

Химия высоких энергий, 2019, T. 53, № 5, стр. 354-362
Влияние мономеров на скорость распада инициаторов полимеризации. Квантово-химический расчет оптимальных структур образующихся комплексов, механизмов их распада и инициирования полимеризации
А. И. Большаков a, Д. А. Гордон a, *, Н. С. Емельянова a, С. И. Кузина a, Д. П. Кирюхин a
a Федеральное государственное бюджетное учреждение науки
Институт проблем химической физики Российской академии наук
142432 Черноголовка, Россия
* E-mail: diliarag@gmail.com
Поступила в редакцию 05.10.2018
После доработки 25.03.2019
Принята к публикации 08.05.2019
Аннотация
Методом ЭПР-спектроскопии установлено, что мономеры влияют на скорость распада инициаторов их полимеризации, образуя с ними комплексы. Методами квантовой химии оптимизировано несколько структур комплексов, определены механизмы их распада, энергии активации реакции инициирования и продолжения цепи полимеризации, а также теплота полимеризации. При отсутствии ингибиторов радикалов (вакуумирование) процесс полимеризации протекает с высокой эффективностью (98%) при комнатной температуре без внешнего энергетического воздействия на систему.
Проблемы полимеризации и сополимеризации виниловых и акриловых мономеров никогда не переставали быть актуальными [1, 2]. Их полимеры обладают ценными свойствами и оптимизация процессов промышленного получения этих полимеров требует дальнейших фундаментальных исследований и поисков новых путей. Множество работ посвящено исследованию механизмов инициирования полимеризации, роста и обрыва цепей полимеров в различных условиях, в том числе при низких температурах [3–12]. Для инициирования процессов полимеризации используют инициаторы, термолиз, фото-, и радиационное облучение. В качестве инициаторов, в основном, используют пероксид бензоила (ПБ), динитрил азоизомасляной кислоты (ДАК), диметоксифенилацетофенон (ДМФА) и др. [4–6, 9]. Обычно, для разложения инициаторов используют энергетическое воздействие на систему: термолиз, фотооблучение светом определенной длины волны, действующим на инициатор и не действующим на мономер, и т.д. [9]. В последнее время в химии свободных радикалов наметилось направление исследования радикалообразующих процессов, не требующих для получения радикалов внешнего энергетического воздействия, свободные радикалы можно получить химическим методом. Радикалы образуются в результате химических реакций, протекающих как при комнатной, так и при низкой температурах, разрывы ковалентной связи происходят за счет внутренней энергии системы. Детальное исследование этого явления позволило установить важную роль полимолекулярных комплексов, в которых происходит одновременный разрыв и образование химических связей с протеканием эндо- и экзотермических стадий в одном элементарном акте с общим экзотермическим эффектом. Результаты низкотемпературного фторирования (вплоть до гелиевых температур) и хлорирования полимер-мономерных систем при 77 К показали, что процессы галогенирования сопровождаются спонтанным образованием высоких концентраций радикалов [7, 10]. В [12] было обнаружено, что при длительном хранении в глицерине накапливаются перекисные соединения, распад которых на радикалы, инициирующие полимеризацию, происходит в результате введения мономера (акриламида) в глицерин и его взаимодействия с перекисью. Метод химического образования радикалов можно применять для инициирования полимеризации [7, 8], модифицирования полимеров и использовать, как источник радикалов в химическом синтезе.
Настоящая работа посвящена исследованию радикальной полимеризации некоторых акриловых и виниловых мономеров, инициированной традиционными инициаторами их полимеризации (ПБ, ДАК, ДМФА) при комнатной температуре без внешнего энергетического воздействия на систему.
МЕТОДИКА ЭКСПЕРИМЕНТА
Мономеры – акриловую кислоту (АК), метилакрилат (МА), нонилакрилат (НА), додецилакрилат (ДДА) очищали вакуумной перегонкой, отбирая фракции, соответствующие температуре кипения чистого вещества. Акриламид (АА) и инициаторы – перекись бензоила (ПБ), динитрил азоизомасляной кислоты (ДАК) и 2,2'-диметокси-2-фенилацетофенон (ДМФА) перекристаллизовывали из растворов в ацетоне и хлороформе соответственно. Растворители – хлористый метилен ХМ, ацетон, этанол (марки ХЧ), дистиллированная вода специальной очистке не подвергались. Полимеризацию проводили в стеклянных ампулах, куда помещали расчетное количество мономера и инициатора. Перемешав реагенты до полного растворения инициатора, образец вакуумировали, ампулу запаивали и помещали на 24 ч в затемненный термостат при температуре 23°С. После этого ампулу вскрывали и выделяли образовавшийся полимер, удаляя непрореагировавший мономер растворением в соответствующих растворителях, в которых образовавшийся полимер не растворяется. Количество образовавшегося полимера определяли гравиметрически. ЭПР-исследования проводили с использованием стабильного нитроксильного радикала в качестве парамагнитного зонда (ПМЗ). Спектры ЭПР регистрировались на ЭПР спектрометре Брукер Elexsys II E 500. Растворы помещали в супрасиловую ампулу с внутренним диаметром 3 мм, длина образца ~25 мм. Число парамагнитных центров в образцах и g-фактор определяли с помощью пакета программ Xepr. Для проверки правильности этих процедур использовали образец CuSO4 ⋅ 5H2O с известной навеской и образец ДФПГ с g-фактором 2.0036. Процедура определения числа парамагнитных центров, используемая в пакете программ Xepr, включает двойное интегрирование спектра. ИК спектры регистрировали на спектрометре “Perkin Elmer” марки Spectrum, а также на ИК-Фурье спектрометре Bruker ALPHA, не требующих приготовления образцов в специальных кюветах, при 300 К, в диапазоне 400–4000 см–1. Спектры оптического поглощения в УФ и видимом диапазоне (200–700 нм) записывали на спектрометре “Shimadzu UV-3101 PC”. Квантово-химические расчеты проводились по программе Gaussian 09 версия D [13] с помощью локального функционала B3LYP и базисного набора 6-311++G**. Расчет частот в оптимизированных геометриях исходных и конечных продуктов показал отсутствие мнимых частот, поэтому все оптимизированные геометрии соответствуют минимумам энергии. Для всех полученных переходных состояний расчет частот показал наличие мнимой частоты. Для установления истинности переходных состояний были проведены IRC расчеты. Для анализа волновой функции методами QTAIM использовался программный пакет AIMAll (Version 15.05.18) [14]. Расчеты волновых функций и топологических характеристик распределения электронной плотности выполнены в тех же приближениях, что и оптимизация геометрии, B3LYP/6-311++G**.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
В обычной практике проведения вещественно-инициированной полимеризации инициаторы термически разлагают на радикалы при температурах 80−140°С. В нашем случае (при проведении реакции в вакууме, т.е. в отсутствие ингибиторов радикалов) реакция полимеризации акрилатов в массе и в растворах в присутствии инициаторов (ПБ, ДАК, ДМФА) не требовала нагревания и эффективно (с выходом до 98%) протекала при комнатной температуре (23°С). Полученные данные приведены в табл. 1.
Таблица 1.
Выход полимеров
| Мономер | Инициатор | Выход полимеров, % | |
|---|---|---|---|
| 1 | Акриловая кислота | ДАК | 98 |
| 2 | Акриловая кислота | ПБ | 94 |
| 3 | Акриловая кислота | ДМФА | 99 |
| 4 | Метилакрилат | ДАК | 98 |
| 5 | Метилакрилат | ПБ | 95 |
| 6 | Метилакрилат | ДМФА | 99 |
| 7 | Акриламид | ДАК | 98 |
| 8 | Винилпирролидон | ДАК | 65 |
| 9 | Стирол | ДАК | 30 |
| 10 | Нонилакрилат | ДАК | 20 |
| 11 | Додецилакрилат | ДАК | 80 |
| 12 | Акрилонитрил | ДАК | 52 |
Перед исследованием полимеризации был проведен ряд экспериментов на отсутствие в исходных мономерах и их растворителях перекисных соединений, которые могли образоваться при хранении реагентов на воздухе, как это имело место в случае глицерина [12]. Экспериментально было установлено, что в использованных мономерах и их растворителях перекисные соединения не присутствуют, в отсутствие инициаторов образования полимеров в них не происходит. Полимер, образующийся при полимеризации АК в присутствии ДАК, не растворяется в собственном мономере и его появление заметно по помутнению раствора. Он представляет собой пористое образование в виде пенопласта (выход достигал 98%). Вспенивание образца и пористость связаны с выделением азота при разложении ДАК, который применяется как пенообразователь в промышленном производстве пено- и поропластов, губчатых уплотнителей. С увеличением концентрации инициатора выход полимера увеличивается и достигает предельного значения (~98%) при содержании инициатора ~1.4 мол. % (рис. 1). Поэтому при исследовании других систем концентрацию инициатора поддерживали постоянной и равной ~1.4 мол. %. Эффективное образование полимера наблюдается в растворах мономеров и с другими инициаторами (табл. 1).
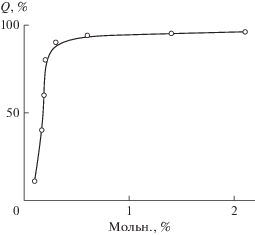
Полимеризацию акриламида, который является кристаллическим веществом, проводили в растворах инициаторов в ацетоне и этаноле, выход полимера достигал 98%. Чтобы выяснить влияние растворителя на реакцию распада инициаторов, полимеризацию исследовали в растворах мономеров. На рис. 2 представлены зависимости выхода полимера для системы [АК в водном растворе] и [МА в хлористом метилене] при содержании ДАК ~1.4 мол. % по отношению к мономеру. С увеличением относительной концентрации АК в воде (или МА в хлористом метилене) выходы полимера увеличиваются до 98%. Следовательно, растворитель не ингибирует реакцию распада инициатора, снижение выхода полимера при концентрации мономера менее 30 мол. % связано с уменьшением концентрации мономера.
Рис. 2.
Зависимость выхода (Q) полимера от относительной концентрации акриловой кислоты в водном растворе (1) и метилакрилата в растворе хлористого метилена (2). Концентрация ДАК относительно мономера 1.4 мол. %.

Ингибиторы радикальной полимеризации − растворенный кислород воздуха, гидрохинон, стабильные нитроксильные радикалы − полностью подавляют процесс, что подтверждает радикальный механизм инициирования и роста полимерных цепей. Образование радикалов для инициирования полимеризации может происходить в результате самораспада инициатора при комнатной температуре. Однако самораспад при комнатной температуре происходит с малой скоростью и не может обеспечить образование полимера с выходом 98% (табл. 1) за время термостатирования в течение 24 ч и менее. Например, для системы МА в ХМ в присутствии ДАК образование полимера наблюдалось в процессе приготовления образцов во время удаления кислорода воздуха из системы. Для АК образование полимера с выходом 98% происходит за ~15–20 мин и реакция протекает в режиме теплового взрыва (рис. 3), по-видимому, из-за высокой скорости распада инициатора и, естественно, повышения температуры. На основании этих экспериментов естественно предположить, что гипотеза о влиянии мономера на скорость распада инициатора, выдвинутая в [12], верна. Для доказательства влияния мономера на скорость распада инициатора при комнатной температуре использовали метод ЭПР-спектроскопии с применением нитроксильного радикала в качестве парамагнитного зонда (ПМЗ). По изменению концентрации нитроксильных радикалов можно судить о скоростях распада инициатора на радикалы до смешивания его с мономером (самораспад) и после смешивания. Для этого в растворитель, хлористый метилен, вводили нитроксильные радикалы и инициатор (ДАК) и следили за изменением концентрации нитроксильных радикалов в результате их взаимодействия с радикалами распада инициатора. За время термостатирования при комнатной температуре в течение 24 ч изменения концентрации нитроксильных радикалов не наблюдалось. Это говорит о том, что концентрация радикалов самораспада ничтожно мала, следовательно, скорость самораспада инициатора мала. При добавлении мономера МА в этот раствор (ХМ + ДАК + + нитроксильные радикалы) концентрация нитроксильных радикалов резко падает до полного исчезновения, при этом образования полимера не наблюдается. Этот эксперимент доказывает, что мономеры влияют на скорость распада инициаторов полимеризации. Наиболее вероятно, что между мономером и инициатором образуются комплексы, в которых ослабляются связи, по которым они распадаются. Действительно, в [15, 16] методами оптической, ИК, ЯМР спектроскопии и квантовой химии было показано образование комплексов между ПБ и мономерами акрилонитрилом и малеиновым ангидридом путем образования межмолекулярных водородных связей. Из полученных данных следовало, что образование комплексов приводит к ослаблению связи О–О в ПБ, т.е. мономеры облегчают через комплексообразование распад ПБ на радикалы, которые инициируют реакцию полимеризации мономеров:
(1)
$\begin{gathered} {\text{П Б }} + {\text{M}} \to {\text{П Б }} \ldots .{\text{M}} \to \\ \to \,\,2{{R}^{\centerdot }} + {\text{M}}\,\,{{\text{и }} \mathord{\left/ {\vphantom {{\text{и }} {{\text{и л и }}}}} \right. \kern-0em} {{\text{и л и }}}}\,\,{\text{R--}}{{{\text{M}}}^{\centerdot }} + {{{\text{R}}}^{\centerdot }} \\ \end{gathered} $Рис. 3.
Изменение температуры реакционной системы [AК + 1.4 мол. % ДАК] при тепловом взрыве от времени термостатирования реакционной системы.

но в работе не были определены ни энергии распада связи О–О в комплексах с образованием радикалов, ни энергии активации реакции инициирования, т.е. реакции присоединения образовавшихся радикалов R. к мономеру, так называемая стадия инициирования:
ни энергии активации реакции роста цепи полимера:
ни теплота полимеризации. Для ответа на эти вопросы мы исследовали методами оптической, ИК спектроскопии, а также квантовой химии систему перекись бензоила + акриловая кислота (ПБ + АК). При записи электронных спектров поглощения растворов ПБ в хлороформе обнаружены полосы индивидуального поглощения для ПБ при 277 и 284 нм. При добавлении небольшого количества (чтобы область поглощения ПБ оставалась прозрачной) АК в раствор ПБ в хлороформе происходит сдвиг полосы поглощения ПБ при 277 до 274.5 нм, что свидетельствует о взаимодействии между АК и ПБ. В табл. 2 приведены данные исследований ИК спектров ПБ, АК, и их смеси ПБ + АК. Из них следует, что в спектре смеси происходит сдвиг полос поглощения валентных колебаний С–H cвязи в группах =СН2 и =СН акриловой кислоты (3057 → 3054 см–1; 2943 → 2937 cм–1), а также сдвиг полосы arC–Hst в ПБ. Кроме того наблюдается сдвиг полос поглощения деформационных внеплоскостных колебаний связи С–Н с 924 до 919 см–1 в АК и с 690 до 703 см–1 в ПБ. Связи СОО–Н обычно соответствуют несколько полос в области 3550–2500 cм–1. Мы видим сдвиг полос с 2886 до 2888 и с 2659 до 2664 cм–1 соответственно. Наблюдается значительный сдвиг полос поглощения валентных колебаний связи С=О в ПБ (обычно для диацилпероксидов это 2 сильные полосы [17]) с 1781 и 1755 до 1790 и 1768 см–1, соответственно, а также сдвиг полосы поглощения внеплоскостных колебаний связи ОС–ОН с 975 до 977 см–1 в АК. Исходя из этих данных можно заключить, что в смеси ПБ + АК происходит взаимодействие между ПБ и АК и в этом взаимодействии участвуют карбоксильные связи С=О перекиси бензоила, С–Н связи как АК, так и ароматического кольца ПБ, а также ОС–ОН, COO–H связи АК. Эти данные позволяют считать, что взаимодействие между пероксидом и АК происходит по типу образования межмолекулярных водородных связей, как и в случае систем, изученных в работах [15, 16].
Таблица 2.
Сдвиг полос ИК-спектров перекиси бензоила ПБ и акриловой кислоты АК в их смеси ПБ + AК
| ν, см–1 | Соответствующие колебания | Δν, см–1 | ||
|---|---|---|---|---|
| ПБ | AК | ПБ + АК | ||
| 3057 | 3054 | =CH2 st | –3 | |
| 3063 | 3067 | ar С–Н st | +4 | |
| 2943 | 2937 | =C–H st | –6 | |
| 2886 | 2888 | COO–H | + 2 | |
| 2659 | 2657 | COO–H | –2 | |
| 1781 | 1790 | С=О ПБ | +9 | |
| 1755 | 1768 | С=О ПБ | +13 | |
| 975 | 977 | ОС–ОН δ оор AК | + 2 | |
| 924 | 919 | =С–Н δ оор АК | –5 | |
| 864 | 861 | =С–Н δ оор АК | –3 | |
| 849 | 847 | аr С–Н δ оор ПБ | –2 | |
| 690 | 703 | аr С–Н δ оор ПБ | +13 | |
С помощью квантово-химического моделирования, а именно DFT и QTAIM мы изучили энергетику происходящих процессов (комплексообразование, распад на радикалы, инициирование полимеризации, полимеризация) и строение образующихся продуктов.
На рис. 4 представлены молекулярные графы комплексов ПБ….М, полученные из топологического анализа волновых функций оптимизированных геометрий (b3lyp/6-311++G**).

Комплексы 1 и 2 представляют собой различные способы координации молекул ПБ и мономера относительно друг друга, однако нельзя исключать и координацию второй молекулы мономера по второй С=О группе ПБ, поэтому мы смоделировали и комплекс 3, как также вероятный в данном процессе. Схема его образования и распада будет следующей
(4)
${\text{П Б }} + 2{\text{М }} \to {\text{М }} \ldots .{\text{П Б }} \ldots .{\text{М }} \to 2{\text{М --}}{{{\text{R}}}^{\centerdot }}.$Наличие критических точек типа (3,-1) и связанных с ними линий атомного взаимодействия [18] на молекулярных графах показывает нам в данном случае, каким образом координируется молекула мономера по отношению к молекуле перекиси бензоила. На рис. 4 приведены три возможных способа. Самой вероятной является связь между кислородом С=О группы ПБ и водородом С–Н группы акриловой кислоты, она присутствует во всех трех структурах. В комплексе 1 помимо нее присутствует связь между кислородом С=О группы акриловой кислоты и водородом бензольного кольца, а в комплексе 2 – между водородом С–Н группы кислоты и углеродом бензольного кольца ПБ. В комплексе 3 первая молекула мономера координируется, как и в комплексе 2, а вторая координируется так, что помимо основной С=О…Н связи имеется и связь между водородом ОН группы АК и углеродом бензольного кольца ПБ. Из анализа критических точек по известной корреляционной формуле [19] мы можем вычислить энергии этих связей
Где EA-B – энергия связи, ${{v}_{{\text{e}}}}$(r) – плотности потенциальной энергии в критической точке этой связи. В табл. 3 приведены результаты этих расчетов.
Таблица 3.
Энергии межмолекулярных связей в молекулярных комплексах (ккал/моль)
| Комплекс | 1 | 2 | 3 |
|---|---|---|---|
| С=О(ПБ)… Н–С(АК) | 1.44 | 1.44 | 1.41, 1.13 |
| С=О(АК)…Н(ПБ) | 1.54 | – | – |
| С(ПБ)…Н–С(АК) | – | 0.44 | 0.44 |
| С(ПБ)…Н–О(АК) | – | – | 0.75 |
| ∑ | 2.98 | 1.88 | 3.73 |
Интересно отметить, что самой прочной оказалась не С=О(ПБ)… Н–С(АК), а связь С=О(АК)…Н(ПБ), хотя и не слишком значительно. Очевидно что самой слабой является связь между атомом углерода кольца ПБ и водородом С–Н группы мономера. Анализируя суммарную энергию связей, можно сделать вывод, что в комплексе 1 мономер прочнее связан с ПБ, чем в комплексе 2. В комплексе 3, несмотря на различия в координации мономеров, суммарная энергия связей одинакова, то есть обе молекулы мономера связаны с ПБ одинаково прочно. Как и следовало ожидать, все реакции образования данных комплексов из молекулы ПБ и мономера являются экзотермическими, то есть образование их энергетически выгодно: 2.38 ккал/моль для комплекса 1; 1.64 ккал/моль для комплекса 2 и 3.06 ккал/моль для комплекса 3. Как мы видим, эти энергии коррелируют со значениями суммарных энергий связей из табл. 3. Возможные структуры продукта распада комплексов по схемам (1) и (4) , а именно структуры комплексных радикалов М R. приведены на рис. 5. Интересно отметить, что независимо от первоначального типа координации мономера относительно ПБ, оптимизация геометрии (b3lyp/6-311++G**) приводит либо к структуре 4, либо к структуре 5. В комплексе 4 имеются две практически эквивалентные 0.94 и 0.88 ккал/моль связи С=О(ПБ)… Н–С(АК). Отметим, что в данном комплексном радикале они слабее, чем были в исходных молекулярных комплексах (таб. 3). В то же время в комплексе 5 связь С=О(ПБ)… Н–С(АК) оказывается несколько сильнее, а именно 1.51 ккал/моль. Прочнее по сравнению с комплексом 1 связь С=О(АК)…Н(ПБ), она равна 1.88 ккал/моль. Это приводит к тому, что комплекс 5 лежит на 1.43 ккал/моль ниже по энергии, чем комплекс 4. Важно, что в обоих этих комплексах еще не происходит перенос спиновой плотности с кислорода на углерод и переход двойной связи АК в одинарную. Спиновая плотность по 0.5 сосредоточена на обоих атомах кислорода ПБ, таким образом, оба кислородных атома в данных комплексах эквивалентны. Длина С=С связи в комплексах 4 и 5 равна 1.333 А, как и в исходном мономере АК.
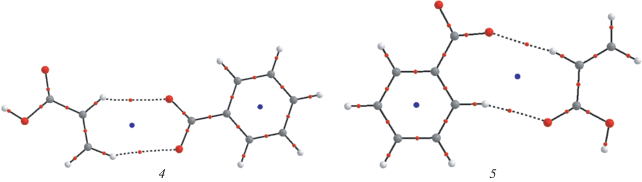
Зная геометрию и энергию конечных продуктов распада молекулярных комплексов перекиси бензоила и мономера, мы можем оценить энергию разрывающейся O–O связи в комплексах и сравнить с энергией O–O связи непосредственно в перекиси бензоила (табл. 4). Из таблицы видно, что образование молекулярного комплекса понижает энергию диссоциации O–O связи, и при координации двух молекул мономера связь ослабевает сильнее, чем при координации одной. Несколько влияет и тип координации – связь O–O будет менее прочной при координации атомами водорода мономера.
Учитывая вышесказанное, схему инициирования и роста цепи можно представить следующим образом (схема 1 ), предположив несколько механизмов реакции распада молекулярных комплексов между ПБ и АК и инициирования реакции полимеризции АК. На схеме 1 представлены оптимизированные геометрии исходных и промежуточных соединений, а также продуктов и переходных состояний. Как было сказано, на первой стадии образуется молекулярный комплекс перекиси бензоила и двух молекул акриловой кислоты. Далее происходит его распад по связи O–O с образованием комплексного радикала 5. Однако, учитывая его строение, а именно то, что один из его кислородов связан не с CH2 группой мономера, к углероду которой должно идти присоединение МR• , а с C–H группой, к углероду которой присоединение радикала затруднено из-за стерического экранирования, создаваемого кислотной группой, следует предположить, что происходит диссоциация О–О с разрывом межмолекулярных связей с образованием R• и M с последующим присоединением радикала R• к мономеру по двойной связи через переходное состояние TS1 [20] (схема 1 ). В данном переходном состоянии расстояние O–C (CH2) составляет 2.022 А, и С–С связь в мономере удлинена и составляет 1.374 А. В переходном состоянии спиновая плотность уже сосредоточена на атоме кислорода, участвующего в образовании новой связи (0.464) и атоме углерода С–H группы мономера (0.496). По своему строению переходное состояние TS1 ближе к исходным реагентам, чем к конечным продуктам (схема 1 ), с мнимой частотой 306 см–1, то есть является “ранним” и его энергия невелика. Таким образом, энергия активации присоединения радикала R• к мономеру по двойной связи в данной системе составляет всего 8.44 ккал/моль. Эта величина хорошо совпадает с принятыми в практике. Значения энергий активации радикальной полимеризации обычно невелики (5–8 ккал/моль) из-за влияния экзотермического эффекта стадии роста цепи [21].

Схема 1 .
На схеме представлен и радикал, образующийся в результате этого присоединения. Длина связи С–С в нем уже 1.478 А, а спиновая плотность (0.913) практически полностью сосредоточена на углероде С–Н группы мономера. В целом, процесс инициирования в данной системе происходит с выигрышем энергии, так как реакция является экзотермической, с энергией 20.43 ккал/моль. Далее происходит рост цепи (схема 1 ), через промежуточное состояние TS2. Расстояние С–С образующейся связи в переходном состоянии 2.278 А, а спиновая плотность распределяется следующим образом: 0.610 на атоме углерода радикала и 0.437 на углероде группы С–H мономера. Про TS3 можно сказать, как и про TS2, что оно ближе к исходным реагентам, и энергия активации процесса роста цепи также невелика и составляет всего 6.67 ккал/моль. Процесс идет с выделением энергии 14.12 ккал/моль. Полученная величина также согласуется с встречающейся в литературе 18.9 ккал/моль [22].
Можно предположить и другой путь реакции, когда образуется комплекс 4 (рис. 5), при трансформации комплексного радикала 5, или при диссоциации молекулярного комплекса 3 (рис. 4) по О–О связи. В комплексе 4, в отличие от 5, существует водородная связь O–H между атомом кислорода радикала и СН2 группой мономера и можно предположить, что присоединение радикала по двойной связи будет происходить внутри этого комплекса через TS3, очень раннее переходное состояние, в котором спиновая плотность на 70 процентов сосредоточена еще на одном из атомов кислорода радикала и длина связи С=С в мономере составляет 1.339. Соответственно, мы имеем еще более низкую энергию активации для трансформирования комплексного радикала MR• 4, в котором спиновая плотность локализуется на атоме кислорода, в радикал R–M•, в котором спиновая плотность сосредоточена на атоме углерода группы СН. Его образованием завершается процесс инициирования реакции полимеризации, энергия активации в этом случае равна всего 4.08 ккал/моль. На схеме 1 видно, что оба эти пути практически одинаковы по энергии, то есть, скорее всего равновероятны.
Проведя квантово-химическое моделирование начальных процессов полимеризации акриловой кислоты в присутствии перекиси бензоила, мы можем заключить:
1. Образование молекулярного комплекса перекиси бензоила и двух молекул мономера понижает энергию диссоциации О–О связи и, скорее всего, предшествует полимеризации;
2. Энергии активации как инициирования (8.44 ккал/моль), так и роста цепи (6.67 ккал/моль) невелики, что связано с характером образующихся переходных состояний;
3. Возможен путь инициирования, при котором растущий радикал RM• образуется непосредственно из комплексного радикала MR•, причем энергия активации этого процесса самая низкая (4.08 ккал/моль);
4. Так как все процессы в данной системе носят радикальный характер, наличие кислорода будет значительно замедлять полимеризацию.
ВЫВОДЫ
Исследованные мономеры способствуют радикальному распаду инициаторов полимеризации этих мономеров и при отсутствии ингибиторов радикалов процесс полимеризации протекает с высокой эффективностью (98%) при комнатной температуре без внешнего энергетического воздействия на систему.
Авторы выражают благодарность Т.Н. Рудневой и Ю.В. Баскаковой за регистрацию ИК спектров, А.В. Куликову за ЭПР исследования.
Работа выполнена при финансовой поддержке Программы фундаментальных исследований Президиума РАН № 32 "Наноструктуры: физика, химия, биология, основы технологий" и темы Государственного задания № 0089-2019-0008.
Список литературы
Bauer C., Becker K., Herrman T., Lilge D., Bush M. // Macromolecular Chemistry and Physics. 2010. V. 211. № 5. P. 510.
Семчиков Ю.Д. // Высокомолекулярные соединения. М.: Академия. 2005.
Sean Ewart, Karjala Thomas W., Demirors Mehmet // J. Polymer Sci. Part A. 2017. V. 55. № 5. P. 861.
Королев Г.В., Березин М.П., Грачев В.П., Зюзин И.Н. // Высокомолек. соед. А. 2007. Т. 49. № 3. С. 421.
Королев Г.В., Березин М.П., Грачев В.П. // Высокомолек. соед. А. 2007. Т. 49. № 6. С. 1145.
Большаков А.И., Кирюхин Д.П. // Высокомолек. соед. Б. 2010. Т. 52. № 6. С. 115.
Gordon D.A., Mikhaylov A.I. // e- Polymer. 2015. V. 6. P. 385.
Гордон Д.А., Эстрина Г.А., Большаков А.И., Михайлов А.И. // Химия высоких энергий. 2015. Т. 49. № 3. С. 138.
Кузина С.И., Большаков А.И., Кирюхин Д.П. // Журн. физической химии. 2017. Т. 91. № 3. С. 482.
Кузина С.И., Куликов А.В., Демидов С.В. и др. // Журн. физ. хим. 2005. Т. 79. № 5. С. 797.
Гордон Д.А., Кичигина Г.А., Эстрина Г.А. // Химия высоких энергий. 2018. Т. 52. № 1. С. 71.
Большаков А.И., Кирюхин Д.П. // Высокомолек. соед. А. 2007. Т. 49. № 9. С. 1621.
Gaussian 09, Revision D.01, Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A., Nakatsuji H., Caricato M., Li X., Hratchian H.P., Izmaylov A.F., Bloino J., Zheng G., Sonnenberg J.L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J.A., Jr., Peralta J.E., Ogliaro F., Bearpark M., Heyd J.J., Brothers E., Kudin K.N., Staroverov V.N., Keith T., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J.C., Iyengar S.S., Tomasi J., Cossi M., Rega N., Millam J.M., Klene M., Knox J.E., Cross J.B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R.E., Yazyev O., Austin A.J., Cammi R., Pomelli C., Ochterski J.W., Martin R.L., Morokuma K., Zakrzewski V.G., Voth G.A., Salvador P., Dannenberg J.J., Dapprich S., Daniels A.D., Farkas O., Foresman J.B., Ortiz J.V., Cioslowski J., and D.J. // Fox, Gaussian, Inc., Wallingford CT, 2013
AIMAll (Version 15.05.18), Todd A. Keith, TK Gri-stmill Software, Overland Park KS, USA, 2015 (aim.tkgristmill.com).
Зайцев С.Ю., Зайцева В.В. // Многофункциональные мономеры. Синтез и полимеризация. 2003. Донецк: Норд Компьютер. 296 с.
Зайцев В.В., Тюрина Т.Г. // Химия. Раздел 1. 2008. С. 70.
Преч Э., Бюльманн Ф., Аффольтер K. // Определение строения органических соединений М.: Мир. 2006.
Бэйдер Р. // Атомы в молекулах. М.: Москва. 2001.
Espinosa E., Molins E., Lecomte C. // Chem. Phys. Lett. 1998. 285. P. 170.
Денисова Т.Г., Емельянова Н.С. // Кинетика и катализ. 2005. Т. 46. № 6. С. 852.
Рафф Р.А., Дак К.В. // Кристаллические полиолефины. Строение и свойства. М.: Химия, 1970. 486 с.
Avons A.G., Tyrral E. // J. Polym. sci. V. 2. P. 367.
Дополнительные материалы отсутствуют.
Инструменты
Химия высоких энергий