

Химия высоких энергий, 2019, T. 53, № 2, стр. 87-94
Кремний-фторорганические олигомеры с концевыми группами, способными к дальнейшей поликонденсации
И. П. Ким a, *, В. М. Мартыненко a, А. В. Черняк a, А. Ф. Шестаков a, В. А. Бендерский a
a Институт проблем химической физики Российской Академии Наук
4 Московская обл., Черноголовка, просп. академика Семенова 1, Россия
* E-mail: ipkim@icp.ac.ru
Поступила в редакцию 29.10.2018
После доработки 30.10.2018
Принята к публикации 30.10.2018
Аннотация
Установлено, что при радикальной полимеризации тетрафторэтилена (ТФЭ) в жидких алкоксисиланах (диметилдиметокси-, триметокси-, метилтриметокси- и тетраметоксисилане) образуются кремний-фторорганические олигомеры с общей формулой R(C2F4)nH, где RH – молекула алкоксисилана, способная к дальнейшей поликонденсации. Состав концевых групп и молекулярно-массовое распределение олигомеров определены из масс-спектров, спектров ЯМР и дифференциальных термогравиметрических кривых. С ростом концентрации ТФЭ в растворах рост длины цепи сопровождается переходом от гомогенного к коллоидному раствору и затем гелю олигомеров. При максимальных концентрациях ТФЭ длина цепи n в коллоидных растворах достигает 10–12. В состав коллоидных частиц входят олигомеры и молекулы растворителя, число которых в расчете на одно мономерное звено минимально в геле и равно 4–6. На основе полученных олигомеров становится принципиально возможным создать покрытия с одновременно высокой гидрофобностью и прочной связью с защищаемой поверхностью.
Амфифильные олигомеры с общей формулой X(C2F4)nR, X = H, F, Cl, в состав которых входит гидрофобная перфторалкильная цепь (n ≥ 4) и гидрофильная концевая группа R, считаются наиболее перспективными веществами для создания гидрофобных антиобледенительных и самоочищающихся покрытий [1–5]. Гидрофобная цепь обеспечивает удаление капель переохлажденной воды, а концевая группа – прочную связь покрытия с защищаемой поверхностью.
В работах [6–8] разработан метод радикальной полимеризации тетрафторэтилена (ТФЭ) в растворах, позволивший получить олигомеры с регулируемой в широких пределах длиной цепи (от 4 до 20) и различными концевыми группами (в том числе карбонилсодержащими и карбоксильными). Слои этих олигомеров, имеющих форму жестких стержней, переходят в ультрагирофобное состояние с наноразмерной шероховатостью, благодаря образованию надмолекулярного каркаса, создаваемого водородными связями концевых групп. Однако, по своим адгезионным характеристикам, покрытия на основе этих олигомеров заметно уступают покрытиям на основе кремнийорганических соединений. Недостатком известных к настоящему времени фторaлкилсиланов ФАС-15, ФАС-17 и ФАС-19 [9–11] (в сокращенном названии указывается число атомов фтора в молекуле) является заметная потеря ультрагидрофобности при длительном пребывании в условиях высокой влажности [12] из-за проникновения молекул воды в объем пористых слоев этих соединений с короткой фторалкильной цепью. Этот нежелательный эффект уменьшается с ростом длины фторалкильной цепи [13], так что получение соединений типа ФАС с более длинной цепью весьма актуально.
В настоящей работе, продолжающей цикл работ [6–8], ФАС получены не химическим синте-зом в виде индивидуальных соединений как в [9–13], а в виде олигомеров с управляемым молекулярно-массовым распределением (ММР) методом радикальной полимеризации, и найдены условия, в которых длина цепи достигает 10–12 (вместо 4 в ФАС-19). В отличие от описанных ранее перфторированных олигомеров кремний-фторорганические олигомеры можно подвергнуть поликонденсации с образованием силиконового каркаса –Si–O–Si– с присоединенными к нему параллельно ориентированными FAn-цепями, обеспечивающими гидрофобность поверхности [13].
Длина цепи олигомеров, образующихся при радикальной полимеризации в жидких растворах, увеличивается с ростом концентрации ТФЭ и определяется отношением констант скорости роста (присоединения ТФЭ к растущему макрорадикалу Rn) и передачи цепи (переноса атома водорода от молекулы растворителя HR0 к макрорадикалу с регенерацией первичного радикала R0, инициирующего рост цепи) [14, 15]
(1)
${{{\text{R}}}_{0}}\xrightarrow{{{{{\text{C}}}_{2}}{{{\text{F}}}_{4}}}}{{{\text{R}}}_{1}} \to ... \to {{{\text{R}}}_{n}}\xrightarrow{{{{{\text{C}}}_{2}}{{{\text{F}}}_{4}}}}{{{\text{R}}}_{{n + 1}}},$(2)
${{{\text{R}}}_{n}}\xrightarrow{{{\text{H}}{{{\text{R}}}_{0}}}}{\text{H}}{{{\text{R}}}_{n}} + {{{\text{R}}}_{0}}.$В органических растворителях, содержащих СН-связи со свободными энергиями разрыва 95–100 Ккал/моль, константа скорости реакции (2), свободная энергия которой лежит в интервале ±2.5 Ккал/моль, на несколько порядков меньше, чем диффузионно-ограниченной сильно экзотермической реакции (1). Отношение этих констант обеспечивает образование олигомеров с длиной цепи ∼102 в растворах ТФЭ с концентрацией ~1 моль/л [7, 15]. Образование олигомеров с длиной цепи такого порядка наблюдалось в растворах ТФЭ в ацетоне, метилэтилкетоне, этилацетате, тетрагидрофуране, окиси пропилена и других кислород содержащих растворителях [16, 17]. Передача цепи, обусловленная переносом атома хлора, определяет длину олигомеров в растворах ТФЭ в тетрахлорметане и фреонах [18]. В растворах с более высокой концентрацией ТФЭ, рост длины цепи становится сублинейным из-за подавления диффузионно-ограниченной реакции (1) при переходе от гомогенного к коллоидному раствору образующихся олигомеров [6, 19]. При образовании геля длина цепи стремится к постоянному значению из-за уменьшения скорости роста цепи, вызванного увеличением вязкости. Поскольку полимеризация сопровождается образованием коллоидного раствора, традиционные методы определения ММР, в частности гель-проникающая хроматография и вискозиметрия, применимы только в гомогенных растворах коротких олигомеров (n ≤ 5). Для определения ММР более длинных олигомеров применялись методы дифференциальных термогравиметрических (ДТГ) кривых и масс-спектрометрии.
Метод определения ММР по измеренным кривым ДТГ и рассчитанным температурам кипения олигомеров развит в работах [17, 20–22]. Поскольку температуры кипения олигомеров возрастают с ростом длины цепи, ДТГ кривые продуктов полимеризации однозначно связаны с ММР в области длин цепей, при которых скорость деструкции остается меньше скорости испарения. Для фторалкильных олигомеров деструкция происходит выше 500°С, при которой испаряются олигомеры с длиной цепи n ≥ 20–25. Температуры кипения олигомеров рассчитываются по аддитивной схеме, как сумма инкрементов функциональных групп, входящих в их состав [20–22]. Для ФАС корреляционные соотношения для расчета температур кипения при нормальном давлении ниже 700 K имеют вид [20]
(3)
$\begin{gathered} {{{\text{T}}}_{b}} = - 94.84 + 1.5577 \cdot {{{\text{T}}}_{{bc}}} - 0.000771 \cdot {\text{T}}_{{bc}}^{2}, \\ {{{\text{T}}}_{{bc}}} = 198.2 + \sum\limits_s {\Delta {{{\text{T}}}_{s}}} , \\ \end{gathered} $ПОЛУЧЕНИЕ ОЛИГОМЕРОВ И МЕТОДЫ ИЗМЕРЕНИЙ
Использованы растворители: (I) диметилдиметоксисилан (CH3)2Si(OCH3)2 фирмы Acros Organics чистотой 95%, (II) триметоксисилан HSi(OCH3)3 фирмы Sigma-Aldrich чистотой 97%, (III) метилтриметоксисилан CH3Si(OCH3)3 и (IV) тетраметоксисилан Si(OCH3)4 – оба фирмы Acros Organic-s чистотой 97% и 99% соответственно. Газообразный ТФЭ очищался от ингибитора в колонке с активированным углем. Растворитель освобождался от газообразного кислорода многократной откачкой при замораживании. Затем в ампулу с твердым охлажденным растворителем конденсировалось требуемое количество ТФЭ. Для радиационно-химического инициирования гамма-квантами 60Со использовалась установка “Гамматок-100”. Облучение реакционной смеси производилось при комнатной температуре и давлении от 1 до 5 атм. Полная конверсия ТФЭ происходит при интегральной дозе облучения 15 кГр. Экспериментальные данные исследования олигомеров приведены для радиационно-инициированных образцов. При химическом инициировании в растворитель перед дегазацией вносилось требуемое количество (от 0.01 до 0.1 моль/л) твердого инициатора (АИБН, ПБ), и полимеризация проводилась при 70, 90°С в течение 6–8 ч. Суммарный выход продуктов полимеризации определялся из отношения массы твердых и жидких продуктов (за вычетом массы не вошедшего в реакцию газообразного ТФЭ) к массе реагентов.
Термогравиметрические измерения проведены на дериватографе “Q-1500D” в стандартных корундовых открытых тиглях. Скорость нагрева 5°С/мин, масса навесок 50–100 мг, эталон – Al2O3. Образцы готовили удалением растворителя из облученных растворов вакуумной отгонкой при комнатной температуре.
Гель-проникающие хроматограммы (ГПХ) измерены на хроматографе Waters GPCV-2000. На входе в колонку была установлена мембрана с размером пор ~1 мкм, так что ГПХ воспроизводят молекулярно-массовое распределение только золь-фракции.
Концевые группы и длина цепи олигомеров идентифицированы по масс-спектрам в области массовых чисел от 10 до 2000 на жидкостном хромато-масс-спектрометре LCMS-2020 фирмы Shimadzu (Japan) с ионизацией электроспрея (ESI) и квадрупольным анализатором масс (FWHM) с разрешением по массовому числу 0.6. В качестве подвижной фазы использовали метанол.
Спектры ЯМР на ядрах 1H и 19F регистрировались спектрометром высокого разрешения AVANCE III 500 MHz, фирмы “Bruker” с рабочими частотами 500, 471 МГц соответственно при 22°С. Калибровку шкалы химических сдвигов осуществляли по сигналу тетраметилсилана (ТМС), как внешнего стандарта (использовали капилляр с смесью DMSO-d6 – ТМС). Расшифровку сигналов проводили с использованием стандартных гомо- и гетероядерных корреляций COSY 19F–19F, HETCOR 19F–1H.
Образование коллоидных частиц наблюдалось методом оптической регистрации кинетических седиментационных кривых [6] с помощью анализатора Horiba-500 при центробежном ускорении ~500g (3000 об./мин). Объемная доля коллоидных частиц найдена из отношения объема осадка после центрифугирования к начальному объему. Молярное отношение олигомер/растворитель в коллоидных частицах и гелях определялось взвешиванием осадков до и после отгонки растворителя.
Для молекулярного моделирования использован новый полуэмпирический метод расчета электронной структуры [24], который обеспечивает хорошую точность межмолекулярных потенциалов и является многообещающим для изучения крупных молекулярных систем. В отличие от традиционных полуэмпирических моделей, этот метод сопоставим по точности с теорией функционала плотности с учетом поправок на дисперсионное взаимодействие при значительно меньших (на ~3 порядка) вычислительных затратах [25].
РЕЗУЛЬТАТЫ ИЗМЕРЕНИЙ
При радиационно-химическом инициировании полимеризации ТФЭ в растворах алкоксисиланов при начальных концентрациях ТФЭ C0 < $C_{0}^{*},$ где $C_{0}^{*}$ составляет ~1.04, 1.7, 0.67, и 0.8 моль/л, об-разуются гомогенные растворы олигомеров в I–IV соответственно. Коллоидные растворы образуются в интервале концентраций $C_{0}^{*}$ < C0 < $C_{0}^{{*{\text{*}}}},$ где $C_{0}^{{*{\text{*}}}}$ = 1.87, 5.9, 1.75, 2.5 для I–IV. При C0 > $C_{0}^{{*{\text{*}}}}$ коллоидный раствор переходит в гель. По данным ГПХ, воспроизводящим ММР в гомогенных растворах, в максимуме ММР длина цепи при C0 ≈ $C_{0}^{*}$ составляет 2–4.
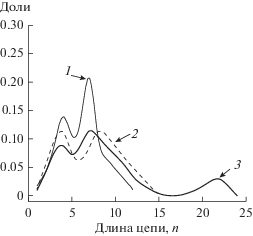
Как следует из табл. 1, в которой приведены рассчитанные по формуле (3) температуры кипения олигомеров I–IV с различной длиной цепи, испарение олигомеров, образующихся в гомогенных растворах, происходит при температурах 200–250°С (температуры пиков испарения на 15–20°С ниже температур кипения [22]). ДТГ кривые при C0 < $C_{0}^{*}$ содержат широкие полосы, относящиеся к испарению олигомеров с длиной цепи n > 3. ММР, расчитанное из этих кривых, согласуется с данными ГПХ. При C0 > $C_{0}^{*}$ ДТГ кривые смещаются в сторону более высоких температур и содержат неразрешенные полосы, относящиеся к испарению олигомеров при T < T1; узкие пики деструкции в области T1 = 330–340°С, которым соответствуют пики на кривых тепловыделения; полосы испарения продуктов частичной деструкции при T > T1; и заканчиваются пиками полной деструкции при T2 = 550 – 570°С (рис. 1). Поскольку пики при T1 не наблюдаются в изученных ранее фторалкильных олигомерах, их следует отнести к отщеплению метоксигрупп. Изменение формы ДТГ кривых с ростом начальной концентрации ТФЭ C0 объясняется изменением доли испаряющихся и деструктирующих олигомеров. При низких концентрациях ТФЭ пик деструкции мал, означая, что образуются олигомеры, температура испарения которых меньше T1 (в частности, золь-фракция при C0 < $C_{0}^{*}$). В области T ≤ T1 испаряются олигомеры с длиной цепи n ≤ 7–8 (табл. 1). Площадь пика при T1 определяется потерей массы более длинных олигомеров с температурами кипения выше 350°С. Широкие полосы в интервале T1 < T < T2 обусловлены испарением продуктов деструкции, в том числе димеров, с температурами кипения, меньшими T2, которым соответствует удвоенная длина фторалкильной цепи, n ≤ 15–17. ММР, рассчитанные из кривых ДТГ и по данным табл. 1, имеет два максимума: низкомолекулярный n1 = 2–4 (близкий к наблюдаемому в гомогенных растворах) и n2 ≈ 5–12 (рис. 2). В модели пространственно неоднородной радикальной полимеризации, предложенной в [19, 23], при бимодальном ММР низкомолекулярная часть ММР относится к гомогенному раствору, а высокомолекулярная к – коллоидному раствору и гелю.
Таблица 1.
Рассчитанные температуры кипения фторалкильных олигомеров I–IV с длиной цепи n при нормальном давлении (°С)*
| Длина цепи, n | I (CH3)2Si(OCH3)2 |
II HSi(OCH3)3 |
III (CH3)Si(OCH3)3 |
IV Si(OCH3)4 |
|---|---|---|---|---|
| 0 | 78.5(81.4**) | 78.9 (81**) | 101.7(103**) | 124.1(121–122**) |
| 1 | 118.8 | 119.1 | 140.5 | 161.4 |
| 2 | 156.4 | 156.8 | 176.8 | 196.2 |
| 3 | 191.6 | 191.9 | 210.4 | 228.4 |
| 4 | 224.1 | 224.4 | 241.5 | 258.1 |
| 5 | 254.2 | 254.4 | 270.1 | 285.2 |
| 6 | 281.6 | 281.8 | 296.1 | 309.7 |
| 7 | 306.5 | 306.7 | 319.5 | 331.6 |
| 8 | 328.8 | 328.9 | 340.3 | 351.0 |
| 9 | 348.5 | 348.7 | 358.6 | 367.9 |
| 10 | 365.7 | 365.8 | 374.3 | 382.2 |
| 11 | 380.3 | 380.4 | 387.5 | 393.9 |
Рис. 1.
ДТГ кривые олигомеров, образующихся при полимеризации ТФЭ в II (триметоксисилане). На панелях сверху вниз начальные концентрации ТФЭ 1.7, 2.5 и 5.9 моль/л соответственно.
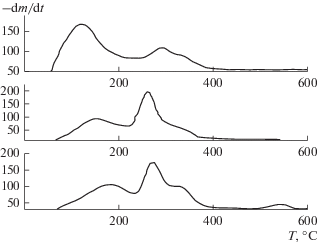
Рис. 2.
ММР олигомеров, образующихся при полимеризации ТФЭ в III (метилтриметоксисилане) при концентрациях ТФЭ 0.67 (кривая 1), 1.04 (2) и 1.75 (3) моль/л.
В спектрах ЯМР 19F золь-фракции олигомера триметоксисилана (II) (С0 = 1.7 моль/л) наблюдается серия из дублетов в области химических сдвигов от –139.1 до –138.9 м. д., которые относятся к фрагментам –СF2-концевой группы HСF2–, в олигомерах состава H(C2F4)nSi(OCH3)3 с n > 1. Дублет при ‒136.54 м. д. относится к концевой группе HCF2– –CF2–Si(OCH3)3, n = 1. Поскольку все олигомеры содержат только одну такую группу, отношение интенсивностей дублетов к общей интенсивности сигналов в спектре 19F непосредственно определяет среднюю длину цепи n = 2.6. Аналогичный метод оценки средней длины фторалкильной цепи описан в работе [26]. Отнесение линий в спектре 19F подтверждает спектр 1H в условиях подавления спин-спинового взаимодействия с ядрами 19F, в котором наблюдается серия из, как минимум, 7 линий. Из-за наложения сигналов концевых групп однозначно удается определить интенсивность только олигомера n = 1 при 6.42 м. д. Все остальные линии не индивидуальны. Отношение интенсивностей линий для групп HCF2 в спектре 1H составляет 1 : 1.6 : 0.3 : 0.9 : : 0.2 : 0.5 : 0.5 (за 1 принята интенсивность HCF2– –CF2–Si(OCH3)3. Для интерпретации линий в спектрах олигомеров H(C2F4)nSi(OCH3)3 были измерены двумерные спектры COSY 19F–19F и HETCOR 19F‒1H. Параметры спектров ЯМР олигомеров II:
n = 1, ЯМР 1H (δ, м.д., J, Гц): 6.42 (–CHF2, т.т., J2(H–F) = 54.0, J3(H–F) = 6.0), 4.3 (–OCH3, c.)
n = 1, ЯМР 19F (δ, м.д., J, Гц): –136.54 (α-CHF2, д., J2(H–F) = 54.0), –131,79 (β-CF2, м.)
n ≥ 2, ЯМР 1H (δ, м.д.,): 6.89–7.04 (–CHF2, м.), 4.0–4.3 (–OCH3, c.)
n ≥ 2, ЯМР 19F (δ, м.д., J, Гц): –139.08–138.87 (α-CHF2, м.), –131.55–130.35 (β-CF2–, м.), ‒124.46–121.82 (–CF2– остальные, м.)
В спектрах ЯМР 19F золь-фракции олигомера III (${{C}_{0}} = $ 0.48 моль/л) наблюдается серия из дублетов в области химических сдвигов от –138 до –142 м. д., которые относятся к фрагментам концевой группы HСF2–. Как и для олигомера II из спектра 19F была определена средняя длина цепи $\bar {n}$ = 2.2. По отношению интенсивностей сигналов концевых групп HСF2– в спектре 1H (с подавлением спин-спинового взаимодействия с ядрами 19F) непосредственно определяем ММР и среднюю длину цепи $\bar {n}$ = 2.2 (что хорошо согласуется с данными, полученными по ЯМР спектру 19F). Полученное мольное отношение для олигомеров составило 1 : 0.73 : 0.39 : 0.21 : : 0.10 : 0.08 для n = 1, 2, 3, 4, 5, 6 соответственно. Измерения двумерных спектров COSY 19F – 19F и HETCOR 19F–1H показало, что линия 6.62 м. д. относится к n = 1, а линия 6.89 м. д. – к n = 2. Следующие линии серии можно отнести к более длинным олигомерам: 6.97 (n = 3), 7.00 (n = 4), 7.01 (n = 5), 7.02 (n = 6). Таким образом, независимые измерения спектров ЯМР ядер 1H и 19F позволяют измерить ММР золь-фракции и определить параметры спектров ЯМР индивидуальных олигомеров III:
n = 1, ЯМР 1H (δ, м.д., J, Гц): 6.62 (–CHF2, т.т., J2(H–F) = 52.9, J3(H–F) = 5.2), 4.62 (–CH2–, т.т., J3(H–F) = 12.7, J4(H–F) = 1.5), 4.07 (–OCH3, c.)
n = 1, ЯМР 19F (δ, м.д., J, Гц): –141.50 (α-CHF2, д.т., J2(H–F) =52.9, J3(F–F) = 4.6), –128.66 (β-CF2–, т. J3(F–F) = 4.6)
n = 2, ЯМР 1H (δ, м.д., J, Гц): 6.89 (–CHF2, м.), 4.76 (–CH2–, м.), 4.07 (–OCH3, c.)
n = 2, ЯМР 19F (δ, м.д., J, Гц): –138.98 (α-CHF2, м.), –131.41 (β-CF2–, м.), –126.21 (γ-CF2–, м.), ‒122,51 (δ-CF2–, м.)
n = 3–6, ЯМР 1H (δ, м.д., J, Гц): 6.97–7.02 (‒CHF2, м.), 4.77–4.78 (–CH2–, м.), 4.07–4.10 (‒OCH3, c.)
n = 3–6, ЯМР 19F (δ, м.д., J, Гц): –138.87–138.77 (α-CHF2, м.), –130.57–130.14 (β-CF2–, м.), –124.27–122.65 (γ-CF2–, м.), –122.50–122.11 (δ-CF2–, м.)
В II идентификация концевой группы не вызывает сомнений, поскольку энергия разрыва связи Si–H на ~15 Ккал/моль меньше, чем связей CH в метоксильных группах и реакция передачи цепи связана с переносом атома водорода фрагмента SiH
(5)
$ - {\text{CF}}_{2}^{\centerdot } + {\text{HSi(OC}}{{{\text{H}}}_{3}}{{)}_{3}} \to - {\text{C}}{{{\text{F}}}_{{\text{2}}}}{\text{H}}{{ + }^{\centerdot }}{\kern 1pt} {\text{Si(OC}}{{{\text{H}}}_{3}}{{)}_{3}}.$По квантово-химическим данным, полученным методом CCSD(T) в базисе $6G({{311}^{ + }},d{\text{**}}),$ энтальпии разрыва связи CH в метильной и метоксильной группах при 298 К составляют 98.7 и 93.8 Ккал/моль соответственно. Значения энтальпий позволяют предположить, что в реакции передачи цепи скорее происходит разрыв связи CH в метоксильной, чем в метильной группе, т.е. образуется олигомер с концевой группой –CH2OSi(OCH3)2CH3, а не ‒CH2Si(OCH3)3. Это предположение подтверждает группа мультиплетов в области 4.6–4.8 м.д. Суммарная интенсивность мультиплетов в 2 раза выше, чем линий CHF2 в области 6 м. д. При подавлении спин-спинового взаимодействия с ядрами 19F мультиплеты превращаются в синглеты. Исходя из значений химических сдвигов, интенсивности и мультиплетности этих линий до и после подавления взаимодействия 19F–1H, эти линии относятся к протонам группы –CH2OSi–, что подтверждает указанную выше структуру концевой группы
(6)
$\begin{gathered} - {\text{CF}}_{2}^{\centerdot } + {\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{Si(OC}}{{{\text{H}}}_{3}}{{)}_{3}} \to - {\text{C}}{{{\text{F}}}_{{\text{2}}}}{\text{H}} + \\ + \,{{\,}^{\centerdot }}{\text{C}}{{{\text{H}}}_{{\text{2}}}}{\text{OSi(OC}}{{{\text{H}}}_{3}}{{)}_{2}}{\text{C}}{{{\text{H}}}_{3}}. \\ \end{gathered} $Масс-спектры анионов, образующихся при диссоциативной ионизации фторалкильных олигомеров [7, 17], содержат линии, плотно заполняющие измеряемый диапазон массовых чисел вплоть до массового числа наиболее длинного олигомера. Наиболее интенсивные линии образуют арифметические прогрессии $a$ + 100n, в которой массовые числа соседних линий отличаются на массовое число ТФЭ, равное 100 (рис. 3, 4). Появление таких прогрессий означает, что рост цепи действительно обусловлен присоединением к растущему радикалу молекулы ТФЭ, в согласие со стехиометрическим уравнением (1). Начальный член прогрессии $a$ и интервал значений n позволяют идентифицировать состав концевых групп и определить по относительным интенсивностям линий в прогрессии ММР олигомеров. Отнесение наиболее интенсивных линий и интервал значений n в наблюдаемых прогрессиях приведены в табл. 2. Помимо прогрессий анионов, в табл. 2 включены наблюдаемые прогрессии димеров, с относительной интенсивностью, большей 0.2 от наиболее интенсивной полосы олигомеров. Образование димеров в ионизационной камере масс-спектрометра обусловлено гидролизом (с участием остаточных молекул воды) и последующей димеризацией [27 ] . Прогрессии димеров, в которых суммарная длина двух фторалкильных цепей (n' + n'') изменяется от 2–4 почти до удвоенной длины цепи в олигомерах, говорит о том, что константа скорости димеризации слабо зависит от длины фторалкильной цепи. Преимущественное образование анионов ${\text{H(}}{{{\text{C}}}_{{\text{2}}}}{{{\text{F}}}_{4}})_{n}^{ - }$ в олигомерах II и III обусловлено разрывом С–С-связи между фторалкильной цепью и фрагментами Si(OCH3)3 и CH3(OCH3)2SiOCH2 соответственно. В олигомерах I происходит разрыв связи Si–C, а в IV анионы образуются без разрыва связи. В олигомерах II наблюдается также последовательность полос, связанных с разрывом связи Si–C. Последовательность полос анионов H(C2F4)n CH2O– с массовыми числами m/z = 31 + 100n в масс-спектрах продуктов полимеризации ТФЭ в I при начальной концентрации ТФЭ 1.02 моль/л показана на рис. 3. Длина цепи олигомера составляет 3–7. ММР хорошо согласуется с данными ДТГ в предположении [27 ] , что вероятность образования анионов не зависит от длины фторалкильной цепи и относительные концентрации анионов пропорциональны концентрациям олигомеров. Последовательности полос анионов H(C2F4)nSi(OCH3)2O– с массовыми числами m/z = 107 + 100n ·в масс-спектрах продуктов полимеризации ТФЭ в II при исходных концентрациях 0.21 и 1.56 моль/л показаны на рис. 4. Анионы образуются при отщеплении метильных групп от молекул олигомера. С ростом исходной концентрации длина цепи олигомера и доля более длинных олигомеров растут. Значения длины цепи n в этих последовательностях удовлетворительно согласуются с ММР, найденными по кривым ДТГ. В обоих случаях наблюдается сдвиг распределения в сторону более длинных цепей с ростом C0. В отличие от прогрессий в масс-спектрах фторалкилсиланов [28], спектры олигомеров I–IV, изученных в настоящей работе, содержат также полосы анионов, в которых метоксигруппы заменены гидроксилами, и полосы димеров FAnSi–O–SiFAn, (FAn – фторалкильная цепь из n мономеров) подтверждающие, что в ионизационной камере спектрометра в присутствие метанола и воды происходят газофазный гидролиз и димеризация фторалкоксисиланов. Следует подчеркнуть, что масс-спектры содержат полосы димеров и продуктов их гидролиза, в которых сохраняются длинные фторалкильные заместители, т.е. отщепление FAn при гидролизе метоксигрупп не происходит.
Рис. 3.
Последовательности полос анионов H(C2F4)nCH2O– c массовыми числами m/z = 31 + + 100n в масс-спектрах продуктов полимеризации ТФЭ в I (диметилдиметоксисилане) при начальной концентрации ТФЭ 1.02 моль/л.
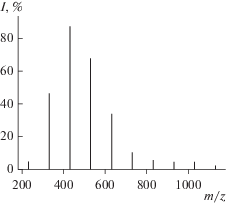
Рис. 4.
Последовательности полос анионов H(C2F4)nSi(OCH3)2O– с массовыми числами m/z = = 107 + 100n в масс-спектрах продуктов полимеризации ТФЭ в II (триметоксисилане) при начальных концентрациях ТФЭ 0.21 и 1.56 моль/л (верхняя и нижняя панель соответственно). Анионы образуются при отщеплении метильных групп от олигомеров.
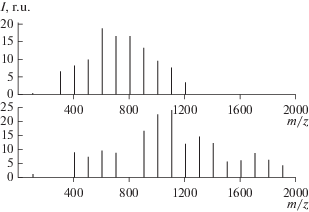
Таблица 2.
Прогрессии массовых чисел анионов, продуктов диссоциативной ионизации олигомеров, образующихся при полимеризации ТФЭ в алкоксисиланах I–IV и последующих гидролизе и димеризации олигомеров, и значения n олигомеров. В скобках указаны начальные концентрации ТФЭ в моль/л
| Анионы, R | m/z | I n, (n' + n'') (1.02) |
II n, (n' + n'') (1.5) |
III n, (n' + n'') (0.67) |
IV n, (n' + n'') (0.66) |
|---|---|---|---|---|---|
| H(C2F4)n | 1 + 100n | 4–9 | 4–11 | ||
| H(C2F4)nCH2O | 31 + 100n | 3–7 | |||
| H(C2F4)nCH2OSiOH | 76 + 100n | 4–12 | |||
| H(C2F4)nCH2OSi(OCH3)(CH3) | 105 + 100n | 5–11 | |||
| H(C2F4)nCH2OSi(OCH3)2O | 107 + 100n | 3–10 | |||
| H(C2F4)nCH2O–Si(OCH3)3 | 153 + 100n | 4–7 | |||
| H(C2F4)nCH2O–Si(CH3)2–O–Si(CH3)2 | 163 + 100n | 4–11 | |||
| H(C2F4)n′Si(OH)2–O–Si(OH)2(C2F4)n'' | 141 + 100 (n' + n'') |
10–15 | |||
| H(C2F4)n′Si(OCH3)2–O–Si(OCH3) (C2F4)n′′ | 167 + 100 (n' + n'') | 1–4 | |||
| H(C2F4)n′CH2OSi(CH3)(OH)–O–Si(CH3)(OH)OCH2(C2F4)n′′ | 195 + 100 (n' + n'') | 1–3 | |||
| (C2F4)n′CH2O–Si(CH3)2–O–Si(CH3)–O–Si(CH3)2–OCH2(C2F4)n′′ | 251 + 100 (n' + n'') | 6–12 | |||
| (C2F4)n′CH2O–Si(OH)(OCH3)–O–Si(OH)2–O–Si(OH)(OCH3)–OCH2(C2F4)n′′ | 305 + 100 (n' + n'') |
2–11 | |||
| H(C2F4) n′CH2O–Si(OH)(OCH3)–O–Si(OH)(OCH3)–O– Si(OH)(OCH3)–OCH2(C2F4) n′′ | 322 + 100 (n' + n'') | 3–12 |
ВЫВОДЫ
Совокупность экспериментальных данных по ДТГ-кривым и масс-спектрам позволяет заключить:
1. длина цепи олигомеров ФАС увеличивается с ростом C0;
2. в гомогенных растворах длина цепи, составляющая 3–5, близка к значениям n в полученных химически индивидуальных соединениях [10–13];
3. в гелях, образующихся при полимеризации I–IV при C0 ~ 1.0 моль/л, длина цепи максимальна и достигает 10–12.
Поскольку короткие олигомеры сосредоточены в гомогенном растворе, а длинные в плотной фазе [19, 23], олигомеры с длиной цепи, меньшей 4–5, можно отделить осаждением коллоидных частиц и получить длинные олигомеры (по сравнению с известными ранее ФАС-17 и ФАС-19) с узким и регулируемым ММР [8].
По сравнению c другими, твердые перфторированные полимеры обладают уникально низкой поверхностной энергией 12–15 эрг/см2, обусловленной параллельно ориентированными FAn-цепями, в которых плотная упаковка атомов фтора обеспечивает дальнодействующий отталкивающий потенциал и, как следствие, гидрофобность поверхности [29]. Эти поверхностные свойства подробно изучены в слоях фторопласта [30] и жидких перфторалканах [31]. Параллельная ориентация FAn-цепей при поликонденсации ФАС с образованием силиконового каркаса –Si–O–Si– с присоединенными к нему FAn-цепями подтверждена рентгено-структурными данными [13].
Теоретический расчет структуры и энергии модельной молекулы [F(C2F4)7]8Si8O12 в различных конформациях показывает очень высокую склонность фторалкильных цепей к агрегированию (рис 5). Рассмотренные изомеры (рис. 5а) с компактным расположением 7 цепей и (рис. 5б) с тремя кластерами из 2, 4 и 2 цепей отличаются по энергии на 138 Ккал/моль (изменение энергии связи двух CF2-фрагметов соседних цепей с различной ориентацией ~5 Ккал/моль). Одна из фторалкильных групп сильно изгибается для того, чтобы создать хороший контакт с основным кластером из 6 цепей. Эти данные указывают, что длинные фторалкильные цепи образуют упорядоченные 2D кластеры при достаточной поверхностной плотности, определяемой силиконовым скелетом. При фиксированных расстояниях между основаниями фторалкильных цепей степень порядка будет увеличиваться с ростом длины цепи. Это показывает целесообразность применения фторалкоксилсиланов с более длинной цепью, чем ФАС-19, для создания гидрофобных покрытий.
Рис. 5.
Структура основного изомера (а) и высокоэнергетического конформера (б) поликонденсированного ФАС Si8O12[(C2F4)7F]8 с псевдокубическим остовом с ребрами –Si–O–Si–. Указаны две проекции структур во взаимно перпендикулярных плоскостях силиконового куба.

Результаты настоящей работы позволяют направленно изменять не только длину цепи олигомеров, но и структуру растворов от разбавленных коллоидных растворов до структурированных гелей, что, в свою очередь, открывает новые возможности для получения защитных покрытий. Описанные в работе кремний-фторорганические олигомеры можно подвергнуть поликонденсации с образованием силиконового каркаса –Si–O–Si– с присоединенными к нему параллельно ориентированными FAn-цепями, обеспечивающими гидрофобность поверхности.
Работа выполнена при финансовой поддержке Программы Фундаментальных Исследований Президиума РАН ПФИ I.8П и по теме Государственного задания № 0089-2014-0025.
Список литературы
Quere D. // Rep.Progr. Phys. 2005. V. 68. P. 2495.
Бойнович Л.Б., Емельяненко А.М. // Успехи химии. 2008. Т. 77. С. 619.
Sarkar D.K., Farzaneh M.J. // Adhesion Science&Technology. 2009. V. 23(9). P. 1215.
Nakajima A. // NPG Asia Materials. 2011. V. 3. P. 49.
Yan Y.Y., Gao N., Barthlott W. // Adv. Colloid interface Sci. 2011. V. 169. P. 80.
Ким И.П., Шестаков А.Ф. // Химия высоких энергий. 2009. Т. 43. № 6. С. 516, 555.
Ким И.П., Мартыненко В.М., Шульга Ю.М., Шестаков А.Ф. // Химия высоких энергий. 2010. Т. 44. № 6. С. 483.
Ким И.П., Мартыненко В.М., Черняк А.В., Бендерский В.А. // Химия высоких энергий. 2017. Т. 51. № 4. С. 300.
Ruppert I., Schlich K., Volbach W. // Tetrahedron. Lett. 1984. V. 25. P. 2195.
Petrov V.A. // Tetrahedron. Lett. 2001. V. 42. P. 3267.
Boyko V.E., Tyutyunov A.A., Don V.I., Igoumnov S.M. // Fluorine notes. 2013. V. 6. P. 91.
Kulinich S.A., Farhadi S., Nose K., Du X.M. // Lagmuir. 2011. V. 27. P. 25.
Marby J.M., Vij A., Iacono S.T., Viers B.D. // Angew. Chem. Int. Ed. 2008. V. 47. P. 4137.
Free radical polymerization / Bemford S.H., Tipper C.F.H. (Eds). Comprehensive Chemical Kinetics. V. 14A. Amsterdam: Elsevier, 1976.
Ким И.П., Бендерский В.А. // Химия высоких энергий. 2011. Т. 45. № 5. С. 406.
Ким И.П., Куница А.А., Черняк А.В. // Журн. физ. химии. 2013. Т.87. № 11. С. 1871.
Ким И.П. // Изв. АН. Cер. хим. 2013. Т. 62. № 9. С. 2065.
Кичигина Г.А., Кущ П.П., Большаков А.И., Кирюхин Д.П., Бузник В.М. // Химия высоких энергий. 2012. Т. 46. № 3. С. 187
Ким И.П., Бендерский В.А. // Журн. физ. химии. 2014. Т. 88. № 11. С. 1776.
Stein S.E., Brown R.L. // J. Chem. Inform. Comput. Sci. 1994. V. 34. P. 581.
Ким И.П. // Журн. физ. химии. 2013. Т.87. № 7. С. 1093.
Ким И.П., Колесникова А.М. // Журн. физ. химии. 2011. Т. 85. № 9. С. 1782.
Ким И.П., Бендерский В.А. // Химия высоких энергий. 2015. Т. 49. № 1. С. 3.
Laikov D.N. // J. Chem. Phys. 2011. V. 135. P. 134120.
Yilmazer N.D., Korth M. // Comp. Struct. Biotehnology J. 2015. V. 13. P. 169.
Ким И.П., Куница А.А., Черняк А.В. // Журн. физ. химии. 2013. Т. 87. № 11. С. 1871. 27.
Ameduri B., Boutevin B. / Well-architectured fluoropolymers: synthesis, properties and applications. Amsterdam. Elsevier, 2004.
Ким И.П., Мартыненко В.М., Черняк А.В., Бендерский В.А. // Химия высоких энергий. 2017. Т. 51. № 4. С. 292.
Dalvi V.H., Rossky P.J. // Proc. Nat. Acad.Sci. USA. 2010. V. 107. P. 13603.
Tsige M., Curro J.G., Grest G.S. // J. Chem. Phys. 2008. V. 129. P. 214901.
Tsige M., Grest G.S. // J.Phys. Chem. C. 2008. V. 112. P. 5029.
Дополнительные материалы отсутствуют.
Инструменты
Химия высоких энергий