипячением над Na и перегоняли в аргоне. Гексан, CНCl3,CDCl3 абсолютировали над СаН2. Другие реагенты и растворители (“Aldrich”) очищали по стандартным методикам.
) использовали без дальнейшей очистки в виде 0.002–0.022 M растворов в хлороформе.
Мета-хлорпербензойную кислоту (ХПБК) (“Aldrich”), ингибитор окисления 2,2´-метилен-бис-(6-трет-бутил-4-метилфенол) (“Aldrich”) применяли также без очистки. Винилэтиловый эфир абсолютировали
кипячением над Na и перегоняли в аргоне. Гексан, CНCl3,CDCl3 абсолютировали над СаН2. Другие реагенты и растворители (“Aldrich”) очищали по стандартным методикам.
Методы измерений
Состав сополимера НБ–ЭЦО и доли диад различного типа в нем находили методом ЯМР 13С для растворов в CDCl3. Спектры ЯМР 1Н и ЯМР 13С{1H} регистрировали на спектрометре “Avance 500TM” фирмы “Bruker” с рабочей частотой 500.13 и 125.77 МГц соответственно. Параметры съемки в случае спектров ЯМР 13С{1H} были следующие – 300 (3 мкс) импульс, время сбора данных 1.3 с, релаксационная задержка 1.4 с, размер фида 64 K, размер реального спектра 32 K. Химические сдвиги (м.д.) определяли для спектров ПМР относительно остаточного сигнала растворителя (7.28 м.д.), для спектров ЯМР 13C – относительно CDCl3 (77.23 м.д.) и пересчитывали к тетраметилсилану. Отнесения сигналов гомополимеров выполняли согласно литературным данным c учетом микроструктуры цепи: транс- (133.01) и цис- (133.8) связи С=С для ПНБ [1], сигналы связи С=С в области 130.1–129.5 м.д. для ПЭЦО [32].
ИК-спектры регистрировали на Фурье-спектрометре “Nicolet 5700”.
Молекулярно-массовые характеристики полимеров измеряли методом гель-проникающей хроматографии на модульном жидкостном хроматографе высокого давления, оснащенном насосом “LabAlliance Series 1500 Constant Flow Pump” (“Scientific Systems”, Великобритания) и детектором “Refractive Index Detector 2142” (LKB, Швеция). Использовали систему последовательно соединенных колонок Waters WAT054460 и Tosoh Biosep G3000HHR со сшитым ПС в качестве наполнителя. Элюентом служил ТГФ со скоростью подачи 1 мл/мин, температура колонки 25°C, концентрация образца 1 мг/мл, объем пробы 100 мкл. Калибровку прибора выполняли по ПС-стандартам (“PolymerLabs”).
ДСК-измерения проводили в калориметре “DSC823e” (“MettlerToledo”). Нагревание и охлаждение образцов осуществляли со скоростью 10 град/мин в атмосфере аргона со скоростью потока 70 мл/мин в диапазоне –100…+150°C. Результаты измерений обрабатывали с помощью сервисной программы STARe, поставляемой в комплекте с прибором. Точность измерения при определении температуры и энтальпии составляла ±0.3°C и ±1 Дж/г соответственно.
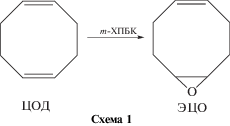
Синтез 5,6-эпоксициклоокт-1-ена (ЭЦО) [33]
В трехгорлую колбу, снабженную магнитной мешалкой и капельной воронкой, загружали ЦОД (42.4 г, 0.3926 моля). Отдельно приготовленный раствор ХПБК (108 г, 0.3755 моля) в хлороформе (900 мл) с помощью капельной воронки приливали к ЦОД, охлаждаемому ледяной баней, в течение 2 ч при интенсивном перемешивании. На следующий день образовавшийся белый осадок отфильтровывали, избыток хлороформа удаляли при пониженном давлении. Органический слой поочередно промывали насыщенными водными растворами NaHCO3 и NaCl (по 2 раза), и оставляли сушить над безводным MgSO4, растворитель удаляли при пониженном давлении. Полученный продукт очищали колоночной хроматографией на силикагеле, в качестве элюента использовали смесь гексана и этилацетата в соотношении 9:1 (по объему), затем перегоняли при пониженном давлении (Tкип = 55°C при 7 мм рт. ст). Получили 23.4 г 5,6-эпоксициклоокт-1-ена (ЭЦО) с выходом 48% и чистотой 99.6% согласно ГЖХ.
ЯМР 1H (CDCl3), δ, м.д.: 5.5 (м, 2H), 3.02 (м, 2H), 2.42 (м, 2H), 2.04 (м, 6H).
Синтез поли(5,6-эпокси-1-октенамера) (ПЭЦО)
В двугорлую колбу, снабженную магнитной мешалкой, помещали катализатор Гр-2 (3.5 мг, 0.00412 ммоля) и абсолютированный хлороформ (0.06 мл). Далее к катализатору при перемешивании приливали мономер ЭЦО (0.2166 г, 1.747 ммоля). Через 1.5 ч добавляли 6 мл хлороформа, винилэтиловый эфир в качестве стоп-агента (1:500 по отношению к катализатору), а также ингибитор окисления. Полученную смесь перемешивали в течение 1 ч до полного растворения полимера. Выделяли ПЭЦО осаждением в гексан, сушили в вакууме до постоянной массы. Выход полимера 0.2 г, 92%. ПЭЦО охарактеризован методами ЯМР, ГПХ, ДСК (табл. 1).
Таблица 1.
Полимеризация ЭЦО и НБ
| Мономер | [Кат] | Мольное соотношение [Кат] : [M] | Время, ч | Mn × 10–3 | Ð | Доля транс связей С=С, % | Tс, °С | Tпл, °С | Выход, % |
|---|---|---|---|---|---|---|---|---|---|
| ЭЦО | Гр-1 | 1 : 350 | 4.5 | –* | –* | 52 | –49 | Нет | 32 |
| ЭЦО | Гр-2 | 1 : 400 | 1.5 | 207 | 2.3 | 72 | –49 | Нет | 90 |
| ЭЦО | Гр-2 | 1 : 460 | 3.0 | 250 | 2.4 | 84 | –48 | 47 | 84 |
| НБ | Гр-1 | 1 : 700 | 1.0 | 181 | 2.0 | 86 | 37 | Нет | 90 |
ЯМР 1Н (600 MГц, CDCl3), δ м.д.: 5.49, 5.44 (2H, С1Н=С2Н), 2.91 (м, 2H, НС5–О–С6Н), 2.19 (м, 4H, С3,8Н2), 1.56 (м, 4H, С4,7Н2).
ЯМР 13C (150.9 MГц, CDCl3) δ м.д.: 130.07 (С1,2транс), 129.52 (С1,2цис); 56.86, 56.83 (С5,6); 29.69 (С3,8транс); 28.01, 27.92 (С4,7); 24.47 (С3,8цис).
Синтез полинорборнена (ПНБ)
В заполненную аргоном круглодонную двугорлую колбу объемом 50 мл, снабженную магнитной мешалкой, помещали навеску катализатора Гр-1 (0.0102 г, 0.0122 ммоля) и добавляли 8.8 мл абсолютированного хлороформа. В охлажденный до 0°С раствор катализатора быстро приливали 1.9 мл (8.6 ммоля) раствора норборнена в хлороформе. Охлаждающую баню убирали сразу же после добавления мономера. Через 1 ч реакционную смесь разбавляли хлороформом (26 мл), добавляли в качестве стоп-агента винилэтиловый эфир и ингибитор окисления. Полимер осаждали этанолом и сушили в вакууме до постоянной массы. Выход ПНБ составил 95% (0.77 г).
Сигналы в спектрах ЯМР 1Н и ЯМР 13С полинорборнена соответствовали литературным данным [1].
Непосредственно перед экспериментами ПНБ очищали от ингибитора и остатков катализатора пропусканием его раствора в хлороформе через колонку с силикагелем, осаждали в этанол и далее тщательно сушили в вакууме.
Кросс-метатезис между ПНБ и ПЭЦО
Методика приведена для мольного соотношения [ПНБ] : [ПЭЦО] : [кат] = 10 : 10 : 1. Гомополимеры ПНБ и ПЭЦО (по 0.25 ммоля в расчете на мономерное звено) помещали в двугорлую круглодонную колбу объемом 10 мл, оборудованную магнитной мешалкой, вакуумировали и заполняли аргоном, добавляли 0.59 мл хлороформа и растворяли полимеры в атмосфере аргона не менее 12 ч. Затем полученный раствор трижды дегазировали и заполняли аргоном, приливали к нему при постоянном перемешивании 0.37 мл раствора катализатора (0.026 ммоля) в хлороформе. Концентрация гомополимеров в растворе составляла 0.5 моль/л. В качестве стоп-агента использовали винилэтиловый эфир, который вводили в мольном отношении к катализатору 500 : 1 по истечении расчетного времени, через 30 мин в реакционную смесь вводили ингибитор окисления. Сополимеры выделяли концентрированием в вакууме или осаждением в этанол с последующей сушкой в вакууме до постоянной массы. Для спектральных исследований реакционную массу концентрировали в вакууме и добавляли CDCl3.
Сополимер НБ–ЭЦО:
ЯМР 1H (500 MГц, CDCl3) δ, м.д.: 5.50–5.20 (4H, ЭЦО=ЭЦО, НБ=НБ, НБ=ЭЦО), 2.90 (2H, НС–О–CH, ПЭЦО), 2.78 (1H, С1Н–СН=СН ПНБ), 2.42 (1H, С4Н–СН=СН ПНБ), 2.23–2.16 (м, 4H, ПЭЦО), 1.85 (м, 1H, ПНБ), 1.76 (м, 2H, ПНБ), 1.56 (м, 4H, ПЭЦО), 1.34 (м, 2H, ПНБ), 1.06 (м, 1H, ПНБ).
ЯМР 13C (CDCl3) δ, м.д.: 136.38–135.73 (C=C, гетеродиады НБ–ЭЦО), 134.08, 133.94, 133.84 (C=C, гомодиады НБ–НБ, цис), 133.20, 133.08, 132.96 (C=C, гомодиады НБ–НБ, транс), 130.05 (C=C, гомодиады ЭЦО–ЭЦО, транс), 129.51 (C=C, гомодиады ЭЦО–ЭЦО, цис), 127.33–126.1 (C=C, гетеродиады ЭЦО–НБ), 56.85 (С5,С6 ПЭЦО); 43.50, 43.26, 43.22 (C1,4 ПНБ); 42.82–41.21 (С7 ПНБ); 38.73, 38.48 (C1,4 ПНБ), 32.99, 32.42, 32.28, 32.16 (C5,6 ПНБ), 29.66, 29.59 (С3,8 ПЭЦО), 28.02, 27.92 (С4,7 ПЭЦО), 24.47 (С3,8 ПЭЦО).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Синтез мономеров и гомополимеров
ЭЦО синтезировали эпоксидированием 1,5-циклооктадиена мета-хлорпербензойной кислотой по описанной в литературе методике [33]
Чистота полученного мономера, согласно ГЖХ, составила 99.6%.
Cинтез гомополимера ПЭЦО по схеме метатезиса при использовании коммерчески доступных катализаторов представляет определенные трудности. Так, в работе [33] полимеризацию ЭЦО под действием катализатора Гр-1 осуществить не удалось. В работах [32, 34] ПЭЦО был получен с выходом 45–80% и Мw = (11.4–13.0) × 104 на специально синтезированном комплексе рутения [RuCl2(p-cymene)]2. Описана сополимеризация ЭЦО с 5,6-(гем-дихлорциклопропан)циклоокт-1-еном [35].
В данной работе гомополимер ПЭЦО получали метатезисной полимеризацией с раскрытием цикла ЭЦО в присутствии катализаторов Гр-1 и Гр-2.
Полимеризацию осуществляли практически без использования растворителя, в массе мономера:
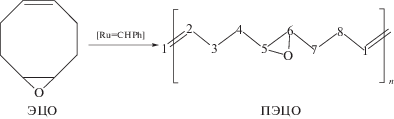
Данный подход позволяет снизить количество циклоолигомеров, образующихся в результате побочной реакции внутримолекулярного метатезиса, повысить выход и ММ полимера.
Гомополимер ПЭЦО, синтезированный с использованием катализатора Гр-1, теряет растворимость после осаждения метанолом или гексаном и непродолжительной (∼20 мин) сушки в вакууме. Такое поведение, вероятно, обусловлено сшивкой ПЭЦО вследствие раскрытия оксиранового кольца под действием метанола, содержащейся в нем воды и рутениевого катализатора Гр-1, который представляет собой слабую кислоту Льюиса. Уменьшение времени реакции с 24 до 4.5 ч существенно не улучшило растворимость ПЭЦО, при этом его выход снизился с 50 до 30%.
В присутствии катализатора Гр-2 в тех же условиях ПЭЦО был получен с выходом 90%. При этом образец ПЭЦО после осаждения гексаном и использования абсолютированного винилэтилового эфира растворился в отличие от ПЭЦО, осажденного метанолом. Время хранения такого ПЭЦО в инертной атмосфере при комнатной температуре без потери растворимости составило до двух недель, его характеристики приведены в табл. 1. При более длительном хранении полимер терял растворимость, возможно, из-за сшивки вследствие раскрытия оксирановых колец, катализируемого остатками катализатора в полимере. Попытки удаления остатков катализатора адсорбционной очисткой на силикагеле приводили к сшивке ПЭЦО.
Интересно, что микроструктура ПЭЦО, получаемого на катализаторах Гр-1 и Гр-2, различна. Под действием катализатора Гр-2 образуется полимер с бòльшим содержанием двойных транс-связей (табл. 1). Такой полимер на термограмме ДСК после первого прогревания демонстрирует два слабых пика плавления, в отличие от ПЭЦО, синтезированного с участием катализатора Гр-1, что свидетельствует о влиянии двойных транс-связей на кристалличность ПЭЦО.
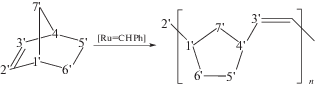
ПНБ получали метатезисной полимеризацией с раскрытием цикла в присутствии катализатора Гр-1 и характеризовали методами ЯМР, ГПХ и ДСК (табл. 1):
Кросс-метатезис между ПНБ и ПЭЦО
Непосредственно перед экспериментами ПНБ очищали от ингибитора и остатков катализатора пропусканием его раствора в хлороформе через колонку с силикагелем, осаждали этанолом и далее тщательно сушили в вакууме. Гомополимер ПЭЦО синтезировали незадолго до эксперимента для предотвращения потери растворимости.
Кросс-метатезис между ПНБ и ПЭЦО проводили в растворе хлороформа, в котором растворимы оба полимера. К раствору гомополимеров добавляли раствор катализатора, реакцию обрывали винилэтиловым эфиром. Конечную смесь вакуумировали через 3–5 ч или на следующий день для удаления легкокипящих компонентов и анализировали методом ЯМР 13С. В отдельных экспериментах сополимер осаждали гексаном (табл. 2, опыт 3). В качестве катализатора использовали Гр-1 по нескольким причинам. Во-первых, он является более доступным; во-вторых, синтезированные в его присутствии сополимеры НБ–ЭЦО не теряли растворимость; в-третьих, ранее полученные сополимеры НБ–ЦО также были синтезированы на Гр-1. После добавления катализатора к смеси гомополимеров вязкость раствора снижалась вследствие уменьшения средней ММ при взаимодействии катализатора с двойными связями полимерных цепей с образованием Ru-карбеновых полимерных комплексов [8]. В результате взаимодействия этих комплексов с ПНБ или ПЭЦО происходит обмен сегментами полимерных цепей с образованием сначала диблок-, а затем мультиблок-сополимеров:
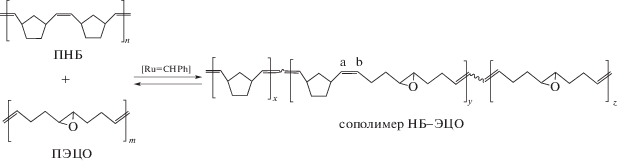
Таблица 2.
Влияние условий реакции кросс-метатезиса между ПНБ и ПЭЦО под действием катализатора Гр-1 на характеристики мультиблок-сополимеров НБ – ЭЦО
| Опыт, № | Сополимер | [Кат], мол. % | Мольное соотношение ПНБ : ПЭЦО | Mn × 10–3 | Ð | Соотношение транс/цис связи С=С в ЭЦО-блоке, % | Длина блока | |
|---|---|---|---|---|---|---|---|---|
| LНБ | LЭЦО | |||||||
| 1 | НБ–ЭЦО | 5 | 1 : 1 | 8.4 | 1.7 | 78/22 | 8.4 | 7.8 |
| 2 | НБ–ЭЦО | 1 | 1 : 1 | н/о | н/о | 83/17 | 24 | 21 |
| 3* | НБ–ЭЦО | 1 | 1 : 1 | 28.9 | 1.6 | 80/20 | 23 | 13 |
| 4** | НБ–ЭЦО | 1 | 1 : 1 | 25.4 | 2.0 | 81/19 | 10 | 10 |
| 5** | НБ–ЭЦО | 1 | 2 : 1 | 40.6 | 1.7 | 82/18 | 12 | 6 |
| 6*** | НБ–ЭЦО | 1 | 1 : 1 | 35.0 | 2.0 | 82/18 | 69 | 51 |
| 7 | НБ–ЭЦО | 1 | 1 : 2 | 31 | 1.9 | 83/17 | 10 | 17 |
| 8 | НБ–ЦО [9] | 5 | 1 : 1 | 6.0 | 1.8 | 80/20 | 2.0 | 2.3 |
| 9 | НБ–ЦО [9] | 1 | 1 : 1 | 19.0 | 2.4 | 80/20 | 11 | 11 |
| 10 | НБ-ЦО [10] | 1 | 2 : 1 | 29.0 | 1.8 | 79/21 | 6.2 | 3.4 |
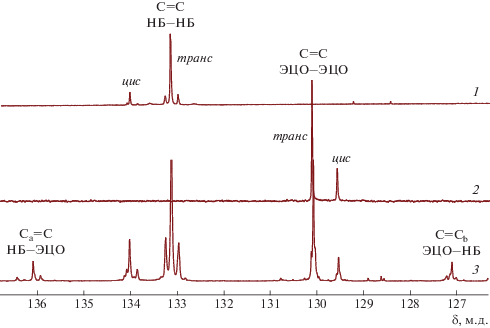
Контроль за протеканием реакции осуществляли по спектрам ЯМР 13С, в которых хорошо разрешены сигналы атомов углерода при двойных связях С=С в гомо- и гетеродиадах сополимеров (рис. 1). Рост содержания гетеродиад в ходе реакции МКМ свидетельствует об уменьшении средней длины блоков и увеличении степени блочности образующихся мультиблок-сополимеров.
Рис. 1.
Спектры ЯМР 13С, область двойных связей: 1 – исходный ПНБ, 2 – исходный ПЭЦО, 3 – сополимер НБ–ЭЦО.
Среднюю длину блоков L в сополимере НБ–ЭЦО рассчитывали, как в работах [2, 9, 10], из соотношений интегральных интенсивностей I сигналов гомо- (НБ–НБ, ЭЦО–ЭЦО) и гетеродиад (НБ–ЭЦО, ЭЦО–НБ) по формулам
Образование сополимеров и степень их блочности устанавливали по появлению в спектрах ЯМР 13С в области связей С=С сигналов гетеродиад с хим. сдвигами 136.44–135.47 м.д., (136.44, 136.28, 136.09, 135.93, 135.63, 135.47 м.д.), относящимися к углероду звена НБ (Сa=), и 127.47–126.84 м.д., соответствующими углероду звена ЭЦО (Cb=) (схема 4, рис. 1). Cигналы углеродов С=С в гомодиадах исходных полимеров характеризуются химическими сдвигами 134.04, 133.92, 133.85 м.д. (цис-С=С), 133.25, 133.12, 132.97 м.д. (транс С=С) для ПНБ [1] и 130.08 м.д. (транс-С=С), 129.53 м.д. (цис-С=С) для ПЭЦО (рис. 1) [33].
С увеличением концентрации катализатора возрастает доля гетеродиад и уменьшается средняя длина блоков L (табл. 2, опыты 1, 2), а ММ сополимера снижается (табл. 2 опыты 1, 3). Для сравнения в табл. 2 (опыты 8–10) приведены результаты кросс-метатезиса ПНБ с незамещенным ПЦО, изученного нами ранее [9, 10]. Видно, что в обоих случаях влияние концентрации катализатора одинаково. В сополимере НБ−ЭЦО, выделенном осаждением в гексан, наблюдается значительная разница между средней длиной блоков LНБ и LЭЦО (табл. 2, опыты 2, 3). Анализ спектров ЯМР 13С показал, что в сополимерах НБ с ЭЦО интегралы сигналов углеродов двойных связей С=С в гетеродиадах НБ–ЭЦО равны между собой, а интегралы сигналов углеродов в гомодиадах ЭЦО−ЭЦО меньше, чем в гомодиадах НБ−НБ. Данный эффект можно объяснить тем, что в ходе межцепного обмена протекает также внутримолекулярный метатезис ПЭЦО, который приводит к образованию циклоолигомеров. Из-за невысокой ММ циклоолигомеры при попытке их выделения отмываются осадителем. Формирование циклоолигомеров хорошо изучено в ROMP норборнена и моноциклоолефинов [1]. В силу жесткости цепи ПНБ циклоолигомеры в ходе полимеризации норборнена практически не образуются [1]. Однако циклоолефины ряда С5−С12 склонны к формированию циклоолигомеров, особенно в разбавленном растворе [1]. Чтобы снизить долю этих продуктов, концентрацию ПНБ и ПЭЦО в реакционной смеси увеличили вдвое (табл. 2, опыт 4). Повышение концентрации отразилось на интенсивности межцепного обмена, и средняя длина блока в мультиблок-сополимере снизилась в 2 раза (табл. 2, опыты 3 и 4).
Рис. 2.
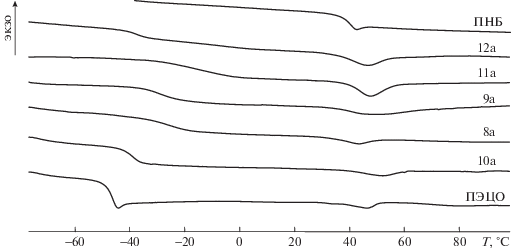
Кривые ДСК гомополимеров ПЭЦО, ПНБ и сополимеров НБ–ЭЦО при первом нагревании. Номера кривых соответствуют номерам сополимеров в табл. 3.
Межцепной метатезис в системе ПНБ–ПЭЦО–Гр-1 протекает менее активно, чем в системе ПНБ–ПЦО–Гр-1 (табл. 2, опыты 1 и 8, 3 и 9, 5 и 10), и в одинаковых условиях блоки в сополимере НБ–ЭЦО оказываются в 2–4 раза длиннее, чем в сополимере НБ–ЦО.
Ранее отмечали, что кросс-метатезис между полибутадиеном и ненасыщенным полиэфиром эффективнее протекает в растворе ТГФ [4]. Как показал наш эксперимент, при использовании ТГФ в кросс-метатезисе между ПНБ и ПЭЦО средняя длина блоков увеличилась, поскольку межцепной обмен происходил намного медленнее (табл. 2, опыт 6). Гетеродиадный состав сополимера удалось определить лишь в случае более разбавленного раствора гомополимеров с концентрацией 0.5 моль/л. При двукратном повышении концентрации полимерных реагентов продукт кросс-метатезиса сшился, и его не удалось проанализировать.
Таким образом, показано, что катализатор Гр-1 дает возможность получить сополимеры НБ–ЭЦО из гомополимеров, в то время как из мономеров на Гр-1 это неосуществимо. Кроме того, в статье сравниваются свойства сополимеров с различным содержанием эпокси-групп. Для корректного сравнения приведены свойства сополимеров, синтезированных на одном и том же типе катализатора. В настоящее время нами проводятся исследования кросс-метатезиса полимеров на других катализаторах, в том числе на катализаторе Гр-2.
Термические свойства полимеров с различным содержанием эпокси-групп
Методом ДСК были изучены термические свойства синтезированных мультиблок-сополимеров НБ–ЭЦО с различной средней длиной блоков. Результаты представлены в табл. 3 для сравнения с термическими характеристиками гомополимеров ПНБ, ПЦО и ПЭЦО, мультиблок-сополимеров незамещенных НБ-ЦО и полностью эпоксидированных сополимеров Э(НБ-ЦО) [31]. Таким образом, был рассмотрен ряд гомо- и сополимеров с различным содержанием эпокси-групп:
Таблица 3.
Термические характеристики гомо- и сополимеров НБ и ЦО c различным содержанием оксирановых звеньев
| Опыт, № |
Полимер (LНБ/LЦО) | Tс, °C | Tпл, °C | ΔH, Дж/г | Опыт, № | ϕ* эпокси, мол. % | Tс, °C | Tпл, °C | ΔH, Дж/г |
|---|---|---|---|---|---|---|---|---|---|
| без эпокси-групп | с эпокси-группами | ||||||||
| 1 [31] | ПНБ | 39 | Нет | Нет | 1а | 100 | 62 | Нет | Нет |
| 2 [11, 31] | ПЦО (68% транс-связей С=С) | –81 | 35 | 58.5 | 2а | 100 | –44 | 35, 67 | 29.5, 44.3 |
| 3 | ПЭЦО (84% транс-связей С=С) | – | – | – | 3а | 100 | –48 | 46, 78 | 1.5**, 0.8** |
| 4 [31] | НБ-ЦО (7/7) | –42, 2 | 33, 41 | 3.0, 20.4 | 4а | 100 | 10 | 32, 55 | 1.5, 15.5 |
| 5 [31] | НБ-ЦО (14/15) | –34 | 46 | 25.7 | 5а | 100 | 9 | 70 | 25.2 |
| 6 [31] | НБ-ЦО (17/31) | –47 | 30, 51 | 16.8, 38.1 | 6а | 100 | 6 | 55, 82 | 18.1, 30.1 |
| 7 [31] | НБ-ЦО (24/10) | –19 | Нет | Нет | 7а | 100 | 16 | 35, 58 | 1.4, 3.8 |
| 8 | НБ-ЭЦО (8/8) | – | – | – | 8а | 50 | –25 | 43 | 1.6** |
| 9 | НБ-ЭЦО (10/10) | – | – | – | 9а | 50 | –27 | 49 | 3.9** |
| 10 | НБ-ЭЦО (10/17) | – | – | – | 10а | 63 | –35 | 52 | 1.9** |
| 11 | НБ-ЭЦО (12/6) | – | – | – | 11а | 36 | –16 | 47 | 4.3** |
| 12 | НБ-ЭЦО (23/13) | – | – | – | 12а | 36 | –38, –6 | 46 | 6.0** |
Гомополимеры
Из табл. 3 следует, что эпоксидирование ПНБ увеличивает Тс полимера, но эпоксидированный полинорборнен (ЭПНБ) остается аморфным [28, 31]. В отличие от ПНБ незамещенный ПЦО обладает кристалличностью, степень которой определяется содержанием двойных транс-связей (опыт 2) [11, 31]. Эпоксидирование двойных связей в ПЦО повышает Тс и Тпл, при этом удельная теплота плавления эпоксидированного полиоктенамера (ЭПЦО) несколько уменьшается (опыт 2а) [31]. В гомополимере ПЭЦО, полученном метатезисной полимеризацией эпоксизамещенного циклооктена, в каждом звене присутствуют как эпокси-группа, так и двойная связь С=С в отличие от ЭПЦО с насыщенной цепью. Значения Тс и Тпл для ЭПЦО и ПЭЦО близки, но значительно выше, чем для ПЦО, что коррелирует с содержанием эпокси-групп в этих полимерах. Удельная теплота плавления ПЭЦО на порядок меньше, чем у ЭПЦО (опыт 2а), причем даже при первом нагревании, выполнявшемся после длительного хранения образцов в холодильнике. Появление кристалличности наблюдается в ПЭЦО, содержащем не менее 84% двойных транс-связей (опыт 3а), при их содержании 72% полимер не имеет Тпл (табл. 1). В то же время как ПЦО, так и ЭПЦО демонстрируют пик плавления на кривых ДСК при содержании двойных транс-связей в ПЦО и транс-эпокси-групп в ЭПЦО, равном 68% (опыты 2 и 2а). Изложенное выше указывает на то, что одновременное наличие двойных связей и эпокси-групп препятствует формированию кристаллической структуры в ПЭЦО, хотя и не делает полимер полностью аморфным.
Мультиблок-сополимеры
Как и для других мультиблок-сополимеров, термические свойства сополимеров НБ–ЦО зависят от средней длины блоков НБ и ЦО и состава сополимера [9–11, 31]. При эпоксидировании происходит замена практически всех двойных связей основной цепи на эпоксидные группы в обоих типах блоков, что приводит к увеличению Тс и Тпл модифицированного сополимера Э(НБ–ЦО) (опыты 4а–7а) [31]. В случае кросс-метатезиса ПНБ и ПЭЦО образуется сополимер НБ–ЭЦО, у которого эпокси-группы присутствуют только в блоке ЭЦО у каждого пятого и шестого атома углерода основной цепи, а через два атома углерода от эпокси-группы располагается двойная связь. Значения Тс сополимеров НБ–ЭЦО (табл. 3, опыты 8а–12а) несколько выше, чем у сополимеров НБ–ЦО (опыты 4–7) с аналогичной средней длиной блока, но ниже, чем у полностью эпоксидированных сополимеров Э(НБ–ЦО) (опыты 4а–7а), что также коррелирует с содержанием эпокси-групп в полимерной цепи.
Интересные данные получены при изучении кристаллических свойств новых сополимеров НБ–ЭЦО. При первом нагревании все они демонстрируют слабый эндотермический пик с удельной теплотой плавления в диапазоне 1.6–6.0 Дж/г (опыты 8а–12а, рис. 2). Изменение средней длины блоков и содержания звеньев НБ и ЭЦО в сополимере в изученных пределах мало сказываются на значении Тпл, которая несколько выше, чем у сополимеров НБ–ЦО с близким строением цепи, и ниже, чем у полностью эпоксидированных сополимеров Э(НБ–ЦО).
Несмотря на условный характер значений ΔH, полученных при первом нагревании, следует отметить, что кристалличность в сополимере НБ–ЭЦО выражена в наибольшей степени для образца 12а, содержащего длинные блоки аморфных норборненовых звеньев. Ранее мы наблюдали обратную зависимость: с увеличением доли НБ-блоков в сополимерах НБ–ЦО и Э(НБ–ЦО) кристалличность исчезала (опыт 7) или ΔH уменьшалась (опыт 7а). Более того, значение ΔH для сополимеров НБ–ЭЦО (опыты 9а, 11а, 12а) выше, чем для гомополимера ПЭЦО (опыт 3а). Можно предположить, что в мультиблок-сополимерах НБ–ЭЦО происходит микрофазное разделение, косвенным свидетельством чего являются две температуры стеклования, фиксируемые для образца 12а. Кристаллизация в условиях пространственных ограничений, накладываемых стенками микродоменов, может протекать совершенно иным образом по сравнению с кристаллизацией в расплаве [36, 37]. Обнаруженные для сополимеров НБ–ЭЦО закономерности требуют более подробного изучения, которое, однако, затруднено из-за малости абсолютных значений ΔH.
Таким образом, сополимеры НБ–ЭЦО по своим термическим свойствам занимают промежуточное положение между сополимерами НБ–ЦО, не содержащими эпокси-группы, и полностью эпоксидированными сополимерами Э(НБ–ЦО), что коррелирует с содержанием эпокси-групп в этих полимерах.
ЗАКЛЮЧЕНИЕ
В работе изучена реакция макромолекулярного кросс-метатезиса между ПНБ и ПЭЦО. С использованием коммерчески доступного Ru-катализатора Граббса первого поколения синтезированы новые статистические мультиблок-сополимеры НБ–ЭЦО различного состава и средней длиной блоков от 8 до 23 мономерных звеньев. Средняя длина блоков определяется соотношениями реагентов, их концентрацией в реакционной смеси и временем реакции. Реакция между ПНБ и ПЭЦО протекает медленнее, чем межцепной обмен между незамещенным ПЦО и ПНБ. Впервые с помощью метатезисной полимеризации с раскрытием цикла 5,6-эпоксициклоокт-1-ена в присутствии катализатора Гр-2 синтезирован гомополимер ПЭЦО. Полимер обладает слабой кристалличностью и не подвержен сшиванию как минимум в течение нескольких дней. Сравнение термических свойств синтезированных мультиблок-сополимеров с различным содержанием и местоположением эпокси-групп показывает, что Тс и Тпл увеличиваются при введении оксиранового фрагмента, и их значения коррелируют с содержанием эпокси-групп в полимере. Наличие двойных цис-связей и эпокси-групп в основной цепи ПЭЦО и сополимеров НБ–ЭЦО препятствует кристаллизации этих полимеров.
Авторы благодарят С.А. Корчагину за проведение анализа методом ГПХ.
Строение полученных соединений изучено с использованием оборудования Центра коллективного пользования ИНХС РАН и Центра исследования строения молекул ИНЭОС РАН.
Работа выполнена в рамках Государственного задания ИНХС РАН при частичной финансовой поддержке Российского фонда фундаментальных исследований (код проекта 16-33-60213).
Список литературы
Ivin K.J., Mol J.C. Olefin Metathesis and Metathesis Polymerization. London: Acad. Press, 1997.
Otsuka H., Muta T., Sakada M., Maeda T., Takahara A. // Chem. Commun. 2009. № 9. P. 1073.
Maeda T., Kamimura S., Ohishi T., Takahara A., Otsuka H. // Polymer. 2014. V. 55. P. 6245.
Ohishi T., Suyama K., Kamimura S., Imato M.S.K., Kawahara S., Takahara A., Otsuka H. // Polymer. 2015. V. 78. P. 145.
Daniele S., Mariconda A., Guerra G., Longo P., Giannin L. // Polymer. 2017. V. 130. P. 143.
Radlauer M.R., Matta M.E., Hillmyer M.A. // Polym. Chem. 2016. V. 7. P. 6269.
Wagner N.L., Timmers F.J., Arriola D.J., Jueptner G., Landes B.G. // Macromol. Rapid Commun. 2008. V. 29. P. 1438.
Denisova Yu.I., Gringolts M.L., Peregudov A.S., Krentsel L.B., Litmanovich E.A., Litmanovich A.D., Finkelshtein E.Sh., Kudryavtsev Y.V. // Beilstein J. Org. Chem. 2015. V. 11. P. 1796.
Gringolts M.L., Denisova Yu.I., Shandryuk G.A., Krentsel L.B., Litmanovich A.D., Finkelshtein E.S., Kudryavtsev Y.V. // RSC Adv. 2015. V. 5. № 1. P. 316.
Denisova Yu.I., Gringolts M.L., Krentsel’ L.B., Shandryuk G.A., Litmanovich A.D., Finkelshtein E.Sh., Kudryavtsev Ya.V. // Polymer Science B. 2016. V. 58. № 3. P. 292.
Shandryuk G.A., Denisova Y.I., Gringolts M.L., Krentsel L.B., Litmanovich A.D., Finkelshtein E.Sh., Kudryavtsev Y.V. // Eur. Polym. J. 2017. V. 86. P. 143.
Denisova Yu.I., Gringolts M.L., Krentsel’ L.B., Shandryuk G.A., Peregudov A.S., Finkelshtein E.Sh., Kudryavtsev Ya.V. // Polymer Science B. 2017. V. 59. № 4. P. 412.
Denisova Y.I., Gringolts M.L., Roenko A.V., Shandryuk G.A., Finkelshtein E.Sh., Kudryavtsev Y.V. // Mendeleev. Commun. 2017. V. 27. P. 416.
Denisova Yu.I., Roenko A.V., Gringolts M.L., Krentsel’ L.B., Peregudov A.S., Shandryuk G.A., Finkelshtein E.Sh., Kudryavtsev Ya.V. // Polymer Science B. 2018. V. 60. № 6. P. 735.
Bates C.M., Bates F.S. // Macromolecules. 2017. V. 50. P. 3.
Lu Y.-X., Tournilhac F., Leibler L., Guan Z. // J. Am. Chem. Soc. 2012. V. 134. P. 8424.
Descour C., Macko T., Schreur-Piet I., Pepels M.P.F., Duchateau R. // RSC Adv. 2015. V. 5. № 13. P. 9658.
Bornand M., Chen P. // Angew. Chem., Int. Ed. 2005. V. 44. № 48. P. 7909.
Naga N., Kikuchi G., Toyota A. // Polymer. 2006. V. 47. № 17. P. 6081.
Ultree A.J. Encyclopedia of Polymer Science and Engineering. New York: Wiley, 1986. V. 6. P. 733.
Bolick J.E., Jensen A.W. Kirk-Othmer Encyclopedia of Chemical Technology. New York: Wiley, 1993. V. 10. P. 624.
Margaritis A.G., Kalfoglou N.K. // Polym. J. 1988. V. 24. P. 1043.
Wang K., Zhu G., Wang Y., Ren F. // J. Appl. Polym. Sci. 2015. V. 132. P. 42045.
Karger-Kocsis J., Kéki S. // Polymers. 2018. V. 10. P. 34.
Pat. 4859746 USA. 1989
Morontsev A.A., Gringolts M.L., Filatova M.P., Finkelshtein E.Sh. // Polymer Science B. 2016. V. 58. № 6. P. 695.
Boyd T.J., Schrock R.R. // Macromolecules. 1999. V. 32. P. 6608.
Belov N.A., Gringolts M.L., Morontsev A.A., Starannikova L.E., Yampolskii Yu.P., Finkelstein E.Sh. // Polymer Science B. 2017. V. 59. № 5. P. 560.
Morontsev A.A., Zhigarev V.A., Nikiforov R.Yu., Belov N.A., Gringolts M.L., Finkelshtein E.Sh., Yampolskii Yu.P. // Eur. Polym. J. 2018. V. 99. P. 340.
Khosravi E., Szymanska-Buzar T. Ring Opening Metathesis Polymerisation and Related Chemistry: Muhlebach A., van der Schaaf P.A., Hafner A. New Materials from Thermal and Photoinduced Ring Opening Metathesis Polymerisation (ROMP/PROMP). London, 2000. P. 129.
Morontsev A.A., Denisova Yu.I., Gringolts M.L., Filatova M.P., Shandryuk G.A., Finkelshtein E.Sh., Kudryavtsev Ya.V. // Polymer Science B. 2018. V. 60. № 5. P. 688.
Demonceau A., Stumpf A.W., Saive E., Noels A.F. // Macromolecules. 1997. V. 30. № 11. P. 3127.
Hillmyer M.A., Laredo W.R., Grubbs R.H. // Macromolecules. 1995. V. 28. P. 6311.
Stumpf A.W., Saive E., Demonceau A., Noels A.F.J. // Chem. Soc., Chem. Commun. 1995. № 11. P. 1127.
Wang J., Kouznetsova T.B., Kean Z.S., Fan L., Mar B.D., Martínez T.J., Craig S.L. // J. Am. Chem. Soc. 2014. V. 136. P. 15162.
Nakagawa S., Tanaka T., Ishizone T., Nojima S., Kamimura K., Yamaguchi K., Nakahama S. // Polymer. 2014. V. 55. P. 4394.
Panthani T.R., Bates F.S. // Macromolecules. 2015. V. 48. P. 4529.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия С)