

Высокомолекулярные соединения (серия С), 2022, T. 64, № 2, стр. 135-147
ПОЛИМЕРИЗАЦИЯ МИЦЕЛЛООБРАЗУЮЩИХ МОНОМЕРОВ
И. М. Зорин a, *, Н. А. Зорина a, П. А. Фетин a
a Санкт-Петербургский государственный университет. Институт химии
198504 Санкт-Петербург, Петергоф, Университетcкий пр., 26, Россия
* E-mail: i.zorin@spbu.ru
Поступила в редакцию 05.04.2022
После доработки 18.05.2022
Принята к публикации 30.05.2022
- EDN: TKFDPA
- DOI: 10.31857/S2308114722700066
Аннотация
Дан анализ работ в области свободнорадикальной мицеллярной полимеризации ионогенных амфифильных мономеров. Описаны основные типы мономеров и особенности процесса их полимеризации в мицеллярном состоянии. Рассмотрены подходы к анализу кинетических закономерностей и механизма мицеллярной полимеризации, показана применимость микроэмульсионной модели Моргана–Калера к описанию количественных закономерностей мицеллярной полимеризации.
Мицеллы поверхностно-активных веществ – уникальный тип молекулярных агрегатов, сохраняющих стабильность формы, размеров, индивидуальные для каждого ПАВ. Мицеллы образуются в растворах при достижении критической концентрации мицеллообразования, при повышении концентрации ПАВ увеличивается не размер мицелл, а их количество. Каждую из мицелл можно рассматривать как отдельную микрофазу, а момент образования всей совокупности мицелл можно считать фазовым переходом второго рода [1]. Величины ККМ, как правило, находятся в области 10–3–10–2 моль/л, концентрация ПАВ в самой мицелле, по всей видимости, приближается к концентрации в массе. По этой причине химические реакции с участием молекул ПАВ в мицеллярном состоянии должны протекать с большой скоростью даже в относительно разбавленных растворах, при условии, что концентрация вещества превышает ККМ. Одна из таких реакций – полимеризация. ИЮПАК определяет мицеллярную полимеризацию как “полимеризацию мицеллообразующего мономера в мицеллярном состоянии”. Подходы химиков к мицеллярной полимеризации выстраиваются сравнительно давно, и причин возникновения интереса к данному типу полимеризации несколько. С одной стороны, это уже упомянутое самопроизвольное концентрирование молекул реагирующего вещества при сохранении его растворенного состояния и небольших брутто-концентраций. С другой стороны, стабильный состав мицелл (примерно постоянное число молекул ПАВ в каждой мицелле) и ориентированное состояние указанных молекул давали надежду на возможность контроля над процессом полимеризации, получения полимеров с хорошо предсказуемыми молекулярно-массовыми характеристиками, а в идеале получение полимеризованной реплики мицелл. Конечно, такое понимание мицеллярной полимеризации является далеким от истины, но оно послужило развитию представлений о протекании полимеризационных процессов в мицеллярных растворах мономеров.
Формально первыми в области мицеллярной полимеризации, следует считать, вероятно, работы A.I. Medalia [2–4]. В 1963 г. [5] появился термин “полимеризованные мицеллы”, который получил широкое употребление после работы C.M. Paleos в 1983 г. [6]. В настоящее время мы предпочитаем называть продукты полимеризации мицеллообразующих мономеров “гребнеобразные полиэлектролиты” или “полиэлектролит-коллоидные комплексы”, в зависимости от молекулярной архитектуры:

Мы рассмотрим свободнорадикальную мицеллярную полимеризацию мономеров, являющихся ионными ПАВ и построенными по классической схеме “полярная голова–гидрофобный хвост”, формирующих прямые мицеллы в воде и обращенные – в неполярных средах.
Наличие в молекуле полимеризующихся ПАВ дополнительной функциональной группы (С=С) неизбежно приводит к существенным отличиям от аналогов, не содержащих таких групп. По сравнению с алкильной цепью даже алкенильная группа обладает заметной полярностью и способностью к образованию водородных связей [7]. В работах [8, 9] на примере ундеканоата и 10-ундеценоата натрия показано увеличение ККМ в 2 раза и числа агрегации в 1.5 раза при введении в структуру молекулы терминальной двойной связи. Еще большего эффекта следует ожидать от введения других функциональных групп, способных к полимеризации (акрилоил-, метакрилоил-, стирил-) [10, 11], сильно изменяющих как геометрию молекулы, так и картину распределения энергии межмолекулярных взаимодействий. По этой причине мицеллообразующие мономеры следует классифицировать по региохимическому признаку – расположению полимеризуемой группы относительно полярной головы молекулы, чем, согласно работе [12], определяется область протекания полимеризации.
А (tail type monomer): полимеризуемая группа локализована в гидрофобной части молекулы мономера – полимеризация в ядре прямой мицеллы.
Б (head type monomer): полимеризуемая группа локализована вблизи полярной части молекулы мономера – полимеризация на периферии прямой мицеллы.
В (polar tail type monomer): полярная полимеризуемая группа локализована в гидрофобной части молекулы – полимеризация, вероятно, на периферии прямой мицелл.
Г – мономеры, в которых реализуется полимеризация противоиона, а получаемый полимер является, по сути, полиэлектролит-коллоидным комплексом (ПЭКК).
Полимеризация мономеров типов А и В приводит к получению гребнеобразных полимеров, содержащих полярную (ионогенную) группу на концах боковых цепей; мономеров типа Б – к получению гребнеобразных полимеров с полярной (ионогенной) группой вблизи точки присоединения боковых цепей к основной цепи; мономеров типа Г – к получению комплексов полиэлектролит–ПАВ (ПЭКК, в которых ионогенные группы полиэлектролита выступают в качестве противоиона по отношению к ионному ПАВ).
К группе А обычно относят мономеры типа терминальных алкенов, важнейшим представителем которых является 10-ундеценовая кислота и ее производные [5, 7, 8, 12–19]. Описаны также мицеллообразующие мономеры со стирильной полимеризуемой группой [3, 12, 14].
Сам факт свободнорадикальной полимеризации 10-ундеценоата натрия при концентрациях в растворе порядка 10–3 моль/л является удивительным. Тем не менее, и в мицеллярной фазе полимеризация требует довольно жестких условий. Радиационно-химическая полимеризация – единственный надежный путь синтеза полимеров 10-ундеценовой кислоты и ее производных (преимущественно N-ундеценоиламинокислот), описанных в работах [13, 17], хотя существуют и альтернативные варианты, такие как фотополимеризация (120–960 Вт, 23–48 ч [20, 21]) или полимеризация с солюбилизированным ДАК [22]. Однако во всех случаях (кроме последнего) выход далек от 100%. Примером успешной полимеризации 10-ундеценоата натрия с вещественным инициированием могут быть работы [23, 24], где описана полимеризация в лиотропной ЖК-фазе, при концентрации мономера 47–59 мас. % (около 2.5 моль/л), с персульфатом аммония. Данный процесс может служить образцом влияния упорядоченности раствора мономера на эффективность полимеризации. В условиях синтеза (60°С) раствор мономера формирует гексагональную фазу, тогда как полимер дает изотропный раствор, при охлаждении которого формируется ламелярная фаза раствора полимера.
Отметим, что радиационное инициирование в водных растворах достигается за счет генерации большого числа радикалов в результате радиолиза воды [25]. Это способствует не только эффективному инициированию, но также и эффективному обрыву цепи, чем и можно объяснить обычно невысокую степень полимеризации образующихся полимеров. В работе [17] описано 15 дипептидных производных ундеценовой кислоты, имеющих число агрегации от 38 до 380. Их радиационная полимеризация приводит к получению полимеров со степенью полимеризации 18–33. Таким образом, при радиационной мицеллярной полимеризации производных ундеценовой кислоты в литературе представлено формирование полимеров со степенью полимеризации от 10 до ~80. На примере таких ПАВ становится ясно, что структура, степень полимеризации, характер агрегации полимеризованных мицелл не совпадает с параметрами мицелл мономера, т.е. полимеризация не обеспечивает сохранение структуры мицелл. Производные ундеценовой кислоты в ряде случаев образуют полимеры, весьма близкие по структуре к мицеллам мономеров, но, вероятнее всего, это не является результатом топохимического характера процесса, а обусловлено механизмом радиационно-химической полимеризации в воде.
Мономеры типа Б содержат полимеризуемую группу в непосредственной близости от полярной группы. В своем большинстве они восходят к производным акрилоил-этаноламина, гидроксиэтилакрилата, аллиламина и чаще всего имеют четвертично-аммонийную ионогенную группу. Такие объекты описаны в работах В.В. Егорова [26–32], D. Sherrington [11, 33] и в ряде других публикаций [34–38]. В мономерах типа Б активный центр локализован вблизи заряженной группы, что понижает его реакционную способность по отношению к одноименно заряженным мономерам [11, 39]. В статье [12] высказано предположение, что реакции обрыва цепи менее вероятны в случае протекания реакции в ядре мицеллы (мономеры А, В), чем на периферии (мономеры Б). Часто полимеризация мономеров типа Б приводит к потере растворимости, поскольку формирующаяся основная цепь полимера, во-первых, существенно ограничивает подвижность полярных групп, непосредственно к ней присоединенных, и во-вторых, изменяет баланс и распределение гидрофильности-гидрофобности. Гидрофобные хвосты, однако, остаются относительно свободными, что только усиливает фазовую сегрегацию в воде, но облегчает растворимость в органических средах. Полимеризация мономера типа Б критически изменяет гидрофильно-липофильный баланс и способность вещества к мицеллообразованию [12]. После полимеризации полярная группа оказывается зажата между гидрофобным хвостом и гидрофобной цепью. По всей видимости, возникает конфликт между оптимальной упаковкой полярных групп и хвостов, а также оптимальной конформацией гидрофобной цепи. Полярные группы уже не могут стабилизировать такую систему; в результате многие из полимеров этого типа утрачивают растворимость в воде, вероятно, при достижении некоторой критической степени полимеризации.
Мономеры типа В соответствуют расположению полярной полимеризуемой группы на конце углеводородного хвоста ПАВ. Наиболее часто встречаются эфиры или амиды акриловой и метакриловой кислоты в качестве полимеризуемых групп, –COO– или четвертичный азот – в качестве ионогенной группы ПАВ. Наличие полярной группы в хвосте дополнительно структурирует мицеллы, формируемые такими мономерами. Для них свойственна высокая скорость полимеризации с конверсией, близкой к количественной. Именно полимеры 11-акрилоилоксиундекановой кислоты (ААУ-Н) впервые были описаны в патенте 1959 г. [4]. Полимеризация этого мономера в деталях впервые описана L.M Gan [40], в дальнейшем опубликовавшим ряд работ по исследованию полимеризации ААУ-Na [41–46]. Полимеризация натриевой соли ААУ-Н (ААУ-Na) в мицеллах при концентрациях мономера от долей процента в воде, с персульфатным инициированием достигает количественной конверсии за 22 мин [45]. Следует упомянуть также работы [47–54], посвященные исследованию структурообразования в растворах мономера ААУ-Na, полимеров на его основе, а также мономеров и полимеров натриевых солей N-(11-акрилоиламиноундеканоил)-L-аминокислот. Общей особенностью этих объектов является неожиданно низкая величина ККМ (4.2 × 10–4 моль/л для ААУ-Na) и большое разнообразие надмолекулярных структур (сферических, цилиндрических мицелл, везикул и трубок), сосуществующих в растворах мономеров при концентрациях, значимых для проведения полимеризации (0.1–0.2 моль/л, что превышает критическую концентрацию мицеллообразования в 500 раз – 500 × ККМ). Это должно затруднять понимание процесса полимеризации ААУ-Na, однако последующие исследования в широком диапазоне концентраций (от 5 × ККМ до 500 × ККМ) выявили наличие общих закономерностей кинетики полимеризации во всем исследованном диапазоне.
Другое значимое семейство мономеров данного типа – акрилоильные и метакрилоильные производные 11-бромундеканола [10–12, 34, 35, 55–59]. В основном это 11-акрилоилоксиундецил-триметиламмоний бромид (АУТА-Br) и его структурные аналоги. Указанные мономеры имеют величину ККМ около 1.6 × 10–2 моль/л и в актуальном для проведения полимеризации концентрационном диапазоне (8 × ККМ–16 × ККМ) формируют сферические мицеллы.
Практически всегда полимеризация мономеров типа В приводит к образованию растворов соответствующих полимеров – гребнеобразных полиэлектролитов. Они обладают выраженной поверхностной активностью и склонностью к ассоциации в растворах, поэтому долгое время в литературе не было адекватных сведений о молекулярных характеристиках продуктов. Проведенные в работах [60–64] детальные исследования позволили установить, что молекулярная масса полимеров ААУ-Na, получаемых при концентрациях 0.1–0.2 моль/л с инициированием персульфатом калия (1 г/л), составляет не более 3 × 105, а величина молекулярной массы чувствительна к концентрации мономера в растворе. Индекс дисперсности подобных полимеров находится в диапазоне 1.3–1.8, т.е. заметно меньше, чем в “обычных” свободнорадикальных процессах, а в сочетании с молекулярными массами порядка 105 это можно считать хорошим результатом.
Особую разновидность мицеллобразующих мономеров (тип Г) составляют ПАВ с полимеризуемым противоионом. Среди таковых есть и катионные, и анионные ПАВ. К полимеризуемым анионам относятся акриловая и метакриловая кислоты [27, 65–69], стиролсульфокислота [70], винилбензойная кислота [71–77], 2-акриламидо-2-метилпропансульфоновая кислота с амфифильными катионами алкиламмония [78–83]. Полимеризуемые катионы являются в основном производными холина – триметил(метакрилоксиэтил)аммоний и аналоги с амфифильным анионом додецилсульфатом [84–87]. Поскольку в данном случае полимеризуемая группа принадлежит противоиону, который обычно в водных растворах имеет степень связывания с мицеллами 0.6–0.7, в растворах таких веществ всегда имеется значимое количество мономера, не связанного в мицеллы, а значит, полимеризация может протекать как в мицеллах, так и с участием несвязанного мономера. Пожалуй, данная ситуация наиболее близко подходит к варианту темплатной полимеризации по pick-up механизму. Часть молекул мономеров группируется вблизи поверхности мицелл (сферических или цилиндрических), часть пребывает в несвязанном состоянии. Однако, каков бы ни был механизм, продуктом является полиэлектролит-коллоидный комплекс (ПЭКК), молекулярная архитектура которого больше всего напоминает цилиндрическую обращенную мицеллу (полиэлектролит, окруженный шубой из гидрофобных противоионов). В связи с этим чаще всего ПЭКК не растворимы в воде, а полимеризация мономеров типа Г происходит в осадительном режиме. Удивительным исключением являются полимеры на основе алкиламмоний винилбензоатов. Они растворимы в воде, как это ни странно, именно благодаря гидрофобности полимерного компонента и слабо ионного характера связи полимер–ПАВ.
Процессы мицеллярной полимеризации в неводных системах (обращенные мицеллы) в ранних публикациях встречаются сравнительно редко [36, 37, 88]. Отмечается, что мономеры типа Б при полимеризации в полярных органических средах формируют сравнительно низкомолекулярные продукты, в неполярных и умеренно полярных молекулярные массы сопоставимы с достигаемыми в воде. Также отмечено, что кинетические кривые при полимеризации в воде и неполярных средах имеют сходную S-образную форму и демонстрируют сильную зависимость от длины алкильного хвоста. В полярных органических растворителях кинетические кривые гладкие, и характер их от длины хвоста не зависит. Приведенные факты свидетельствуют об общем характере полимеризации в прямых (вода) и обращенных (неполярные среды) мицеллах мономера и значимых отличиях от полимеризации в немицеллярных растворах.
В большинстве работ, посвященных мицеллярной полимеризации, отмечается, что полимеризация в мицеллах мономера, протекает в целом эффективнее, чем полимеризация в гомогенной среде. Это проявляется в высоких скоростях полимеризации, высоких конверсиях за короткий промежуток времени и в возможности полимеризации при низких концентрациях.
Главное требование для успешной реализации полимеризации – ее проведение выше ККМ, но в большинстве рассматриваемых в литературе случаев ККМ не превышает 2 × 10–2 моль/л, и низкая эффективность полимеризации при более низких концентрациях не вызывает удивления.
Основной вопрос состоит в том, является ли мицеллярная полимеризация самостоятельным типом полимеризационного процесса или представляет собой частный (предельный) случай эмульсионной либо гомогенной полимеризации.
Раствор мицеллообразующего мономера термодинамически стабильный, однако концентрация молекулярно-дисперсного вещества не превышает ККМ. Вещество сверх ККМ находится в ассоциированном состоянии. Мицеллы представляют собой отдельную псевдофазу, характеризующуюся собственной микрополярностью, микровязкостью, они отделены от фазы раствора двойным электрическим слоем. Наличие отдельной дисперсной фазы, содержащей мономер, характерно для эмульсионной полимеризации.
Существование мицелл, обладающих определенным размером и числом агрегации N поддерживается равновесным процессом обмена молекулами ПАВ между мицеллой и раствором. Это равновесие характеризуется двумя временами релаксации τ1 и τ2. Первое соответствует равновесному процессу добавления одной молекулы ПАВ в мицеллу из фазы раствора и обратно, второе – равновесному процессу образования мицеллы с числом агрегации N и ее полной диссоциации на молекулы ПАВ [89]. При концентрации много большей, чем ККМ, среднее время жизни одной мицеллы приблизительно равно N×τ2 и лежит в диапазоне от миллисекунд до десятков секунд. Среднее время пребывания одной молекулы в составе мицеллы имеет величину порядка 10-8 с.
Таким образом, молекулы мономера, хотя и не находятся в истинно-дисперсном состоянии, не принадлежат конкретным частицам, а находятся в состоянии постоянных перескоков из одной мицеллы в другую, не минуя фазы молекулярно дисперсного раствора. В секундном и минутном диапазоне времени поведение молекулы мицеллообразующего мономера не сильно отличается от поведения молекулы истинно растворимого вещества. По этой причине мицеллярную полимеризацию можно рассматривать как гомогенную. Кроме того, в большинстве случаев продуктом мицеллярной полимеризации является раствор или осадок полимера, но не латекс, характерный для эмульсионного процесса. В эмульсионной полимеризации латекс в некоторой степени представляет собой слепок дисперсной фазы мономера, результатом ее некоторой структурной перестройки, т.е. имеет место сохранение дисперсной фазы как сущности. В чисто мицеллярном процессе, если считать мицеллы фазой, в результате полимеризации происходит гибель этой фазы, поскольку на выходе формируется молекулярно-дисперсный раствор полимера. И даже если в рассматриваемом растворе в результате внутримакромолекулярной ассоциации формируются гидрофобные домены, он остается гомогенным, так как нельзя считать фазовой частицей отдельную молекулу, тем более ее часть.
Одним из подробных исследований кинетики и особенностей полимеризации в мицеллярных растворах следует считать цикл работ В.В. Егорова [26–32], в которых исследована полимеризация мономеров, главным образом, типа Б, с маслорастворимым солюбилизированным инициатром (ДАК) или персульфатом калия [28]. Было установлено, что полимеризация в сферических и ламелярных мицеллах в воде действительно протекает с большими скоростями, локализованно в мицеллах ПАВ, где концентрация приближается к блочной. Константа скорости роста в таких условиях несущественно отличается от наблюдаемой при полимеризации в истинном растворе (в этаноле) [29], хотя общая скорость полимеризации в мицеллярном растворе существенно выше, чем в истинном [26]. Полимеризация, как правило, идет с индукционным периодом, после чего скорость достигает максимальных значений и остается постоянной до конверсии 10–15% [32]. Отмечается, что приведенная скорость полимеризации резко снижается при переходе через ККМ-2, т.е. при формировании анизотропных мицелл. Это явление воспроизводится для разных мономеров, в том числе и для смешанных мицелл с неполимеризуемыми ПАВ [32]. Снижение общей скорости полимеризации может быть обусловлено в том числе уменьшением количества сферических мицелл и солюбилизированного в них маслорастворимого инициатора. Полимеризация же непосредственно в анизотропных (цилиндрических) мицеллах затруднена по причинам топологического характера. Однако утверждение о неэффективности полимеризации в анизоторпных мицеллах в литературе подтверждения не находит. Цикл работ L.M. Walker и S.R. Kline [72, 74], посвященный полимеризации в червеобразных мицеллах цетилтриметиламмоний винилбензоата, показывает, что этот мономер полимеризуется с конверсией, близкой к количественной.
Вопросам соотношения условий полимеризации и молекулярных характеристик продуктов, как и в целом кинетике мицеллярной полимеризации, существенное внимание уделяется в работах [90–93]. Для мицеллярной полимеризации авторы [90] предлагают два механизма.
1. Полимеризация мономера с длинным хвостом – низкой ККМ, числом агрегации 50–100 и большим временем жизни мицеллы (tm ≈ N × τ2 > > 5 × 10–2 с). В этом случае практически весь мономер содержится в мицеллах, и гидрофобный инициатор также полностью солюбилизирован в мицеллах. Радикал, образующийся при распаде инициатора в мицелле мономера, с учетом времени, необходимого для одного акта роста tp ~ 10–3 с, за время жизни мицеллы tm успевает присоединить tm/tp ≈ 20–100 звеньев, в то время как мицелла остается стабильной, но ее число агрегации флуктуирует. Образовавшийся макрорадикал стабилизирует мицеллу и она уже не распадается на мономеры. Гидрофобность олигомера выше, чем соответствующего мономера, это отмечали и в работе [26]. Поскольку время прибавления одной молекулы ПАВ к мицелле обычно не более 10–5 с, имеет место рост макрорадикала до тех пор, пока не произойдет обрыв за счет попадания в мицеллу второго радикала или ее слияние с другим макрорадикалом. Таким образом, формируются макромолекулы со степенью полимеризации больше числа агрегации мицелл мономера.
2. Полимеризация мономера с коротким хвостом, имеющего высокую ККМ, мицеллы малого размера с небольшим временем жизни (порядка 10–3 с). В этих условиях доля мицеллизованного мономера может быть весьма мала. Инициирование в данном случае происходит в молекулярно-дисперсном растворе, либо в мицеллах, но малое время жизни мицелл не позволяет сформировать достаточно гидрофобный макрорадикал для их стабилизации. В таких условиях рост преимущественно протекает в водной фазе, особенно на поздних стадиях, когда мицеллярная фаза представлена уже исключительно заполимеризованным веществом, а непрореагировавший мономер находится в молекулярно-дисперсном растворе. Продукт полимеризации может иметь высокую степень полимеризации лишь в том случае, если реакционная способность мономера очень высока.
С точки зрения возможностей синтеза полимеров с заданными характеристиками наибольшие вопросы вызывает не столько механизм формирования макромолекулы в мицеллярной полимеризации, сколько взаимосвязь молекулярной массы полимера с числом агрегации мицелл мономера. Молекулярная масса (степень полимеризации) в некоторых случаях может мало зависеть от начальной концентрации мономера: в работе [91] для короткохвостного мономера (С8) в концентрационной области до и вблизи ККМ наблюдалась сильная зависимость ММ полимера от концентрации мономера, выше ККМ зависимость вырождалась. В работе [72] так же отмечено, что молекулярная масса полимеризованных мицелл не зависит от концентрации мономера, но зависит от концентрации инициатора.
Простая идея о возможности зафиксировать все молекулы, составляющие мицеллу, путем их полимеризации, как видно из приведенных выше данных, является несостоятельной. Первая критика концепции полимеризованных мицелл появилась уже в 1986 году в работе D. Sherrington [33]. Скорость протекания полимеризации акриловых мономеров, точнее, продолжительность одного акта присоединения молекулы мономера к активному центру tp = (Kp[M])–1 ≈ 10–6–10–2 с. Характеристическое время обмена одной молекулы ПАВ между мицеллой и раствором составляет 10–5–10–9 с. Очевидно, что скорость обмена молекул ПАВ между мицеллой и молекулярным раствором может быть на порядки больше, чем скорость роста макромолекулы. Таким образом, за время роста макромолекулы большое число молекул мономера могут “успеть” присоединиться к мицелле, давая возможность в ходе полимеризации сформироваться частице, более крупной, чем исходная мицелла мономера. Степень полимеризации в такой системе определяется в том числе соотношением упомянутых скоростей. Именно поэтому полимеризованные мицеллы в общем случае не являются репликами мицелл мономера. Образование полимеров со степенью полимеризации меньшей или равной числу агрегации мицелл обусловлено, скорее всего, не топохимическим характером процесса, а реакциями передачи цепи.
В работе [94] был проведен анализ процесса мицеллярной полимеризации мономеров типа В – акрилоиламиноалкановых кислот (в Н- и Na-форме). Реакционная способность акриламидной группы в гомологическом ряду ω-N-акрилоиламиноалкановых кислот мало зависит от числа n углеродных атомов скелета ω-аминокислоты. Но удлинение алифатической цепи критическим образом влияет на агрегацию и мицеллообразование. В связи с этим влияние мицеллообразования можно проследить при изучении кинетики полимеризации в ряду Na-солей N-акрилоилзамещенных аминокислот 3-аминопропановой (ААП-Na), 6-аминогексановой (ААГ-Na), 11-аминоундекановой (ААУ-Na) и 12-аминододекановой кислот (ААД-Na), из которых ААП-Na не является мицеллообразующим, ААГ-Na образует мицеллы при С > 0.17 моль/л, а ААУ-Na и ААД-Na агрегированы во всем доступном диапазоне концентраций от 4 × 10–4 моль/л.
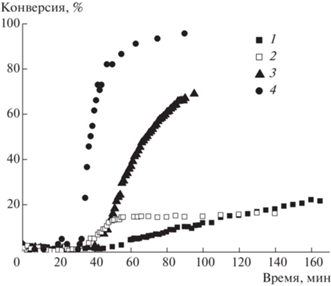
На рис. 1 приведены кривые конверсии нескольких акриламидных мономеров при полимеризации в разбавленном растворе. Хорошо видно, что форма кривых резко различается для акрилоиламиноалканоатов натрия и акриламида, не имеющего ни заряженного, ни гидрофобного фрагмента, присоединенного к атому азота. Анионогенные мономеры с гидрофобным хвостом характеризуются выраженным S-образным видом кинетической кривой, что наводит на мысль о близком к эмульсионному характере процесса, т.е. гетерогенной полимеризации [95–97].
Рис. 1.
Зависимость конверсии от времени при полимеризации акриламидных мономеров с различной длиной углеводородной цепи. С = 0.005 моль/л; K2S2O8 = 1 г/л; 1 – акриламид, 2 – ААП-Na, 3 – ААГ-Na, 4 – ААУ-Na. Здесь и на рис. 2–4 – по данным фотометрии. Цветные рисунки можно посмотреть в электронной версии.
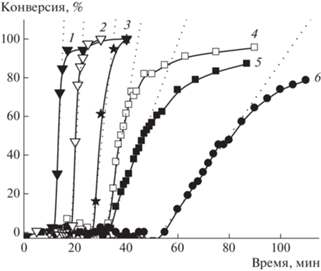
Вид кривых конверсии (рис. 2), имеющих три характерных участка – индукционный период, область примерно постоянной скорости и область замедления реакции, полностью идентичен наблюдаемому для микроэмульсионной полимеризации гексилметакрилата в мицеллах додецилтриметиламмоний бромида [98]. Начальный участок кривых конверсии с почти нулевой скоростью авторы связывают с процессом гомогенной нуклеации, т.е. инициированием и ростом цепей в фазе гомогенного раствора, где концентрация мономера крайне мала. При накоплении достаточного количества макромолекул, имеющих критическую степень полимеризации, процесс переходит в мицеллярный режим и скорость резко возрастает, поскольку концентрация мономера в мицеллах весьма велика. При полимеризации ААП-Na и ААГ-Na такой режим полимеризации может достигаться за счет агрегации мономера на растущих полимерных цепях.
Рис. 2.
Зависимости конверсии мономера от времени в ходе полимеризации ААУ-Na при концентрации 0.18–0.20 (1), 0.10–0.11 (2), 0.045–0.050 (3) , 0.0087 (4), 0.0045 (5) и 0.0022 моль/л (6).
Рассмотрим полученные данные с позиций кинетики микроэмульсионной полимеризации, характерная особенность которой – повышение скорости полимеризации от начала реакции до довольно больших значений конверсии, несмотря на убыль брутто-концентрации мономера в реакционной смеси мономера [97]. В работе [99] приведены теоретические выкладки, описывающие кинетику микроэмульсионной полимеризации и экспериментальные данные, подтверждающие их справедливость для полимеризации гексилметакрилата в присутствии цетилтриметиламмоний бромида. Если опустить промежуточные выкладки, зависимость скорости полимеризации от конверсии мономера р определяется уравнением
в котором $A = \frac{{{{k}_{p}}{{C}_{0}}{{\rho }_{0}}}}{{{{M}_{0}}}}$.В отсутствие обрыва скорость генерирования радикалов ρ0= 2fkd[I], где f – эффективность инициирования.
Из этого уравнения следует, что максимум скорости полимеризации достигается при конверсии 39%.
Величины, входящие в выражение для параметра А – константы скорости распада инициатора, скорости реакции роста цепи, начальная брутто-концентрация мономера M0 и локальная концентрация мономера в частицах в момент их образования С0 – это начальные параметры реакционной смеси.
Для оценки применимости модели были построены экспериментальные зависимости скорости полимеризации от конверсии для акрилоиламиноалканоатов натрия в воде. На рис. 3 кинетические данные полимеризации акрилоил-аминоалканоатов [94] представлены в координатах r(p). Там же сплошными линиями показаны расчетные кривые.
Рис. 3.
Зависимости скорости полимеризации от конверсии при полимеризации акрилоиламиноалканоатов натрия в воде (большие точки – из необработанных экспериментальных данных, маленькие – аппроксимация В-сплайном) и теоретические зависимости (кривые). а – ААД-Na + персульфат аммония, концентрация ААД-Na 0.01 (1), 0.02 (2), 0.05 (3), 0.10 (4) и 0.20 моль/л (5); б – ААУ-Na + персульфат аммония, концентрация ААУ-Na 0.002 (1), 0.004 (2), 0.008 (3), 0.05 (4), 0.10 (5) и 0.20 моль/л (6); в – ААГ-Na + персульфат калия, концентрация ААГ-Na 0.02 (1), 0.05 (2) и 0.10 моль/л (3); г – ААП-Na + персульфат калия, концентрация ААП-Na 0.02 (1), 0.05 (2) и 0.10 моль/л (3); д – ААГ-Na + персульфат аммония, концентрация ААГ-Na 0.015 (1), 0.025 (2) и 0.050 моль/л (3).

Максимальная скорость полимеризации действительно наблюдается при конверсиях около 30–40%, а расчетные кривые хорошо описывают экспериментальные данные на начальных участках (до конверсии около 40–50%). Теоретические кривые были построены по уравнению $\frac{{\partial p}}{{\partial t}}$ = = ${{(1 - p)}^{\alpha }}\sqrt { - 2A{\kern 1pt} {\text{*}}\ln (1 - p)} $, где А* и α выступали в качестве подгоночных параметров. Смысл эмпирического параметра А* соответствует описанному выше А, параметр α имеет значения от 1 до 2 и связан с эффективностью инициирования и переноса радикалов из раствора в мицеллы (при f = 1 и быстром захвате радикалов мицеллами α = 1).
Сопоставление данных, полученных при исследовании полимеризации ω-акрилоиламиноалканоатов натрия в воде, свидетельствует об отсутствии качественных различий в протекании полимеризации мицеллобразующих и немицеллообразующих мономеров данной серии. Явным образом проявляется лишь тенденция к повышению скорости полимеризации при увеличении длины углеводородной развязки между акриламидной и карбоксилатной группой, т.е. известный из литературы “эффект конденсации мономера” [100]. Тем не менее, способность к полимеризации в весьма разбавленных (10–2 моль/л) растворах с высокими скоростями и конверсиями оказалась общим свойством ω-акрилоиламиноалканоатов, не связанным с образованием мицелл мономерами. Это обусловлено, с одной стороны, общей склонностью замещенных мономерных акриламидов к агрегации в воде и, с другой стороны, агрегированным состоянием полимеров. Возможность внутримакромолекулярной агрегации образующегося полимера стабилизирует и агрегаты мономера, что способствует увеличению скорости протекания полимеризации. Такой механизм стабилизации полимер-мономерных частиц характерен для микроэмульсионной полимеризации, стартующей с гомогенной фазы [95, 97]. Для эмульсионных и микроэмульсионных процессов характерны две особенности: первая – наличие довольно продолжительного стационарного периода с высокой скоростью полимеризации вплоть до конверсии 40–60%; вторая – максимальная скорость полимеризации достигается не в начальный период (при максимальной брутто концентрации мономера), а при некоторой довольно значительной величине конверсии (39% в модели Моргана–Калера). Это обусловлено именно стабилизацией частиц и солюбилизацией в них большого количества мономера.
Исходя из представленных экспериментальных фактов, можно сделать вывод, что полимеризация акрилоиламиноалканоатов натрия в воде осуществляется по механизму, близкому к микроэмульсионному. Это означает, что инициирование и нуклеация протекают в фазе раствора на протяжении всего времени реакции, одновременно происходит формирование мицелл на растущих макрорадикалах за счет динамического обмена с мицеллами мономера или за счет солюбилизации и ассоциации молекул немицеллообразующего мономера на олигомерных радикалах со степенью полимеризации выше пороговой. Рост цепи при этом происходит локализовано в мицеллах, содержащих макрорадикал и солюбилизированные молекулы мономера. Механизм полимеризации можно описать следующим образом.
1. Распад инициатора приводит к образованию первичных радикалов ∙${\text{SO}}_{4}^{ - }$ и ∙ОН–. Данные о концевых группах полимера, полученные методом МАЛДИ-масс-спектрометрии [94, 101], свидетельствуют о том, что основным инициирующим радикалом является (в случае ААУ-Na) ∙ OH–, образующийся при взаимодействии первичных радикалов с водой (поскольку концентрация мономера в растворе мала, а одинаковый заряд первичных радикалов ∙${\text{SO}}_{4}^{ - }$ и мицелл препятствует их взаимодействию). Оба радикала заряжены отрицательно, поэтому их взаимодействие непосредственно с мицеллами мономера маловероятно; инициирование происходит в фазе гомогенного раствора при взаимодействии радикалов с одиночными молекулами мономера. В случае не образующих мицеллы мономеров ААГ-Na и ААП-Na это единственно возможный вариант.
2. Рост цепи может начинаться в растворе и продолжаться именно в растворе некоторое время, либо инициированная молекула мономера может захватываться в мицеллу в результате динамического обмена. Первый вариант реализуется для ААГ-Na и ААП-Na, второй более вероятен для мицеллообразующих ААУ-Na и ААД-Na.
3. В результате полимеризации, протекающей в растворе, формируются олигомеры (олигорадикалы). Их макромолекулы обладают большей гидрофобностью по сравнению с молекулами мономера (за счет алифатической основной цепи). Образующиеся олигорадикалы формируют мицеллоподобные частицы, которые принимают участие в ассоциации. Молекулы мономера ассоциируются с олигорадикалами – в случае ААУ-Na и ААД-Na возможность этого не вызывает сомнений, в случае ААГ-Na и ААП-Na представляется вполне вероятной.
4. Рост цепи продолжается в частице, содержащей олигорадикал и ассоциированные молекулы мономера, находящиеся в состоянии динамического обмена с вмещающим раствором и мицеллами мономера (если они есть). Внедрение в мицеллу или генерирование в ней одного олигорадикала со степенью полимеризации, например, около 20, что по порядку сопоставимо с числом агрегации сферических мицелл, способствует значительному снижению в образовавшемся агрегате доли несвязанных молекул ПАВ и уменьшению скорости их диффузии из агрегата, тогда как скорость диффузии мономера в агрегат остается прежней. Это приводит к преобладанию процесса диффузии молекул мономера в агрегат до тех пор, пока их содержание не станет равновесным с содержанием их в дисперсионной среде. Таким образом, с повышением степени полимеризации цепи, содержание в агрегатах не связанных в цепь молекул ПАВ стремится оставаться на одном уровне, соответствующем их содержанию в мицелле мономерного ПАВ. Вместе с тем доля связанных в полимерную цепь в качестве мономерных звеньев молекул ПАВ постоянно увеличивается, и агрегат превращается в макромолекулу с сорбированным внутри клубка мономером, за счет полимеризации которого продолжается рост цепи.
Если по своей природе мономерное ПАВ склонно формировать цилиндрические мицеллы, например ПАВ с гидротропным противоионом, то получающаяся макромолекулярная цепь со степенью полимеризации существенно выше числа агрегации исходной мицеллы, формирует подобную цилиндрическую унимакромолекулярную мицеллу, но со значительно большей длиной [77].
Если же тенденция к образованию цилиндрических мицелл отсутствует, то сформировавшаяся макромолекула при достижении степени полимеризации, превышающей предельное число агрегации для сферических мицелл (10–170), принимает самостоятельные конформации, отличные от конформации, сходной со сферической мицеллой. Это может быть конформация “жемчужного ожерелья”, в которой длинная макромолекулярная цепь формирует множество мицеллоподобных агрегатов, связанных воедино. Альтернативой могут быть конформации развернутого статистического клубка, свойственные заряженным водорастворимым полимерам (разрушение мицеллярной структуры, полимер остается в растворе), или плотной глобулы (разрушение мицеллярной структуры, сопровождающееся осаждением полимера). В случае полиакрилоиламиноалканоатов реализуется вариант “жемчужного ожерелья”, что подтверждается высокими степенями полимеризации по сравнению с числами агрегации сферических мицелл, наличием в растворе гидрофобных доменов, солюбилизирующих пирен и красители, отсутствием выраженного эффекта полиэлектролитного набухания при разбавлении раствора.
5. Прекращение роста цепи происходит либо по реакции передачи цепи (на полимер, на мономер), либо в результате обрыва. Бимолекулярный обрыв при взаимодействии двух макрорадикалов близкой степени полимеризации маловероятен, поскольку макрорадикалы представляют собой заряженные частицы, стабилизированные двойным электрическим слоем. Более вероятен обрыв при захвате частицей, содержащей макрорадикал, коротких олигорадикалов или инициированных мономерных молекул из раствора.
Для подтверждения того, что именно агрегатообразование влияет на характер протекания полимеризации, была осуществлена полимеризация акрилоиламиноалкановых кислот в немицеллизующей среде – водно-диоксановой смеси (20 : 80). Соответствующие кривые конверсии представлены на рис. 4б в сравнении с данными мицеллярной полимеризации в воде (рис. 4а). Вид кривых конверсии в воде соответствует микроэмульсионному режиму [98], в водном диоксане – гомогенной полимеризации. В органическом растворителе наблюдается менее продолжительный индукционный период и более плавное начало полимеризации. Значительное замедление реакции происходит уже при конверсии 40%, тогда как в мицеллярном или микроэмульсионном режиме высокая скорость полимеризации поддерживается до конверсии 70–80%.
Рис. 4.
Зависимости конверсии мономеров от времени в ходе полимеризации при С = 0.1 моль/л в воде (а) и в смеси вода 20% + диоксан 80% (б). а: 1 – ААУ-Na, 2 – ААГ-Na, 3 – ААП-Na; б: 1 – ААУ-Н, 2 – ААГ-Н, 3 – ААП-Н [94].

Гладкие кинетические кривые свидетельствуют о том, что полимеризация ААУ-Н и ААГ-Н в водном диоксане протекает в гомогенном режиме: максимальная скорость полимеризации наблюдается в начальный период времени и затем процесс осуществляется с постепенным понижением скорости. Полимеризация ААП-Н в водном диоксане, вероятно, протекает в обращенном микроэмульсионном режиме.
Гомогенный режим полимеризации ААУ-Н и ААГ-Н в водно-диоксановой смеси обусловлен приблизительно одинаковым сродством карбоксильных групп, алифатических хвостов и растущих цепей к растворителю, следствием чего является отсутствие агрегации как мономера, так олигомеров. В случае ААП-Н соотношение полярных и гидрофобных частей молекулы мономера способствует формированию мономер-олигомерных агрегатов со структурой обращенных мицелл.
Таким образом, показано, что полимеризация акрилоиламиноалканоатов натрия в воде реализуется в микроэмульсионном режиме вне зависимости от длины углеводородной развязки между акриламидной и карбоксилатной группой. Это обусловлено агрегацией мономера на растущих макрорадикалах. Полимеризация акрилоиламинокарбоновых кислот в водно-диоксановых смесях может протекать как в гомогенном, так и в микроэмульсионном режиме в зависимости от длины углеводородной развязки: в гомогенном режиме в случае гидрофобных мономеров, в микроэмульсионном – в случае полярных.
ЗАКЛЮЧЕНИЕ
Мицеллярная полимеризация ионных мицеллообразующих мономеров в водных и неводных растворах – эффективный путь синтеза гребнеобразных полиэлектролитов и полиэлектролит-коллоидных комплексов. Мицеллярная полимеризация кинетически эквивалентна микроэмульсионной полимеризации и может быть описана в рамках кинетической модели Моргана и Калера. Мицеллярный режим полимеризации реализуется не только для мицеллообразующих мономеров, но и в растворах сильно ассоциированных мономеров.
Работа выполнена при финансовой поддержке Российского научного фонда (проект 21-73-10132).
Список литературы
Rusanov A.I. // Colloid J. 2014. V. 76. P. 121.
Sinha S.K., Medalia A.I. // J. Am. Chem. Soc. 1957. V. 79. № 1. P. 281.
Freedman H.H., Mason J.P., Medalia A.I. // J. Org. Chem. 1958. V. 23. № 1. P. 76.
Medalia A.I., Freedman H.H. // Pat. 2874151 USA. 1959.
Hyde A.J., Robb D.J.M. // J. Phys. Chem. 1963. V. 67. № 10. P. 2089.
Paleos C.M., Stassinopoulou C.I., Malllaris A. // J. Phys. Chem. 1983. V. 87. № 2. P. 251.
Rodrıguez J.L., Schulz P.C., Pieroni O., Vuano B. // Colloid Polym. Sci. 2004. V. 282. P. 734.
Larrabee C.E., Sprague E.D. // J. Colloid Interface Sci. 1986. V. 114. № 1. P. 256.
Sprague E.D., Duecker D.C., Larrabee C.E. // J. Colloid Interface Sci. 1983. V. 92. P. 416.
Samakande A., Hartmann P.C., Sanderson R.D. // J. Colloid Interface Sci. 2006. V. 296. P. 316.
Hamid S.M., Sherrington D.C. // Polymer. 1987. V. 28. № 2. P. 325.
Aoki S., Morimoto Y. // Polym. Bull. 1996. V. 37. № 6. P. 777.
Larrabee C.E., Spargue E.D. // J. Polym. Sci., Polym. Lett. 1979. V. 17. P. 749.
Wu H., Kawaguchi S., Ito I. // Colloid. Polym. Sci. 2004. V. 282. P. 1365.
Glatzhofer D.T., Cho G., Lai C.L., O’Rear E.A., Fung B.M. // Langmuir. 1993. V. 9. P. 2949.
Wang J., Warner I.M. // Anal. Chem. 1994. V. 66. № 21. P. 3773.
Billiot F.H., Mccarrol M., Billiot E.J., Rugutt J.K., Morris K., Warner I.M. // Langmuir. 2002. V. 18. № 8. P. 2993.
Arai K.,Yamazaki K. // Polym. J. 1993. V. 25. № 11. P. 1169.
Tundo P., Kippenberger D.J., Klahn P.L., Prieto N.E., Jao T.-C., Fendler J.H. // J. Am. Chem. Soc. 1982. V. 104. № 2. P. 456.
Dobashi A., Hamada M., Dobashi Y., Yamaguchi J. // Anal. Chem. 1995. V. 67. P. 3011.
Arai K., Sugita J., Ogiwara Y. // Makromol. Chem. 1987. V. 188. № 11. P. 2511.
Tian L., Yam L., Wang J., Tat H., Uhrich K.E. // J. Mater. Chem. 2004. V. 14. P. 2317.
Friberg S.E., Thundathil R., Stoffer J.O. // J. Polym. Sci. 1980. V. 18. P. 2629.
Chu D.Y., Thomas J.K. // Macromolecules. 1991. V. 24. P. 2212.
Henley E.J., Johnson E.R. Chemistry and Physics of High Energy Reactions. Washington: Univ. Press, 1969.
Егоров В.В., Зубов В.П. // Успехи химии. 1987. V. 56. № 12. P. 2076.
Egorov V.V. // J. Polym. Sci., Polym. Chem. 1995. V. 33. № 10. P. 1727.
Егоров В.В., Батракова Е.В., Зубов В.П. // Высокомолек. соед. A. 1988. V. 30. № 9. P. 1859.
Егоров В.В., Батракова Е.В., Зубов В.П. // Высокомолек. соед. A. 1990. V. 32. № 5. P. 927.
Егоров В.В., Батракова Е.В., Ксенофонтова О.Б., Зубов В.П. // Высокомолек. соед. A. 1988. V. 30. № 9. P. 1854.
Егоров В.В., Зайцев P.Ю., Зубов В.П. // Высокомолек. соед. A. 1991. V. 33. № 8. P. 1587.
Егоров В.В., Ксенофонтова О.Б. // Высокомолек. соед. A. 1991. V. 33. № 8. P. 1780.
Hamid S., Sherrington D. // J. Chem. Soc., Chem. Commun. 1986. P. 936.
Dreja M., Pyckhout-Hintzen W., Tieke B. // Macromolecules. 1998. V. 31. P. 272.
Michas J., Paleos C.M., Dais P. // Liq. Cryst. 1989. V. 5. № 6. P. 1737.
Nagai K., Ohishi Y. // J. Polym. Sci., Polym. Chem. 1987. V. 25. P. 1.
Nagai K., Ohishi Y., Inaba H., Kudo S. // J. Polym. Sci., Polym. Chem. Ed. 1985. V. 23. P. 1221.
Gutierrez-Hijar D.P.D.J., Becerra F., Puig J.E., Soltero-Martınez J.F.A., Sierra M.B., Schulz P.C. // Colloid Polym. Sci. 2004. V. 283. P. 74.
Мономерные и полимерные карбоксилаты металлов / Под ред А.Д. Помогайло, Г.И. Джардималиева. М. : Физматлит, 2009.
Gan L.M., Chew C.H. // J. Dispersion Sci. Technol. 1984. V. 5. № 2. P. 179.
Gan L.M., Yeoh K.W., Chew C.H., Koh L.L., Tan T.L. // J. Appl. Polym. Sci. 1991. V. 42. № 1. P. 225.
Imae T., Chew C.H., Gan L.M. // Colloids Surf. 1991. V. 61. P. 75.
Yeoh K.W., Chew C.H., Gan L.M., Koh L.L. // Polym. Bull. 1989. V. 22. P. 123.
Yeoh K.W., Chew C.H., Gan L.M., Koh L.L., Ng S.-C. // J. Macromol. Sci. A. 1990. V. 27. № 6. P. 711.
Yeoh K.W., Chew C.H., Gan L.M., Koh L.L., Teo H.H. // J. Macromol. Sci. A. 1989. V. 26. № 4. P. 663.
Yeoh K.W., Chew C.H., Tan T.L., Koh L.L. // Environment. Monitoring Assessment. 1991. V. 19. № 1–3. P. 215.
Roy S., Dey J. // Langmuir. 2003. V. 19. № 23. P. 9625.
Nayak R.R., Roy S., Dey J. // Polymer. 2005. V. 46. P. 12401.
Nayak R.R., Roy S., Dey J. // Colloid Polym. Sci. 2006. V. 285. № 2. P. 219.
Roy S., Dey J. // Langmuir. 2005. V. 21. № 23. P. 10362.
Roy S., Dey J. // Bull. Chem. Soc. Jpn. 2006. V. 79. № 1. P. 59.
Roy S., Dey J. // J. Colloid Interface Sci. 2007. V. 307. № 1. P. 229.
Roy S., Mohanty A., Dey J. // Chem. Phys. Lett. 2005. V. 414. № 1–3. P. 23.
Roy S., Nayak R.R., Dey J. // Colloids Surf. A. 2006. V. 290. P. 62.
Paleos C.M., Margomenou-Leonidopoulou G., Malliaris A. // Molec. Cryst. Liq. Cryst. 1988. V. 161. № 1. P. 385.
Zhang X., Wang M., Wu T., Jiang S., Wang Z. // J. Am. Chem. Soc. 2004. V. 126. P. 6572.
Hamid S.M., Sherrington D.C. // Polymer. 1987. V. 28. № 2. P. 332.
ummers M., Eastoe J. // Langmuir 2003. V. 19. № 16. P. 6357
Joynes D., Sherrington D.C. // Polymer. 1996. V. 37. № 8. P. 1453.
Tsvetkov N.V., Andreeva L.N., Zorin I.M., Bushin S.V., Lebedeva E.V., Strelina I.A., Bezrukova M.A., Lezov A.A., Makarov I.A., Bilibin A.Yu. // Polymer Science A. 2011. V. 53. № 5. P. 355.
Tsvetkov N.V., Andreeva L.N., Lebedeva E.V., Strelina I.A., Lezov A.A., Podseval’nikova A.N., Mikusheva N.G., Ivanova V.O., Makarov I.A., Zorin I.M., Bilibin A.Yu. // Polymer Science A. 2011. V. 53. № 8. P. 666.
Tsvetkov N.V., Lebedeva E.V., Lezov A.A., Podseval’nikova A.N., Akhmadeeva L.I., Mikhailova M.E., Zorin I.M., Makarov I.A., Bilibin A.Yu. // Colloid Polym. Sci. 2014. V. 292. P. 2727.
Yazhgur P.A., Akent’ev A.V., Bilibin A.Yu., Zorin I.M., Noskov B.A. // Colloid J. 2012. V. 74. № 6. P. 743.
Tsvetkov N.V., Lebedeva E.V., Lezov A.A., Podseval’nikova A.N., Akhmadeeva L.I., Zorin I.M., Bilibin A.Yu. // Polymer. 2014. V. 55. № 7. P. 1716.
Hartmann P.C., Dieudonné P., Sanderson R.D. // J. Colloid Interface Sci. 2005. V. 284. P. 289.
Fukuda H., Diem T., Stefely J., Kezdy F.J., Regen S.L. // J. Am. Chem. Soc. 1986. V. 108. № 9. P. 2321.
Lerebours B., Perly B., Pileni M.P. // Chem. Phys. Lett. 1988. V. 147. № 5. P. 503.
Lerebours B., Perly B., Pileni M.P. // Progr. Colloid Polym. Sci. 1989. V. 79. P. 239.
Hartmann P.C., Sanderson R.D. // Macromol. Symp. 2005. V. 225. P. 229.
Higashi N., Adachi T., Niwa M. // Macromolecules. 1990. V. 23. № 5. P. 1475.
Summers M., Eastoe J., Davis S., Du Z., Richardson R.M., Heenan R.K., Steytler D., Grillo I. // Langmuir. 2001. V. 17. № 17. P. 5388.
Kline S.R. // Langmuir. 1999. V. 15. P. 2726.
Gerber M.J., Kline S.R., Walker L.M. // Langmuir. 2004. V. 20. № 20. P. 8510.
Gerber M.J., Walker L.M. // Langmuir. 2006. V. 22. № 3. P. 941.
Kline S.R. // J. Appl. Cryst. 2000. V. 33. № 3. P. 618.
Kuntz D.M., Walker L.M. // J. Phys. Chem. B. 2007. V. 111. P. 6417.
Walker L.M., Kuntz D.M. // Current Opinion Colloid Interface Sci. 2007. V. 12. P. 101.
Bilibin A.Yu., Scherbinina T.M., Kondratenko Ju.A., Zorina N.A., Zorin I.M. // Colloid. Polym. Sci. 2015. V. 293. P. 1215.
Zorin I.M., Shcherbinina T.M., Mel’Nikov A.B., Molchanov V.S., Bilibin A.Yu. // Colloid. J. 2014. V. 76. № 3. P. 314.
Bilibin A.Yu., Sukhanova T.M., Kondratenko Yu.A., Zorin I.M. // Polymer Science B. 2013. V. 55. № 1–2. P. 22.
Bilibin A.Yu., Shcherbinina T.M., Girbasova N.V. Lebedev V.T., Kulvelis Yu.V., Molchanov V.S., Zorin I.M. // Design. Monomers Polymers. 2016. V. 19. № 5. P. 369.
Tsvetkov N.V., Mikusheva N.G., Lezov A.A., Gubarev A.S., Mikhailova M.E., Podseval’nikova A.N., Akhmadeeva L.I., Lebedeva E.V., Zorin I.M., Shcherbinina T.M., Bilibin A.Yu. // Eur. Polym. J. 2016. V. 75. P. 251.
Zorin I.M., Shcherbinina T.M., Demidov E.I., Mechtaeva E.V., Zorina N.A., Fetin P.A., Bilibin A.Yu. // Colloid Polym. Sci. 2019. V. 297. P. 1169.
Shulevich Y.V., Dukhanina E.G., Bykov D.S., Navrotskii A.V., Novakov I.A., Zakharova Yu.A., Tolstoi P.M., Vovk M.A. // Polymer Science B. 2019. V. 61. № 6. P. 715.
Shulevich Y., Dukhanina E., Navrotskii A., Novakov I. // Colloid Polym. Sci. 2018. V. 296. P. 871.
Novakov I.A., Shulevich Y.V., Zakharova Y.A., Chang L.T.D., Dukhanina E.G., Navrotskii A.V. // Russ. Chem. Bull. 2015. V. 64. P. 597.
Shulevich Y.V., Kovaleva O.Y., Navrotskii A.V., Zakharova Yu.A., Zezin A.B., Novakov I.A. // Polymer Science A. 2007. V. 49. № 12. P. 1284.
Bezzaoucha F., Lochon P., Jonquieres A., Fischer A., Brembilla A., Ainad-Tabet D. // Eur. Polym. J. 2007. V. 43. P. 4440.
Patist A., Oh S.G., Leung R., Shah D.O. // Colloids Surf. A. 2001. V. 176. № 1. P. 3.
Cochin D., Zana R., Candau F. // Macromolecules. 1993. V. 26. № 21. P. 5765.
Cochin D., Candau F., Zana R. // Macromolecules. 1993. V. 26. № 21. P. 5755.
Kim T.-H., Choi S.-M., Kline S.R. // Langmuir. 2006. V. 22. P. 2844.
Zhu Z., Gonzalez Y.I., Xu H., Kaler E.W., Liu S. // Langmuir. 2006. V. 22. P. 949.
Zorin I.M., Podolskaya E.P., Bilibin A.Yu. // Eur. Polym. J. 2019. V. 110. P. 355.
Nomura M., Tobita H., Suzuki K. // Adv. Polym. Sci. 2005. V. 175. P. 1.
Schork F.J., Luo Y., Smulders W., Russum J.P., Butté A., Fontenot K. // Adv. Polym. Sci. 2005. V. 175. P. 129.
Chow P.Y., Gan L.M. // Adv. Polym. Sci. 2005. V. 175. P. 257.
Katime I., Arellano J., Schulz P. // J. Colloid Interface Sci. 2006. V. 296. P. 490.
Morgan J.D., Lusvardi K.M., Kaler E.W. // Macromolecules. 1997. V. 30. № 7. P. 1897.
Tajima K., Aida T. // Chem. Commun. 2000. P. 2399.
Зорин И.М., Макаров И.А., Поляков Н.Б., Подольская Е.П., Билибин А.Ю. // Научное приборостроение. 2008. Т. 18. № 4. С. 73.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия С)