

Химия высоких энергий, 2020, T. 54, № 1, стр. 19-28
Переключаемая люминесценция новых соединений на основе имидазолов и азометинов
И. Р. Мардалейшвили a, Г. В. Любимова a, А. В. Любимов a, Л. С. Кольцова a, А. И. Шиенок a, П. П. Левин a, b, А. С. Татиколов b, Н. Л. Зайченко a, *
a Федеральное государственное бюджетное учреждение науки, Институт химической физики
им. Н.Н. Семенова, Российской академии наук
119991 Москва,
ул. Косыгина, 4, Россия
b Федеральное государственное бюджетное учреждение науки Институт биохимической физики
им Н.М. Эмануэля Российской академии наук
119334 Москва, Россия
* E-mail: marli2007@yandex.ru
Поступила в редакцию 19.12.2018
После доработки 19.12.2018
Принята к публикации 20.08.2019
Аннотация
Исследованы спектрально-люминесцентные свойства соединений А3–СА, А4–о–СА и А4–о–К, структура которых включает в качестве фрагментов трифенилимидазол А3, тетрафенилимидазол А4 и гидрокиазометины – салицилиденанилин СА и кумаринзамещенный азометин К. В то время как в соединении А3–СА существует электронное сопряжение фрагментов и переключаемая люминесценция проявляется только в спиртовых растворах, при соединении этих люминофоров через кислородный мостик наблюдается независимое поведение фрагментов и существует возможность переключения люминесценции изменением длины волны возбуждения в растворах и в полимерных средах.
В продолжение наших работ по дизайну и синтезу соединений [1, 2], отличающихся множественной люминесценцией, синтезированы и исследованы спектрально-люминесцентные свойства соединений, состоящие из имидазола и азометина при разных способах соединения этих люминофорных фрагментов. В качестве люминесцирующих фрагментов выбраны гидрокитрифенил и гидрокситетерафенлимидазол А3 и А4 и гидроксиазометины – СА (салицилиденанилин) и К. В каждом из фрагментов возможен внутримолекулярный перенос протона, приводящий к образованию люминесцирующей кето-формы.
В А4–о–СА и А4–о–К соединение двух фрагментов происходит через кислородный мостик, а в А3–СА азометин присутствует как заместитель в фенольном кольце трифенилимидазола. В А4–о–К структура СА фрагмента усложнена введением кумариновой структуры.
Совмещая в одной молекуле фрагменты – люминофоры с люминесценцией в разных спектральных областях рассчитывали на получение в спектре люминесценции двух полос эмиссии с возможностью переключения между ними изменением длины волны возбуждения. В соединении А3–СА, структура которого допускает два пути внутримолекулярного переноса протона, можно ожидать образования смеси продуктов переноса протона в обоих фрагментах, характеризующихся различными спектрами люминесценции.

МЕТОДИКА ЭКСПЕРИМЕНТА
Соединение А3–СА получено в соответствии с методикой, описанной в [1], а А4–о–СА и А4–о–К – по методике, представленной в приложении к работе [3].
Для приготовления растворов исследуемых соединений (10–6–10–4 моль/л) использовали органические растворители фирмы Acros марки “для спектроскопии”. Полимерные пленки ПММА и ПВБ с добавкой исследуемых соединений приготовлены из совместных растворов полимеров и добавок в метиленхлориде и метаноле соответственно. Спектры поглощения растворов регистрировали на спектрофотометре MultiSpec-1501. Люминесцентные измерения выполнены на спектрофлуориметре Perkin Elmer–LS-55 или Флюорат 02–Панорама, спектры скорректированы на спектральные характеристики канала возбуждения. Оценка квантовых выходов люминесценции в растворах или полимерной матрице проводилась с использованием в качестве стандарта раствора родамина 6Ж в этаноле (0.95) [4, 5] или в пленке ПММА при концентрации 5 × 10–4 моль/л (квантовый выход люминесценции при этой концентрации 0.66) [6]. Исследование промежуточных продуктов проводили на установке наносекундного лазерного фотолиза (N2-лазер, длительность импульса 1 нс, длина волны излучения 337 нм).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Ранее в работе [1] было показано, что в растворах соединения А3–СА при присоединении к А3 фрагмента СА наблюдается более длинноволновое поглощение и более длинноволновая люминесценция 490–500 нм по сравнению с модельным гидроксиимидазолом А3 (428–461 нм, спирт – МЦГ). При этом в спектрах люминесценции в неполярных растворителях А3–СА присутствует дополнительно слабая более коротковолновая полоса 450 нм. В спиртовых средах присутствуют обе полосы, причем коротковолновая полоса эмиссии становится основной. На основании различий спектров возбуждения для наблюдаемых полос эмиссии предположили наличие двух изомеров в основном состоянии и изменение соотношения между ними в зависимости от растворителя, не детализируя структуру возможных изомеров [1].
Полосы эмиссии люминесценции в слабополярных растворах А3–СА, как и в модельном гидроксиимидазоле А3, характеризуются аномально большим стоксовым сдвигом относительно полос поглощения (для соединения А3–СА 5100–7200 см–1 и 7000–8200 см–1 для А3), что может свидетельствовать об образовании люминесцирующего продукта в результате внутримолекулярного переноса протона в возбужденном состоянии (ВППВС), что предполагалось ранее для гидрокситриарилмидазолов [7, 8].
Структура соединения А3–СА допускает при фотовозбуждении возможность переноса протона на атом азота имидазола или азот азометинового мостика азометина и, соответственно, наблюдаемую длинноволновую полосу эмиссии можно считать исходящей от возможных изомеров (КимЕам)* или от (ЕимКам)*, образующихся в результате ВППВС или при прямом возбуждении кето-формы в имидазольной (Ким) или азометиновой части (Кам) молекулы А3–СА

Квантовый выход люминесценции А3–СА в органических растворителях в длинноволновой полосе люминесценции 490–500 нм при возбуждении λ = 365 нм составляет 0.03–0.09, что ниже чем в модельном гидрокситрифенилимидазоле А3 [8], и существенно выше значения, характерного для салицилиденанилина (CA) (менее 0.001) в растворах при 293 К [9, 10].
Положение полосы эмиссии люминесценции для растворов А3–СА приходится на более коротковолновую спектральную область, чем люминесценция, характерная для растворов салицилиденанилина. Так в растворах СА, например, в хлороформе [11, 12] максимум полосы Em продукта ВППВС при возбуждении в полосе Е-формы – 532 нм и 525 нм при прямом возбуждении К-формы.
При лазерном фотолизе (λ = 337 нм) растворов после 1 нсек импульса не наблюдается сигналов от промежуточных продуктов со временем жизни длиннее 10 нс, что может свидетельствовать об отсутствии продуктов фотопереноса протона в азометиновом фрагменте, наличие которых регистрируется в растворах салицилиденанилина и родственных соединений в исследуемом временном диапазоне.
Эти экспериментальные факты приводят к выводу о преобладании в слабополярных средах люминесценции от продукта переноса протона в имидазольной части молекулы (КимЕам)*.
Люминесценция соединения А3–СА отличается от люминесценции похожего по структуре гибридного соединения на основе гидрокситиазола и гидроксиазометина, исследуемого в работе [13], для которого при понижении температуры растворов, а также в полимерной среде, в результате процессов ВППВС проявляются две полосы эмиссии около 500 нм и 538 нм, наличие которых, по-видимому, обусловлено возможностью установления двух типов внутримолекулярной водородной связи в силу присутствия двух ОН-групп – в тиазольной и в азометиновой частях молекулы.

В случае А3–СА люминесценция в пленке ПММА аналогична наблюдаемой в неполярных растворителях – полоса эмиссии в области 490–500 нм с соответствующей ей длинноволновой полосой спектра возбуждения Ex с λмакс = 380–390 нм. Охлаждение пленки ПММА до 77 К приводит к увеличению интенсивности этой полосы и не выявляет новых полос, что означает преобладание как в неполярных растворителях, так и в полимерной среде изомера (КЕ)*, образованного в результате внутримолекулярного переноса протона (ВППВС) на атом N имидазольного цикла.
В спиртовых растворах А3–СА наблюдаются две полосы эмиссии – коротковолновая Em с максимумом 450 нм, с соответствующим спектром Ex с максимумом 330 нм, и более слабая длинноволновая люминесценции 500 нм, с Ex с λмакс = 380–400 нм, которая как и длинноволновая волоса в спектре поглощения, ослабевает при хранении спиртового раствора. В случае разбавления раствора А3–СА в диоксане спиртом или при добавлении капель воды наблюдается ослабление длинноволновой полосы эмиссии 500 нм и относительное усиление коротковолновой полосы с максимумом 450 нм. При обратном процессе-разбавлении спиртового раствора А3–СА диоксаном наблюдается усиление интенсивности длинноволновой полосы в спектре возбуждения и появление спектра Em, характерного для раствора в диоксане (рис. 1).
Рис. 1.
Спектры люминесценции раствора А3–СА в метаноле до (1, 2, 5, 6) и после (3, 4, 7, 8) разбавления диоксаном 1 : 5. Em: λ возб = 330 (1, 3), λвозб = 380 нм (2, 4); Ex: λрег = 450 (5, 7), λ = 500 (6) и 520 нм (8).

Аналогично, при приготовлении пленки ПВБ с А3–СА при испарении растворителя (спирт) и при превращении вязкого полимерного раствора в твердую полимерную пленку происходит изменение голубой (450 нм) люминесценции на зеленую (500 нм) (рис. 2). Tаким образом, полоса люминесценции с максимумом 490–500 нм, независимо от длины волны возбуждения, наблюдается не только в пленке ПММА, полученной из слабополярных растворителей, но и в пленке ПВБ, полученной с использованием полярного растворителя. В то же время при высыхании этилцеллюлозной пленки (с большим содержанием ОН-групп), способной долго удерживать следы растворителя после ее пропитки спиртовым раствором А3–СА, наблюдается перекрывание двух полос эмиссии с преобладанием голубой люминесценция, через год хранения цвет люминесценции пленки меняется на зеленый.
Рис. 2.
Спектры люминесценции А3–СА в ПВБ до (1, 2) и после (3–5) высыхания полимерного слоя, Em: λвозб = 330 (1), 380 (3), Ex: λрег = 450 (2), 500 нм (4).

Возможно двоякое объяснение причин возникновения коротковолновой полосы люминесценции в полярных средах. Можно предположить, что наблюдаемый при увеличении полярности растворителя переход от полосы Em соединения А3–СА с максимумом 500 нм в неполярных растворителях к полосе с максимумом около 450 нм в метаноле связан с отсутствием ВППВС в связи с разрывом внутримолекулярной водородной связи в имидазольной части молекулы и образованием межмолекулярных связей с растворителем. В работе [14] предполагалось, что полярные растворители способствуют ослаблению внутримолекулярной водородной связи в гидроксиимидазолах и стабилизации ротамера, образованного в результате прототропии или поворота на 180° вокруг связи, соединяющей имидазольный и фенольный циклы, и для которого перенос протона невозможен.

С другой стороны влияние азометинового фрагмента на люминесценцию А3–СА может также меняться за счет различного состояния этого фрагмента в разных средах, (протонирования атома N азометинового мостика в спиртовой среде или изомеризации).

Таким образом, в растворах А3–СА в органических растворителях присутствуют по крайней мере два изомера соединения А3–СА с разными спектрами люминесценции, соотношение между которыми определяется свойствами среды. В А3–СА азометиновый фрагмент выступает в роли заместителя к имидазолу А3, что в сравнении с А3 (полосы эмиссии 375 и 450 нм) приводит к смещению полос эмиссии в красную область до 450 и 500 нм. В слабополярных растворителях преобладает люминесценция, обусловленная продуктом ВППВС в имидазольной части молекулы. В полимерных пленках ПММА и ПВБ наблюдается только длинноволновая люминесценция с максимумом эмиссии около 500 нм. В спиртовых средах в зависимости от длины волны возбуждения можно регистрировать коротковолновую 440–450 нм и более длинноволновую люминесценцию 500–510 нм.
В соединениях А4–о–СА и А4–о–К в отличие от А3–СА можно ожидать слабое взаимовлияние фрагментов, соединенных через кислородный мостик. Так, в работах [3, 15] показано, что для бисимидазольной диады с аналогичным кислородным мостиком, реализуется почти полная перпендикулярность фенильных колец в дефинильном фрагменте, препятствующая электронному взаимодействию между фрагментами.
При независимом поглощении света фрагментами в соединениях А4–о–СА и А4–о–К соотношение двух полос эмиссии от двух фрагментов должно определяться как поглощением на длине волны возбуждения, так и величиной квантового выхода люминесценции фрагментов – люминофоров.
Спектры поглощения А4 в органических растворителях характеризуются максимумом на 318–325 нм и коэффициентом экстинкции в максимуме поглощения 1.0 –1.7 × 104 л моль–1 см–1, в то время как для СА аналогичные параметры 337–347 нм и 1.1–1.2 × 104 л моль–1 см–1 [16]. Граница поглощения А в спектрах поглощения около 370 нм.
В растворах наблюдается одна полоса эмиссии люминесценции с максимумом – 472–454 нм (МХ, толуол, АСN, этанол), в пленках ПММА и ПВБ около 450 нм. Положение максимума флуоресценции А4 зависит от растворителя, смещаясь с ростом полярности растворителя в короткую область. Наблюдаемая люминесценция исходит от продукта внутримолекулярного фотопереноса протона [2]. В отличие от А3 в растворах А4 отсутствует люминесценция от изомера с разорванной внутримолекулярной связью, для которого исключен внутримолекулярный фотоперенос протона. Возможно, это обусловлено присутствием в структуре тетраимидазола дополнительного бензольного кольца, что способствует упрочнению внутримолекулярной водородной связи [3]. По данным лазерного фотолиза фотовозбуждение А4 не приводит к образованию каких-либо промежуточных продуктов, поглощающих во временном диапазоне ≥20 нс.
В спектрах люминесценции А4–о–СА в растворах наблюдается полоса эмиссии с максимумом на 450–470 нм от имидазольного фрагмента А и не регистрируется эмиссия от СА. Это связано с низким квантовым выходом люминесценции СА в растворах [9, 10] и косвенно свидетельствует об отсутствии электронного взаимодействия между фрагментами. В полимерной матрице ПМ-МА при 20°С флуоресценция А4‒о–СА представляет собой суперпозицию полос эмиссии – основного сигнал на 450 нм от имидазола А4 и слабого на 510–530 нм от СА (рис. 3), проявляющихся при λ возбуждения длиннее 370 нм. Как видно из рис. 4 при λвозб = 370 нм при 20°С в ПМ-МА в условиях преобладании поглощения от СА наблюдаются две сравнимые по интенсивности полосы на 450 нм и 510–530 нм, понижение температуры пленки до 77 К приводит к увеличению интенсивности полосы 510–530 нм в 4 раза.
Рис. 3.
Спектры люминесценции А4–о–СА в пленке ПММА при 20 С (1) и 77 К (2–5), Em: λвозб = 370 нм при 20°С (1) и 77 К (2), λ возб = 330 нм при 77 К (3), Ex: λ рег = 460 нм при 77 К(4) и λ рег = 560 нм при 77 К (5).

Рис. 4.
Спектры люминесценции раствора А4–о–К в метиленхлориде Em: λвозб = 305 (1), 370 (2), 490 нм (3), Ex: λрег = 450 (4), 550 нм (5).
Положение длинноволновой полосы эмиссии в спектральной области с максимумом 510–530 нм свидетельствует о наличии ряда изомеров, известных в литературе для СА в твердой фазе [17] и также указывает на независимое поведение фрагмента СА в молекуле А3–СА. На это указывают также данные лазерного фотолиза. При исследовании пленок ПММА с А–СА (с = 1.5 × 10–3 моль/л) методом лазерного фотолиза (337 нм) фиксируется промежуточный продукт с максимумом λ = 470 нм и временем жизни, соответствующим литературным данным об образовании долгоживущей Ктранс формы СА и подтверждающей наличие фотопереноса протона в СА-фрагменте [12, 18, 19]. При повышении концентрации А4–о–СА в пленках ПММА до 1.5 × 10–2 моль/л обе полосы эмиссии в спектре люминесценции сохраняются. Увеличение интенсивности сигнала эмиссии от СА при возбуждении с λ = 370 нм с ростом концентрации линейно.
Квантовый выход люминесценции А4–о–СА в длинноволновой полосе эмиссии в ПММА при возбуждении на λ = 365 нм, оцененный с использованием в качестве стандарта пленки ПММА с родамином 6Ж, равен 0.007. Поскольку длинноволновая полоса эмиссии приходится на зеленую область спектра, отвечающую максимальной чувствительности человеческого глаза, несмотря на невысокую эффективности этой полосы, можно визуально наблюдать эффект переключения с сине-зеленой эмиссии от А4 при возбуждении светом с λ = 330 нм на зеленую от СА при возбуждении λ = 370 нм.
Так как люминесценция А4–о–СА даже при больших концентрациях остается достаточно слабой, фрагмент СА заменили на К. Присутствием кумарина в соединении А4–о–К достигается значительно более эффективная длинноволновая люминесценция, чем в А4–о–СА и небольшой сдвиг полосы эмиссии в красную область.
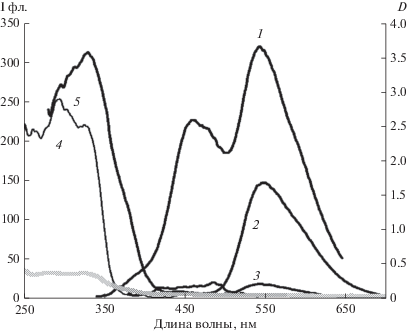
Поскольку квантовые выходы люминесценции фрагментов А4 (в растворах 0.15 [15]) и фрагмента К (оценка в толуоле 0.05,) в растворах сравнимы, а в области спектра поглощения длиннее λ = 370 нм наблюдается слабое поглощения от А, в растворах А4–К (рис. 5) можно наблюдать переход от двух полос эмиссии с максимумами на 460 нм и 545 нм при коротковолновом возбуждении короче λ = 370 нм к одной полосе на 545 нм при возбуждении длиннее λ = 370 нм.
Рис. 5.
Спектры поглощения (7, 8) и люминесценции (1–6) А4–о–К в пленке ПММА, EM: λвозб = 330 (1, 3), 370 нм (2, 4), Ex: λ рег = 450 (5), 520 нм (6), концентрации с = 6.7 × 10–4 моль/л (1, 2, 7) и с = 3.4 × 10–3 моль/л (3–6, 8), толщина пленок 60 мкм.

Сравнение А4–о–К с люминесценцией модельных соединений А4 и К, позволяет отнести наблюдаемые полосы эмиссии к структурам с переносом протона в обоих фрагментах. При лазерном фотолизе при возбуждении светом с λ = 337 нм растворов А4–о–К и К в толуоле, СН2Cl2 и EtOH фиксируются относительно долгоживущий промежуточный продукт с максимумом около 500 нм, аналогичный спектру поглощения промежуточного продукта, характерному для соединения К. Спектр поглощения и кинетическое поведение соответствуют кето-таутомеру, образованному при переносе протона в К-части молекулы А4–о–К. Более короткоживущие продукты от переноса Н в имидазольной части А4 не фиксируются.
В полимерных пленках наличие двух полос эмиссии сохраняется (рис. 5), при этом эффективность люминесценции А4–К в полимерных пленках существенно выше, чем в растворах. Квантовый выход эмиссии при возбуждении 365 нм, оцененный при концентрации А4–К с = = 6.7 × 10–4 моль/л в пленке ПММА (60 мкм) равен 0.37.
При повышении концентрации в спектре поглощения растворов А4–о–К и в пленке ПММА становится заметным смещение границы поглощения до 500 нм, аналогично в спектре возбуждения люминесценции наблюдается полоса 400–500 нм. Это поглощение характерно и для растворов К и обусловлено присутствием кето-формы фрагмента К в основном состоянии. При возбуждении в этой спектральной области наблюдается та же эмиссия на 550 нм, как и при возбуждении Е-формы соединения К.
В спектрах люминесценции в растворах и пленках при переходе к большим концентрациям и оптическим плотностям наблюдается увеличение относительного вклада длинноволновой полосы эмиссии на 540 нм. Это может быть вызвано эффектами экранирования и перепоглощения света.
ВЫВОДЫ
1. В растворах соединении А3–СА только в полярных средах возможно переключение люминесценции изменением длины волны возбуждения.
2. В А4–о–СА переключение люминесценции от длины волны возможно, но вклад эмиссии от СА фрагмента даже в полимерной матрице слабый. Сопоставимые по интенсивности сигналы наблюдаются только при 77 К в полимерной матрице.
3. В спектрах люминесценции А4–о–К вклад от К-фрагмента более существенен чем в предыдущих соединениях, полосы эмиссии от структур с фотопереносом протона в А- и К-части сравнимы по интенсивности. Можно варьировать соотношение полос эмиссии на 450 и 550 нм изменением длины волны возбуждения. Возможность переключения люминесценции существует как в растворах, так и в полимерных средах.
Список литературы
Мардалейшвили И.Р., Любимов А.В., Зайченко Н.Л., Кольцова Л.С., Шиенок А.И., Татиколов А.С. // Химия высоких энергий. 2016. Т. 5. № 4. С. 286.
Мардалейшвили И.Р., Любимова Г.В., Кольцова Л.С., Шиенок А.И., Левин П.П., Татиколов А.С., Зайченко Н.Л. // Химия высоких энергии 2018. Т. 52. № 3. С. 219.
Park S., Kwon J.E., Kim S.H., Seo J., Chung K., Park S.-Y., Jang D-J., Medina B.M., Gierschner J., Park S.Y. // JACS. 2009. № 131. P. 14043.
Паркер С. Фотолюминесценция растворов / Под ред. Васильева Р.Ф. М.: Мир, 1972. С. 252.
Kubin R.F., Fletcher A.N. // J. Luminescence. 1982. V. 27. P. 455.
Kurian A., George N.A., Paul B., Nampoori V.P.N., Vallabhan C.P.G. // Lazer chemistry. 2002. V. 20. № 2–4. P. 99.
Шиенок А.И., Кольцова Л.С., Зайченко Н.Л., Маревцев В.С. // Изв. АН. сер. хим. 2002. № 11. С. 1894.
Fridman N., Kaftory M., Eichen Y., Speiser S. // J. Mol. Structure. 2009. V. 917. P. 101.
Красовицкий В.М., Болотин Б.М. Органические люминофоры. M.: Химия, 1984. С. 336.
Vargas V. // J. Phys. Chem. A. 2004. V. 108. P. 281.
Alarcon S.H., Pagani D., Bacigalupo J., Olivieri A.C. // J. Mol. Structure. 1999. V. 475. P. 233.
Becker R.S., Lenoble C., Zein A. // J. Phys. Chem. 1987. V. 91. P. 3509.
Sun W., Li S., Hu R., Qian Y., Wang S., Yang G. // J. Phys. Chem. A. 2009. V. 113. P. 5888.
Rodembusch F.S., Buckup T., Segala M., Tavares L., Correia R.R.B., Stefani V. // Chemical Physics 2004. V. 305. P. 115.
Park S., Kwon J.E., Seo J., Park S.-Y. // Phys. Chem. Chem. Phys. 2012. V. 14. P. 8878.
Dudek G.O., Dudek E.P. // J. Am. Chem. Sos. 1966. V. 88. P. 2407.
Knyazhansky M.I., Metelitsa A.V., Kletskii M.E., Millov A.A., Besugliy S.O. // J. Mol. Structure. 2000. V. 526. P. 65.
Мардалейшвили И.Р., Кольцова Л.С., Зайченко Н.Л., Шиенок А.И., Левин П.П., Татиколов А.С. // Химия высоких энергии 2013. Т. 47. № 5. С. 331.
Kownacki K., Mordzinski A., Wilbrandt R., Grabowska A. // Chemical Physics Letters 1994. V. 227. P. 270.
Дополнительные материалы отсутствуют.
Инструменты
Химия высоких энергий