

Химия высоких энергий, 2020, T. 54, № 6, стр. 455-461
Квантово-химическое моделирование спектров масс-селективной пороговой фотоионизации ферроцена и кобальтоцена
С. Ю. Кетков a, *, Е. А. Рычагова a, Г. Ю. Жигулин a, Ш. Ю. Цзэн b, В. Б. Цзэн b
a Институт металлоорганической химии им. Г.А. Разуваева Российской Академии наук
603950 Нижний Новгород, Россия
b Institute of Atomic and Molecular Sciences, Academia Sinica
10617 Taipei, Taiwan
* E-mail: sketkov@iomc.ras.ru
Поступила в редакцию 05.07.2020
После доработки 05.07.2020
Принята к публикации 06.07.2020
Аннотация
В рамках теории функционала плотности проведено моделирование вибронной структуры в спектрах масс-селективной пороговой ионизации ферроцена и кобальтоцена. На примере (C5H5)2Co показано, что предложенные уровни расчета позволяют воспроизвести положение и относительные интенсивности в экспериментальном спектре высокого разрешения. Это дает возможность прецизионного прогнозирования изменений молекулярной структуры и колебательных частот при ионизации металлоценов в газовой фазе.
ВВЕДЕНИЕ
За десятилетия, которые прошли с открытия сэндвичевой структуры ферроцена (Cp)2Fe (Cp = = η5-C5H5) [1, 2] металлоцены превратились в один из важнейших классов металлоорганических соединений. Существенно возросший в последние годы интерес к этим металлокомплексам обусловлен их уникальными свойствами, открывающими перспективы использования в таких актуальных областях как медицинская химия [3], молекулярная электроника [4], катализ [5]. Многие их этих свойств связаны со способностью металлоценов образовывать стабильные или, наоборот, реакционноспособные ионы [3–6]. Поэтому особенно важным становится детальное изучение изменений молекулярного и электронного строения сэндвичевых систем, сопровождающих процессы отрыва электрона.
Прорыв в экспериментальных исследованиях ионизации многоатомных молекул, наблюдающийся в последние годы, связан, в частности, с развитием методов пороговой ионизации, использующих лазерное возбуждеиие [7]. В методах фотоэлектронной спектроскопии нулевой кинетической энергии электронов (zero electron kinetic energy, ZEKE) и масс-селективной пороговой ионизации (mass-analyzed threshold ionization, MATI) молекулы под действием лазерных импульсов переходят на высокие ридберговские уровни, лежащие вблизи соответствующих электронно-колебательных состояний катионов, и затем ионизируются электрическим импульсом. Спектры, полученные при регистрации образующихся электронов (ZEKE) или молекулярных ионов (MATI), позволяют определить наиболее точные на сегодняшний день значения потенциалов ионизации (ПИ) многих нейтральных молекул и колебательных частот свободных катионов.
Применение методов пороговой спектроскопии к исследованию сэндвичевых комплексов представляет собой сложную экспериментальную задачу из-за низкой летучести соединений, их легкого окисления в присутствии воздуха и влаги, а также сверхкоротких времен жизни промежуточных электронно-возбужденных состояний, не позволяющих эффективно использовать многофотонные схемы возбуждения, Тем не менее, удалось получить однофотонные ZEKE/MATI спектры ряда бис-ареновых комплексов, имеющие четко выраженную колебательную структуру [8–14]. Оказалось, что положение и относительные интенсивности вибронных компонент в этих спектрах хорошо описываются на основе расчетов колебательных частот и факторов Франка−Кондона в рамках теории функционала плотности (DFT) [8–10, 13, 14].
Поскольку вибронная структура фотоионизационных спектров определяется структурными изменениями молекул при отрыве электрона, экспериментальные ZEKE/MATI данные и результаты DFT дополняют друг друга при анализе процессов ионизации бис-ареновых комплексов. Пороговая спектроскопия высокого разрешения служит прецизионным инструментом для проверки качества DFT-моделей, предсказывающих геометрию и частоты нормальных колебаний свободных молекул. В свою очередь, DFT-расчеты позволяют прогнозировать структуру ZEKE/MATI спектров для комплексов, экспериментальное исследование которых представляет существенные сложности.
К таким комплексам относятся металлоцены. Их ПИ, как правило, превышают 6 эВ [15]. Поэтому ионизация этих соединений в однофотонном эксперименте требует использования мощных источников монохроматического излучения в вакуумном УФ диапазоне, что недоступно большинству исследовательских групп, занимающихся ZEKE/MATI спектроскопией. Исключением является кобальтоцен с более низким ПИ, для которого был получен MATI спектр, имеющий богатую вибронную структуру [16]. Этот спектр дает уникальную возможность верификации DFT расчетов для нейтральных металлоценов и их ионов. Найдя оптимальное сочетание функционала и базисного набора, можно с высокой надежностью определить характер структурных изменений при ионизации других металлоценов и рассчитать параметры неизвестных еще спектров пороговой ионизации. В первую очередь, конечно же, интересен спектр главного представителя данного класса – ферроцена. В настоящей работе было проведено DFT-моделирование MATI спектров (Cp)2Co и (Cp)2Fe.
РАСЧЕТНЫЕ МЕТОДЫ
В расчетах использовались гибридный (B3PW91 [17, 18]) и “чистый” (BPW91 [19, 20]) функционалы в сочетании с трехкратно расщепленными (triple-ζ) базисными наборами TZVP [21] и 6-311++G(d,p) [22, 23]. Уровень B3PW91/6-311++G(d,p) хорошо зарекомендовал себя при моделировании MATI спектров бис-ареновых комплексов [14, 24], а сочетание BPW91/TZVP ранее было с успехом использовано в MATI исследовании кобальтоцена [16], ограниченном рассмотрением только интенсивностей компонент, отвечающих полносимметричному валентному колебанию металл−кольцо. Для расчетов использовался программный пакет Gaussian 09 [25]. Проводилась полная оптимизация нейтральных молекул и ионов металлоценов в основном электронном состоянии с последующим вычислением частот нормальных колебаний. Затем выполнялся расчет факторов Франка−Кондона и строились модельные MATI спектры. При моделировании исключались низкочастотные крутильные колебания лигандов и ян-теллеровские компоненты с мнимыми частотами. Для ферроцена были также проведены расчет иона в возбужденном состоянии, соответствующем отрыву электрона с несвязывающей орбитали a1(dz2), и моделирование структуры соответствующего перехода MATI спектре.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Характер верхних занятых молекулярных орбиталей (МО) исследуемых металлоценов хорошо известен (рис. 1). Кобальтоцен представляет собой “19-электронный” комплекс с электронной конфигурацией …[e2(${{{{d}_{{xy,{{x}^{2}} - {{y}^{2}}}}}} \mathord{\left/ {\vphantom {{{{d}_{{xy,{{x}^{2}} - {{y}^{2}}}}}} \pi }} \right. \kern-0em} \pi }$)]4[a1(${{d}_{{{{z}^{2}}}}}$)]2[e1(dxz,yz/π)]1 (для обозначения симметрии МО, электронных состояний и колебаний здесь и далее использованы неприводимые представления точечной группы D5). Неспаренный электрон находится на вырожденной МО, поэтому основное электронное состояние молекулы 2E1 также вырождено. Из-за эффекта Яна−Теллера минимум на поверхности потенциальной энергии (ППЭ) смещен вдоль нормальных координат, отвечающих колебаниям e2-симметрии [16, 26].
Рис. 1.
Граничные орбитали изоэлектронных металлоценов (Cp)2Co+ и (Cp)2Fe. Изоповерхности соответствуют значению волновой функции 0.03.
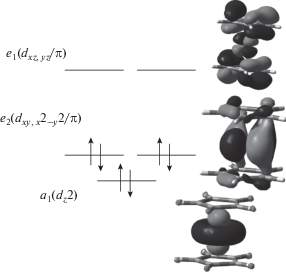
Отрыв электрона с разрыхляющей МО e1 приводит к образованию симметричного (D5h) “18-электронного” иона с закрытой оболочкой 1A1. При этом расстояние между металлом и лигандами сокращается (табл. 1). Как следствие этого, MATI спектр (Cp)2Co [16] содержит протяженную прогрессию по симметричному валентному a1-колебанию металл-кольцо ν4 (в работе используется общепринятая нумерация колебаний металлоценов [27]). Кроме этого, наблюдаются вибронные компоненты, связанные с возбуждением внеплоскостного деформационного e2-колебания ν28, имеющего наибольшую ян-теллеровскую активность в нейтральной молекуле [26]. Формы нормальных колебаний иона (Cp)2Co+ приведены на рис. 2. В спектре также присутствуют компоненты, соответствующие скелетным модам ν22e1-типа [16]. При понижении симметрии нейтральной молекулы из-за смещения минимума ППЭ e1-колебания расщепляются на две компоненты, одна из которых становится полносимметричной, и соответствующие вибронные переходы в MATI спектре разрешены правилами отбора. Их появление указывает на псевдоэффект Яна–Теллера [16]. Значения адиабатического ПИ кобальтоцена, рассчитанные на уровнях BPW91/TZVP и B3PW91/6-311++G(d,p), составляют 5.256 и 5.557 эВ соответственно. Экспериментальный ПИ, найденный с высокой точностью из MATI спектра, равен 5.3275 эВ [16]. Таким образом, вычисления с “чистым” функционалом несколько занижают (на 1.4%), а с гибридным – завышают (на 4.3%) ПИ. Тем не менее, DFT расчеты, как оказалось, способны адекватно воспроизвести относительные положения и интенсивности вибронных компонент a1- и e2-типов в MATI спектре (Cp)2Co.
Таблица 1.
Расстояния металл–центр кольца r(M-Cp) и частоты отдельных нормальных колебаний ν в нейтральных и ионных формах кобальтоцена и ферроцена, рассчитанные с функционалами BPW91/B3PW91. В скобках приведены типы электронных состояний
| Параметр | (Cp)2Co (2E1) | (Cp)2Co+ (1A1) | (Cp)2Fe (1A1) | (Cp)2Fe+ (2E2) | (Cp)2Fe+ (2A1) |
|---|---|---|---|---|---|
| r(M-Cp), Å | 1.735/1.737 | 1.659/1.644 | 1.654/1.651 | 1.730/1.716 | –/1.676 |
| ν2 , см–1 | 775/810 | 843/877 a | 811/840 | 842/842 | –/857b |
| ν4 , см–1 | 267/266 | 303/315a, c | 304/306 | 287/286b | –/307b |
| ν16 , см–1 | 375/395 | 355/376 | 366/381 | 340/357b | –/363 |
| ν22 , см–1 | 141/143 | 162/169c | 164/170 | 140/154b | –/115b |
| ν28 , см–1 | 574/613 | 560/605a,c | 572/611 | 577/610b | –/307 |
a – Колебание проявляется в расчетном MATI спектре (Cp)2Co. b – Колебание проявляется в расчетном MATI спектре (Cp)2Fe. c – Колебание проявляется в экспериментальном MATI спектре (Cp)2Co [16].
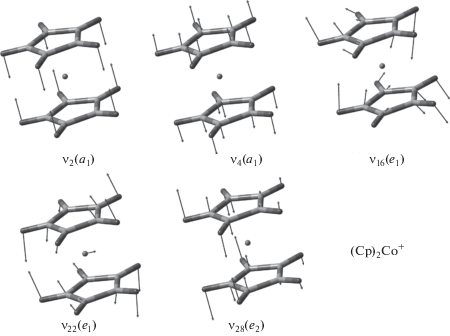
Частоты колебаний катиона (Cp)2Co+ ν2, ν4, ν22 и ν28, вычисленные на уровнях BPW91/TZVP и B3PW91/6-311++G(d,p) (табл. 1), хорошо согласуются с частотами, найденными из MATI эксперимента [16] (851, 309, 158, 589 см–1 соответственно). Сопоставление расчетных и экспериментальных параметров MATI спектра кобальтоцена (рис. 3) показывает, что моделирование с использованием “чистого” функционала BPW91 (рис. 3а) лучше воспроизводит распределение интенсивности в прогрессии по колебанию металл−кольцо ν4. При этом относительные интенсивности e2-компонент оказываются несколько завышенными, что указывает на преувеличенное расчетом ян-теллеровское искажение нейтральной молекулы. Кроме этого, в теоретическом спектре присутствуют достаточно сильные пики, отвечающие возбуждению симметричного зонтичного колебания CH ν2(a1). Эти компоненты в эксперименте проявляются в виде слабых полос, перекрывающихся с более сильными сигналами других колебаний.
Рис. 3.
Модели MATI спектра кобальтоцена, полученные на уровне DFT BPW91/TZVP (a) и B3PW91/6-311++G(d,p) (б). Экспериментальные положения и относительные интенсивности MATI пиков [16] приведены для сравнения (в). Обозначены компоненты, соответствующие различным нормальным колебаниям катиона (Cp)2Co+ в основном электронном состоянии 1A1.
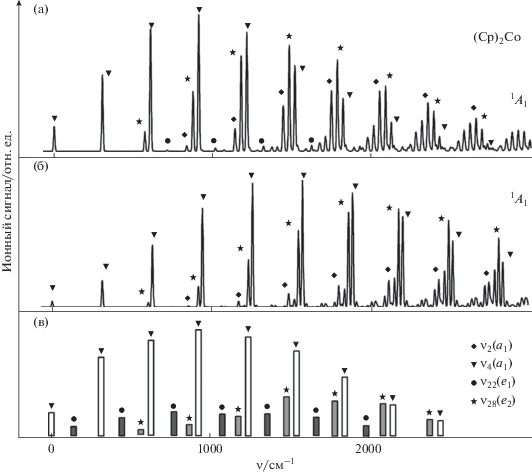
Применение гибридного функционала B3PW91 в сочетании с базисом, включающим диффузные функции (рис. 3б) дает более близкие к эксперименту отношения интенсивностей ν4(a1)- и ν28(e2)-компонент и слабым ν2(a1)-полосам. При этом увеличенная протяженность прогрессии по ν4 свидетельствует о завышенной теоретической оценке изменения расстояния металл−кольцо при ионизации. Тем не менее, в целом, оба функционала, используемых в данной работе, позволяют воспроизвести общий вид экспериментального MA-TI спектра кобальтоцена, включая вибронные компоненты, отвечающие не только полносимметричным, но и e2-колебаниям иона. Низкочастотное деформационное e1-колебание металл−кольцо ν22 проявляется в виде очень слабых пиков в модели B3PW91/6-311++G(d,p), указывая на заниженную DFT-оценку искажения вдоль соответствующей нормальной координаты (Cp)2Co.
Сопоставление геометрических параметров нейтральной молекулы и иона кобальтоцена, рассчитанных на уровне BPW91/TZVP и B3PW91/6-311++G(d,p) (табл. 1) показывает, что разная протяженность прогрессии по ν4 в расчетных спектрах (рис. 3) связана с очень небольшими различиями в изменениях равновесных расстояний металл−лиганд при ионизации. Эти расстояния уменьшаются на 0.076 и 0.093 Å при описании отрыва электрона от (Cp)2Co функционалами BPW91 и B3PW91 соответственно. Таким образом, расчеты вибронной структуры MATI спектров в рамках DFT служат чрезвычайно чувствительным инструментом для исследования структурных изменений сэндвичевых молекул при ионизации.
В отличие от кобальтоцена, где электрон отрывается с разрыхляющей МО e1 (рис. 1), при образовании иона (Cp)2Fe+ в основном электронном состоянии ионизируется слабосвязывающая МО e2 ферроцена. Кроме того, если для (Cp)2Co вырождено основное электронное состояние нейтральной молекулы, а состояние иона полносимметрично (1A1), то в случае ферроцена, наоборот, нейтральная молекула находится в состоянии 1A1, тогда как ион – в вырожденном состоянии 2E2. Поэтому молекула нейтрального (Cp)2Fe имеет высокую симметрию (D5h), а геометрия иона искажается из-за эффекта и псевдоэффекта Яна−Теллера.
Aдиабатические BPW91/TZVP и B3PW91/6-311++G(d,p) ПИ ферроцена равны, соответственно, 6.650 и 6.983 эВ. Высокоточные экспериментальные данные для (Cp)2Fe отсутствуют. ПИ, определенные различными методами, варьируются от 6.61 [28] до 6.99 [29] эВ. В соответствии с природой ионизирующейся МО, при отрыве e2-электрона от (Cp)2Fe происходит заметное удлинение расстояний металл−лиганд (табл. 1). Это приводит к появлению протяженной прогрессии по колебанию ν4 в расчетных MATI спектрах (рис. 4а, б). В отличие от ионизации (Cp)2Co, в случае ферроцена более заметное изменение геометрии (табл. 1) и больший сдвиг максимума интенсивности вибронных компонент MATI относительно $0_{0}^{0}$-перехода (рис. 4) предсказывается на уровне BPW91/TZVP. Кроме интенсивных пиков основной прогрессии, в расчетных MATI спектрах присутствуют слабые полосы, соответствующие возбуждению колебаний ν16, ν22 и ν28 иона (Cp)2Fe+. Замена Co на Fe в молекуле металлоцена приводит к тому, что в модельных MATI спектрах компоненты, связанные с возбуждением e1-колебаний ν16 и ν22 становятся более интенсивными, чем e2-компоненты (рис. 3, 4). Это результат более значительных смещений ППЭ иона (Cp)2Fe+ вдоль соответствующих нормальных координат, которые можно связать с формой МО e2 (рис. 1), имеющей максимум в плоскости, параллельной кольцам и проходящей через атом металла. В отличие от этого, для МО e1 кобальтоцена данная плоскость является узловой (рис. 1). Колебания типа e1 неактивны в эффекте Яна–Теллера первого порядка, но могут проявляться из-за псевдоэффекта Яна−Теллера [30]. Соответствующие компоненты хорошо видны в экспериментальном MATI спектре кобальтоцена [16].
Рис. 4.
Модели MATI спектра ферроцена, полученные на уровне DFT BPW91/TZVP (a) и B3PW91/6-311++G(d,p) (б) для катиона в основном электронном состоянии 2E2. Для сравнения приведена модель B3PW91/6-311++G(d,p) для MATI спектра (Cp)2Fe, соответствующего отрыву электрона с МО a1 и образованию катиона (Cp)2Fe+ в электронно-возбужденном состоянии 2A1. Обозначены компоненты, соответствующие возбуждению нормальных колебаний иона (Cp)2Fe+.
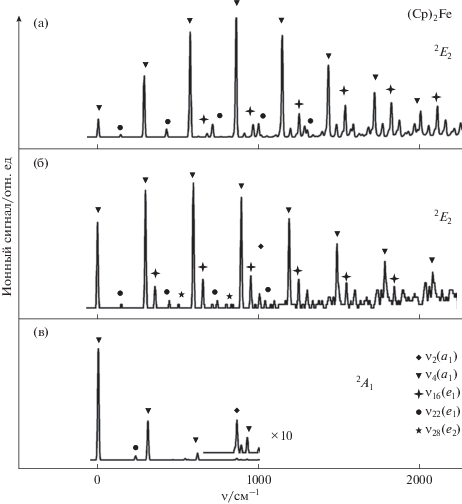
Зонтичные CH колебания ν2 (рис. 2), имеющие заметную интенсивность в расчетных спектрах (Cp)2Co (рис. 3а, 3б), в случае фероцена не проявляются из-за перекрывания с более интенсивными компонентами, соответствующими основной прогрессии (рис. 4а, 4б). Слабый ν2-пик можно видеть в расчетном спектре (Cp)2Fe, соответствующем отрыву a1-электрона (рис. 4в). В этом случае ионизируется несвязывающая ${{d}_{{{{z}^{2}}}}}$ орбиталь металла, и в расчетном MATI спектре присутствует короткая прогрессия по колебанию ν4 с наиболее интенсивным пиком, отвечающим $0_{0}^{0}$‑переходу. Аналогичную структуру имеют расчетный и экспериментальный MATI спектры изоэлектронного бис(бензол)хрома [31], где также ионизируется a1-орбиталь. Похожее распределение интенсивностей в прогрессии по ν4 было найдено для ридберговского перехода a1 → R4px,y в спектре резонансной многофотонной ионизации (REMPI) ферроцена [32, 33]. В последнем случае сходство обусловлено тем, что ППЭ ридберговского и ионного состояния близки по форме.
Удаление электрона с МО a1 ферроцена дает катион в полносимметричном состоянии 2A1. Поэтому в соответствующем расчетном MATI спектре (рис. 4в) нет компонент, соответствующих однократному возбуждению колебаний e1- и e2-типа. Тем не менее, в нем есть слабый пик, отвечающий двукратному возбуждению e1-моды ν16. Результирующее вибронное состояние содержит полносимметричную компоненту, что делает такой переход разрешенным правилами отбора. Малоинтенсивные вибронные компоненты, отвечающие аналогичному колебанию в молекуле (η6-C6H6)2Cr, присутствуют в экспериментальном и расчетном MATI спектрах [31].
ЗАКЛЮЧЕНИЕ
В целом, данное исследование показало, что DFT моделирование MATI сигналов кобальтоцена и ферроцена позволяет получить вполне реалистичную вибронную структуру спектров. Это подтверждается сопоставлением с экспериментальными данными для (Cp)2Co, опубликованными ранее [16]. Такой результат трудно было предсказать заранее, учитывая участие вырожденных электронных состояний в процессе ионизации. Полученные модельные спектры ферроцена (рис. 4) будут крайне полезны при будущих экспериментальных исследованиях этого соединения методами пороговой ионизационной спектроскопии.
Список литературы
Wilkinson G., Rosenblum M., Whiting M.C., Woodward R.B. // J. Am. Chem. Soc. 1952. V. 74. P. 2125.
Fischer E.O., Pfab W. // Z. Naturforsch. 1952. V. 7. P. 377.
Albada B., Metzler-Nolte N. // Chem. Rev. 2016. V. 116. P. 11797.
Plachida P., Evans D.R., Solanki R. / In: Nanoelectronic Device Applications Handbook. Eds. J.E. Morris, K. Iniewski. Boca Raton: CRC Press. 2013. P. 409.
Bochman M. // Organometallics and Catalysis: An Introduction. Oxford: Oxford University Press. 2015. 432 p.
Comprehensive Organometallic Chemistry II: a Review of the Literature 1982–1994. Eds. E.W. Abel, F.G.A. Stone, G. Wilkinson. Oxford, N.Y.: Pergamon. 1995. V. 5–9.
Schlag E.W. // ZEKE Spectroscopy. Cambridge: Cambridge University Press. 1998.
Choi K.-W., Kim S.K., Ahn D.-S., Lee S. // J. Phys. Chem. A. 2004. V. 108. P. 11292.
Sohnlein B.R., Yang D.-S. // J. Chem. Phys. 2006. V. 124. P. 134305/1.
Yang D.-S. // J. Phys. Chem. Lett. 2011. V. 2. P. 25.
Ketkov S.Y., Selzle H.L., Cloke F.G.N. // Angew. Chem. Int. Ed. 2007. V. 46. P. 7072.
Ketkov S.Y., Selzle H.L., Cloke F.G.N., Markin G.V., Schevelev Y.A., Domrachev G.A., Schlag E.W. // J. Phys. Chem. A. 2010. V. 114. P. 11298.
Ketkov S.Y., Markin G.V., Tzeng S.Y., Tzeng W.B. // Chem. Eur. J. 2016. V. 22. P. 4690.
Ketkov S.Y., Tzeng S.-Y., Wu P.-Y., Markin G.V., T-zeng W.-B. // Chem. Eur. J. 2017. V. 23. P. 13669.
Green J.C. // Structure and Bonding. 1981. V. 43. P. 37.
Ketkov S.Y., Selzle H.L. // Angew. Chem. Int. Ed. 2012. V. 51. P. 11527; Ketkov S.Y., Selzle H.L. // Angew. Chem. 2012. V. 124. P. 11695.
Becke A.D. // J. Chem. Phys. 1992. V. 98. P. 5648.
Perdew P., Burke K., Wang Y. // Phys. Rev. B. 1996. V. 54. P. 16533.
Becke A.D. // Phys. Rev. A. 1988. V. 38. P. 3098.
Perdew J.P., Chevary J.A., Vosko S.H., Jackson K.A., Pederson M.R., Singh D.J., Fiolhais C. // Phys. Rev. B. 1992. V. 46. P. 6671.
Schaefer A., Huber C., Ahlrichs R. // J. Chem. Phys. 1994. V. 100. P. 5829.
Krishnan R., Binkley J.S., Seeger R., Pople J.A. // J. Chem. Phys. 1980. V. 72. P. 650.
Clark T., Chandrasekhar J., Spitznagel G.W., Schle-yer P. v. R. // J. Comp. Chem. 1983. V. 4. P. 294.
Ketkov S. // Dalton Trans. 2020. V. 49. P. 569.
Frisch M.J. et al. GAUSSIAN 09 (Revision B.01), Gaussian Inc., Wallingford CT. 2010.
Zlatar M., Schläpfer C.-W., Fowe E.P., Daul C. // Pure Appl. Chem. 2009. V. 81. P. 1397.
Lippincott E.R., Nelson R.D. // Spectrochimica Acta. 1958. V. 10. P. 307.
Barfuss S., Emrich R.-H., Hirschwald W., Dowben P.A., Boag N.M. // J. Org. Chem. 1990. V. 391. P. 209.
Foffani A., Pignataro S., Distefano G., Innorta G. // J. Organometal. Chem. 1967. V. 7. P. 473.
Bersuker I.B. Jahn – Teller Effect. Cambridge: Cambridge University Press. 2006.
Choi K.-W., Choi S., Baek S.J., Kim S.K. // J. Chem. Phys. 2007. V. 126. P. 034308.
Ketkov S.Y., Selzle H.L., Schlag E.W. // Mol. Phys. 2004. V. 102. P. 1749.
Ketkov S.Yu., Selzle H.L., Schlag E.W., Domrachev G.A. // J. Phys. Chem. A. 2003. V. 107. P. 4041.s
Дополнительные материалы отсутствуют.
Инструменты
Химия высоких энергий