

Химия высоких энергий, 2023, T. 57, № 1, стр. 80-88
Влияние ионизации и спиновых переходов на делокализацию электронов в молекулах сэндвичевых комплексов переходных металлов
С. Ю. Кетков a, *, Е. А. Рычагова a
a Институт металлоорганической химии им. Г.А. Разуваева Российской Академии наук
603950 Нижний Новгород, Россия
* E-mail: sketkov@iomc.ras.ru
Поступила в редакцию 08.07.2022
После доработки 02.09.2022
Принята к публикации 04.09.2022
- EDN: DDWGZK
- DOI: 10.31857/S0023119323010072
Аннотация
В рамках модели электронной плотности делокализованных связей проведено квантово-химическое исследование строения симметричных сэндвичевых комплексов 3d металлов с бензольными и циклопентадиенильными лигандами. Рассмотрены нейтральные и ионизированные молекулы в различных спиновых состояниях. Показано, что последовательное заполнение d-электронной оболочки при варьировании атома металла в ряду однотипных комплексов, как правило, ведет к уменьшению степени делокализации электронной плотности. Отрыв электрона от нейтральных молекул также снижает число делокализованных электронов в сэндвичевой системе, однако вклад атома металла в делокализацию в большинстве случаев при этом возрастает. Синглет-триплетные переходы в металлоценах и бис-бензольных комплексах понижают электронную плотность делокализованных связей, но в меньшей степени чем в свободных лигандах C5${\text{H}}_{5}^{ - }$ и C6H6.
ВВЕДЕНИЕ
В 2022 г. отмечается 70-летний юбилей установления сэндвичевой структуры ферроцена [1, 2], которое открыло новую эпоху в области металлоорганической химии [3, 4]. В настоящее время металлоцены и родственные им бис-ареновые комплексы переходных металлов составляют один из важнейших классов металлоорганических соединений. Заметно возросший в последние годы интерес к сэндвичевым системам связан с их уникальными физико-химическими свойствами, открывающими широкие перспективы для использования в таких актуальных наукоемких областях, как катализ [5], наноэлектроника [6], биомедицина [7] и молекулярные машины [8]. Один из наиболее интересных аспектов строения и реакционной способности сэндвичевых соединений связан с представлениями об их ароматичности. Само название “ферроцен” было предложено группой Р. Вудворта на основе аналогии c аценами, чтобы подчеркнуть ароматические свойства сэндвичевого комплекса [9]. Сэндвичевые соединения сыграли ключевую роль в исследованиях природы делокализованных связей металл-лиганд [10, 11].
Концепции ароматичности и электронной делокализации имеют ключевое значение в современной химии, объясняя относительную стабильность и особенности химического поведения систем с сопряженными связями [12]. В состав сэндвичевых молекул входят два циклических лиганда, которые можно рассматривать как отдельные ароматические фрагменты [13]. Однако электронное сопряжение охватывает и всю сэндвичевую систему в целом, что приводит к способности таких комплексов служить “проводниками” ароматичности при их введении в качестве мостиков в макроциклы большего размера [14, 15].
Металлоцены и родственные соединения представляют собой очень удобные модели для изучения влияния состава и строения металлоорганических молекул на делокализацию электронов, поскольку можно изменять электронную конфигурацию системы, замещая центральный атом металла или карбоциклический лиганд. Кроме того, большинство сэндвичевых комплексов легко окисляются, образуя соответствующие катионы, а близкие энергии верхних занятых молекулярных орбиталей (МО) обеспечивают наличие низколежащих электронных состояний различной мультиплетности. Очевидно, что ионизация сэндвичевых молекул и спиновые переходы, приводящие к изменению заселенностей МО, должны влиять на степень делокализации электронов. Несмотря на огромное количество публикуемых статей, посвященных строению и свойствам металлоценов и их аналогов, систематические исследования такого влияния, по нашим данным, пока не проводились.
В настоящей работе для оценки влияния отрыва электрона и изменения спиновой мультиплетности на характер распределения электронов в сэндвичевых молекулах, содержащих циклопентадиенильные или бензольные лиганды, использован предложенный недавно подход, основанный на анализе электронной плотности делокализованных связей (electron density of delocalized bonds, EDDB) [16, 17]. К преимуществам данного метода можно отнести отсутствие необходимости построения искусственных модельных систем, независимость рассчитываемых параметров от размера циклов, удобную визуализацию (граничные поверхности или контурные карты, описывающие делокализованную электронную плотность) и интерпретацию (количество электронов, участвующих в делокализации) результатов расчета, а также короткое время, требуемое для вычислений. В качестве объектов были выбраны нейтральные комплексы 3d-металлов (Cp)2M (Cp = = η5-C5H5, M = Cr, Mn, Fe, Co) и (BZ)2M (BZ = = η6-C6H6, M = Ti, V, Cr), а также катионы (Cp)2M+ (M = Mn, Fe, Co) и (BZ)2M+ (M = Ti, V, Cr, Mn). Все они представляют собой известные системы, полученные ранее экспериментально [3, 11, 18].
РАСЧЕТНЫЕ МЕТОДЫ
Вычисление молекулярных параметров в рамках модели EDDB [16, 17] основано на использовании результатов DFT-расчетов, включающих анализ естественных связывающих орбиталей (NBO) [19]. Поэтому после оптимизации геометрии каждой сэндвичевой молекулы в заданном зарядовом и спиновом состоянии с помощью программного пакета Gaussian 09 [20] проводился NBO-анализ. Для того, чтобы оценить влияние выбора функционала и базисного набора на результаты, в расчетах использовались гибридный функционал B3PW91 и метагибридные функционалы M06-2X, M06 в сочетании с трехкратно расщепленными (triple-ζ) базисными наборами TZ-VP, def2-TZVP и 6-311+G(d,p), включенными в пакет Gaussian 09.
Функционал B3PW91 и базис TZVP ранее хорошо зарекомендовали себя при моделировании отрыва электрона от сэндвичевых молекул [21–24]. Вклады Хартри–Фока в функционалы B3PW91 (20%) и M06 (27%) сравнительно близки, тогда как в случае M06-2X этот вклад значительно больше (57%). Функционал M06-2X изначально разрабатывался для соединений непереходных элементов и не предназначался для расчетов комплексов переходных металлов [25]. Из-за большого вклада Хартри–Фока модели связывания, рассчитанные на основе M06-2X, содержат повышенную ионную составляющую. Тем не менее оказалось, что применение данного функционала дает хорошие результаты при оценке энергетических параметров реакций с участием цирконоценов [26]. Поэтому он был использован в данной работе для исследования влияния повышенного вклада Хартри–Фока на результаты расчетов делокализованной электронной плотности в сэндвичевых молекулах.
Матрица плотности в базисе естественных атомных орбиталей, полученная после NBO-анализа, была использована для расчета функций EDDB(r) с помощью кода RunEDDB [27]. В соответствии с рекомендациями авторов метода, вычислялись функции, описывающие электронную плотность делокализованных связей без участия атомов водорода (EDDBH(r)) и делокализованную плотность электронов вдоль циклического контура, соответствующего 5- или 6-членному карбоциклу, (EDDBP(r)). Для сравнения с сэндвичевыми комплексами были рассчитаны функции EDDB(r) для свободных органических молекул и ионов ${{{\text{C}}}_{{\text{5}}}}{\text{H}}_{5}^{ - }$, C5H5, C6H6 и ${{{\text{C}}}_{{\text{6}}}}{\text{H}}_{6}^{ + }$.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Согласно общепринятому подходу, при расчете формального числа валентных электронов “классических” сэндвичевых соединений 3d‑элементов с двумя плоскими параллельными карбоциклическими лигандами учитываются 4s- и 3d‑электроны атома металла и π-электроны карбоциклов. Поэтому ферроцен (Cp)2Fe и бис(η6-бензол)хром (BZ)2Cr рассматриваются как “18‑электронные” комплексы. В соответствии с “правилом 18 электронов” [28], они характеризуются повышенной устойчивостью. d-Электронная конфигурация сэндвичевых молекул в общем случае имеет вид [e2(dxy,x2‑y2/π)]m[a1(dz2)]n × × [e1(dxz,yz/π)]k (m = 2–4, n = 0–2, k = 0–2). Для обозначения симметрии МО здесь, как и в большинстве других работ, используются неприводимые представления точечных групп D5 (металлоцены) и D6 (бис-бензольные комплексы). МО a1 представляет собой несвязывающую dz2 орбиталь металла, тогда как орбитали e2 и e1 содержат вклад π-уровней лигандов.
Из-за близких энергий верхних заполненных МО изоэлектронные сэндвичевые комплексы могут иметь различную электронную конфигурацию и разную спиновую мультиплетность [10, 18]. Например, “17-электронные” молекулы (BZ)2Cr+, (Cp)2Fe+ и (BZ)2V находятся в низкоспиновом (2S + + 1 = 2) основном состоянии, а изоэлектронный манганоцен – в высокоспиновом (2S + 1 = 6) [18]. При этом электронные конфигурации ионов различаются. В катионе (BZ)2Cr+ неспаренный электрон заселяет МО a1, а в (Cp)2Fe+ – e2. Как отрыв электрона от сэндвичевой молекулы, так и спиновые переходы приводят к изменению заселенностей МО, что должно влиять на характер делокализации электронов. Это подтверждают квантово-химические исследования, проведенные в настоящей работе.
Расчеты функций EDDBH(r) с использованием функционалов B3PW91 и M06 приводят к очень близким характеристикам делокализации электронов. Так, общее число делокализованных электронов $N_{e}^{{{\text{tot}}}}$ для (BZ)2Cr, вычисленное на уровнях B3PW91/TZVP и M06/def2-TZVP, составляет 14.55 и 14.58 соответственно, для (Cp)2Co –10.77 и 10.77 соответственно. Замена базисного набора def2-TZVP на 6-311+G(d,p) также не привела к существенным изменениям расчетных параметров. Однако при переходе к функционалу M06-2X с повышенным вкладом Хартри–Фока характеристики делокализации для некоторых комплексов заметно меняются. Поэтому результаты для полного ряда исследованных объектов были сопоставлены по расчетам на уровнях B3PW91/TZVP и M06-2X/def2-TZVP (табл. 1, 2).
Таблица 1.
Формальное количество валентных электронов ($N_{e}^{{\text{V}}}$), заряды (q), d-электронные конфигурации и спиновая мультиплетность (2S + 1) металлоценов с указанием общего количества делокализованных электронов $N_{e}^{{{\text{tot}}}}$, рассчитанных по функциям EDDBH (r) жирным шрифтом на уровнях DFT B3PW91/TZVP и M06-2X/def2-TZVP. Приведены вклады атома металла ($N_{e}^{{\text{M}}}$), атома углерода ($N_{e}^{{\text{C}}}$) и циклопентадиенильного лиганда в целом ($N_{e}^{{\text{L}}}$). Для сравнения представлены значения, рассчитанные для свободного C5H5 в анионной и нейтральной формах
| Молекула | $N_{e}^{{\text{V}}}$ (q) | d-электронная конфигурация | 2S + 1 | $N_{e}^{{{\text{tot}}}}$ ($N_{e}^{{\text{M}}}$, $N_{e}^{{\text{C}}}$, $N_{e}^{{\text{L}}}$) B3PW91/TZVP | $N_{e}^{{{\text{tot}}}}$ ($N_{e}^{{\text{M}}}$, $N_{e}^{{\text{C}}}$, $N_{e}^{{\text{L}}}$) M06-2X/def2-TZVP |
|---|---|---|---|---|---|
| (Cp)2Co | 19 (0) | [e2]4[a1]2[e1]1 | 2a | 10.77 (2.08, 0.87, 4.35) | 10.77 (1.36, 0.94, 4.71) |
| (Cp)2Fe | 18 (0) | [e2]4[a1]2 | 1a | 11.29 (2.72, 0.86, 4.29) | 10.95 (1.79, 0.92, 4.58) |
| (Cp)2Fe | 18 (0) | [e2]3[a1]2[e1]1 | 3 | 10.79 (1.74, 0.91, 4.53) | 10.70 (1.45, 0.93, 4.63) |
| (Cp)2Fe+ | 17 (+1) | [e2]3[a1]2 | 2a | 10.74 (2.84, 0.79, 3.95) | 10.90 (2.35, 0.86, 4.28) |
| (Cp)2Co+ | 18 (+1) | [e2]4[a1]2 | 1a | 10.61 (2.77, 0.78, 3.92) | 10.85 (2.33, 0.85, 4.26) |
| (Cp)2Co+ | 18 (+1) | [e2]3[a1]2[e1]1 | 3 | 9.72б (2.10, 0.76б, 3.81б) | 9.38б (1.32, 0.81б, 4.03б) |
| (Cp)2Mn | 17 (0) | [e2]4[a1]1 | 2 | 11.37 (2.81, 0.86, 4.28) | 11.25 (2.01, 0.92, 4.62) |
| (Cp)2Mn | 17 (0) | [e2]2[a1]1[e1]2 | 6 a | 10.72 (0.83б, 0.99в, 4.95в) | 10.88 (0.75б, 1.01б, 5.07в) |
| (Cp)2Mn+ | 16 (+1) | [e2]4 | 1 | 10.89 (2.94в, 0.80, 3.98) | 11.13 (2.47в, 0.87, 4.33) |
| (Cp)2Mn+ | 16 (+1) | [e2]3[a1]1 | 3a | 11.08 (2.55, 0.85, 4.27) | 11.08 (2.14, 0.89, 4.47) |
| (Cp)2Cr | 16 (0) | [e2]4 | 1 | 11.66в (2.09, 0.96, 4.79) | 11.60в (2.30, 0.93, 4.65) |
| (Cp)2Cr | 16 (0) | [e2]3[a1]1 or [e2]2[a1]2 | 3 a | 11.80 (2.42, 0.94, 4.69)г | 10.86 (1.31, 0.96, 4.78)д |
| ${{{\text{C}}}_{{\text{5}}}}{\text{H}}_{5}^{ - }$ | 6 (–1) | – | 1 a | 5.94 (–, 1.19, –) | 5.96 (–, 1.19, –) |
| ${{{\text{C}}}_{{\text{5}}}}{\text{H}}_{5}^{ - }$ | 6 (–1) | – | 3 | 2.10 (–, 0.42, –) | 2.01 (–, 0.40, –) |
| C5H5 | 5 (0) | – | 2 a | 3.09 (–, 0.62, –) | 3.14 (–, 0.63, –) |
Таблица 2.
Формальное количество валентных электронов ($N_{e}^{{\text{V}}}$), заряды (q), d-электронные конфигурации и спиновая мультиплетность (2S + 1) бис-бензольных комплексов с указанием общего количества делокализованных электронов $N_{e}^{{{\text{tot}}}}$, рассчитанного по функциям EDDBH (r) жирным шрифтом на уровнях DFT B3PW91/TZVP и M06-2X/def2-TZVP. Приведены вклады в делокализованную электронную плотность атома металла ($N_{e}^{{\text{M}}}$), атома углерода ($N_{e}^{{\text{C}}}$) и бензольного лиганда в целом ($N_{e}^{{\text{L}}}$). Для сравнения представлены значения, рассчитанные для свободного C6H6 в нейтральной и катионной формах
| Молекула | $N_{e}^{{\text{V}}}$ (q) | d-электронная конфигурация | 2S + 1 | $N_{e}^{{{\text{tot}}}}$ ($N_{e}^{{\text{M}}}$, $N_{e}^{{\text{C}}}$, $N_{e}^{{\text{L}}}$) B3PW91/TZVP | $N_{e}^{{{\text{tot}}}}$ ($N_{e}^{{\text{M}}}$, $N_{e}^{{\text{C}}}$, $N_{e}^{{\text{L}}}$) M06-2X/def2-TZVP |
|---|---|---|---|---|---|
| (BZ)2Cr | 18 (0) | [e2]4[a1]2 | 1a | 14.55 (3.36, 0.93, 5.60) | 14.27 (3.08, 0.93, 5.60) |
| (BZ)2Cr | 18 (0) | [e2]4[a1]1[e1]1 or [e2]3[a1]2[e1]1 | 3 | 14.29 (2.74, 0.96, 5.78)б | 13.42 (1.85, 0.96, 5.79)в |
| (BZ)2Cr+ | 17 (+1) | [e2]4[a1]1 | 2a | 13.85 (3.37, 0.87, 5.24) | 13.60 (2.70, 0.91, 5.45) |
| (BZ)2Mn+ | 18 (+1) | [e2]4[a1]2 | 1a | 13.61 (3.25, 0.86г, 5.18г) | 12.79 (2.24, 0.88г, 5.28г) |
| (BZ)2Mn+ | 18 (+1) | [e2]4[a1]1[e1]1 or [e2]3[a1]2[e1]1 | 3 | 13.18г (2.48, 0.89, 5.35)б | 12.25г (1.49г, 0.90, 5.38)в |
| (BZ)2V | 17 (0) | [e2]4[a1]1 | 2a | 15.02 (3.23, 0.98, 5.90) | 14.77 (2.85, 0.99, 5.96) |
| (BZ)2V+ | 16 (+1) | [e2]4 | 1 | 14.57 (3.56д, 0.92, 5.51) | 14.44 (3.09д, 0.95, 5.68) |
| (BZ)2V+ | 16 (+1) | [e2]3[a1]1 | 3a | 13.65 (2.59, 0.92, 5.53) | 13.02 (1.93, 0.93, 5.55) |
| (BZ)2Ti | 16 (0) | [e2]4 | 1a | 15.36д (3.10, 1.02д, 6.13д) | 15.49д (2.92, 1.05д, 6.29д) |
| (BZ)2Ti | 16 (0) | [e2]3[a1]1 | 3 | 14.06 (2.29г, 0.98, 5.89) | 14.13 (2.13, 1.00, 6.00) |
| (BZ)2Ti+ | 15 (+1) | [e2]3 | 2a | 13.96 (2.64, 0.94, 5.66) | 14.01 (2.35, 0.97, 5.83) |
| C6H6 | 6 (0) | – | 1a | 5.60 (–, 0.93, –) | 5.60 (–, 0.93, –) |
| C6H6 | 6 (0) | – | 3 | 3.01 (–, 0.50, –) | 1.71 (–, 0.29, –) |
| ${{{\text{C}}}_{{\text{6}}}}{\text{H}}_{6}^{ + }$ | 5 (+1) | – | 2a | 4.08 (–, 0.68, –) | 3.36 (–, 0.56, –) |
Как правило, при переходе к M06-2X наблюдается уменьшение значений $N_{e}^{{{\text{tot}}}}$ и вкладов атома металла $N_{e}^{{\text{M}}}$ в функцию EDDBH(r). Это можно объяснить более ионным характером связей в молекулярных моделях, построенных с помощью этого функционала. В ряде триплетных систем (например, (Cp)2Cr, (BZ)2Cr, (BZ)2Mn+) увеличение вклада Хартри–Фока в функционал приводит к инверсии заселенностей МО, что также влияет на делокализацию электронов (табл. 1, 2). В этих молекулах триплетные состояния характеризуются более низкой симметрией, что приводит к смешиванию близких по энергии МО а1 и е2. Поэтому описание электронных конфигураций с использованием данных обозначений МО в этих случаях весьма условно.
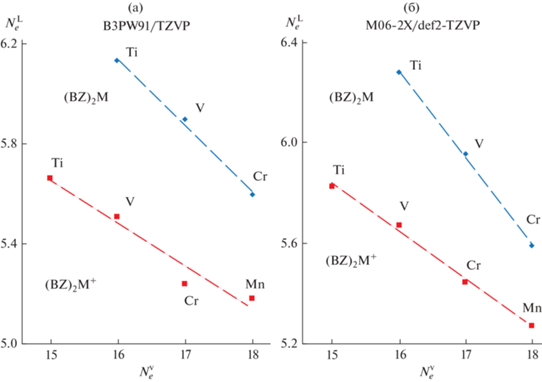
Несмотря на заметные различия в отдельных параметрах, полученных из EDDBH(r), расчеты на уровнях B3PW91/TZVP и M06-2X/def2-TZVP выявляют схожие тенденции в изменении числа делокализованных электронов. Так, наибольшее и наименьшее значение $N_{e}^{{{\text{tot}}}}$ в ряду металлоценов отвечает нейтральному (Cp)2Cr и катиону (Cp)2Co+ соответственно (табл. 1), а в ряду бис-ареновых комплексов – нейтральному (BZ)2Ti и катиону (BZ)2Mn+ соответственно (табл. 2). Оба уровня расчета указывают на уменьшение степени делокализации при увеличении атомного номера металла (и, соответственно, числа электронов на МО a1(dz2) и e1(dxz,yz/π)) в рядах низкоспиновых комплексов одного типа. Эта тенденция наиболее отчетливо проявляется на значениях $N_{e}^{{{\text{tot}}}}$ для металлоценов (рис. 1). С увеличением заселенности а1- и е1-орбиталей, как правило, уменьшается и вклад лиганда $N_{e}^{{\text{L}}}$ в EDDBH(r), что особенно хорошо видно на примере низкоспиновых бис-бензольных комплексов (рис. 2).
Рис. 1.
Общее количество делокализованных электронов $N_{e}^{{{\text{tot}}}}$ в нейтральных молекулах и ионах металлоценов в низкоспиновых состояниях с различным формальным числом валентных электронов $N_{e}^{{\text{V}}}$, рассчитанное на уровнях DFT B3PW91/TZVP (a) и M06-2X/def2-TZVP (б).
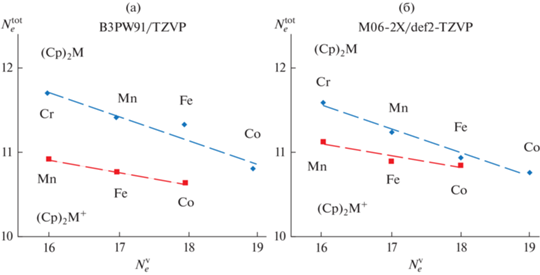
Рис. 2.
Вклады бензольного лиганда NeL в общее число делокализованных электронов $N_{e}^{{{\text{tot}}}}$ в комплексах (BZ)2M и (BZ)2M+ (M = Ti, V, Cr, Mn) в низкоспиновых состояниях с различным формальным числом валентных электронов $N_{e}^{{\text{V}}}$, рассчитанные на уровнях DFT B3PW91/TZVP (a) и M06-2X/def2-TZVP (б).
Данные, приведенные в табл. 1, 2 и на рис. 1, 2, показывают, что при ионизации сэндвичевых молекул делокализованная электронная плотность обычно уменьшается, но изменение значения $N_{e}^{{{\text{tot}}}}$, как правило, меньше 1. Исключение составляют (BZ)2V и (BZ)2Ti, ионизация которых приводит к уменьшению $N_{e}^{{{\text{tot}}}}$ на ~1.5, что приближается к аналогичной величине для свободного бензола (табл. 2). Следует отметить, что взаимодействие карбоциклического лиганда с металлом кардинально изменяет топологию функции EDDBH(r). Если в свободных лигандах C5H$_{5}^{ - }$ и C6H6 присутствуют две отдельные области, разделенные плоскостью карбоцикла, то в нейтральных и катионных сэндвичевых комплексах EDDBH(r) образует единую область, охватывающую всю молекулу (рис. 3, 4).
Рис. 3.
Контурные карты функции EDDBH(r) для (BZ)2Ti (a), (BZ)2Ti+ (б), C6H6 (в) и ${{{\text{C}}}_{{\text{6}}}}{\text{H}}_{6}^{ + }$ (г) в плоскости, перпендикулярной карбоциклам и проходящей через центр связи С–С. Интервал значений EDDBH(r) 0.01–0.05 ат. ед., шаг 0.005 ат. ед. Обозначены максимальные значения функции в области экстремумов. Для титановых комплексов показан один из бензольных лигандов. Уровень расчета B3PW91/TZVP.
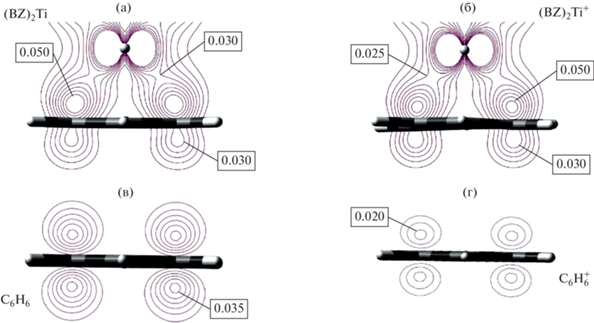
Рис. 4.
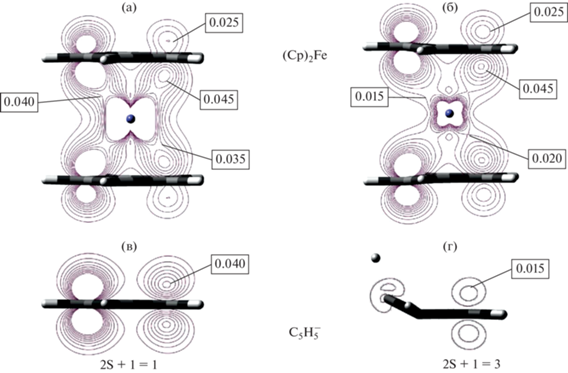
Граничные поверхности EDDBH(r) для низкоспиновых (Cp)2Mn (a) и (Cp)2Mn+ (б) в сравнении с анионом ${{{\text{C}}}_{{\text{5}}}}{\text{H}}_{5}^{ - }$ (в) и радикалом ${{{\text{C}}}_{{\text{5}}}}{\text{H}}_{5}^{\centerdot }$ (г). Уровень расчета B3PW91/TZVP. Значение функции 0.02 ат. ед.
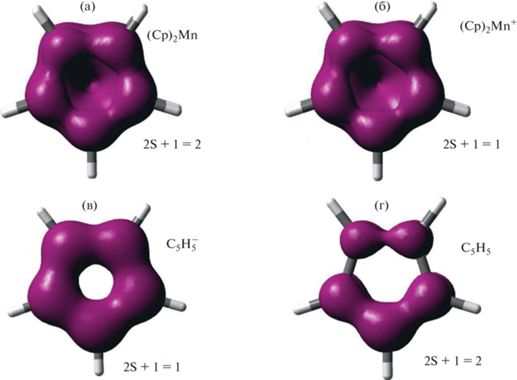
При ионизации сэндвичевых молекул EDDBH(r) изменяется неоднородно. В частности, отрыв электрона от (BZ)2Ti приводит к некоторому уменьшению этой функции в окрестностях атома металла и около плоскости бензольного кольца в межлигандном пространстве (рис. 3а, б). В то же время с внешней стороны плоскости карбоцикла делокализованная плотность даже несколько увеличивается. Совершенно иное поведение функции EDDBH(r) характерно для свободных лигандов, когда при ионизации происходит просто сильное уменьшение ее значений с обеих сторон от плоскости цикла (рис. 3в, г). Такое сильное различие связано с особенностями ионизации сэндвичевых комплексов, где отрывается электрон, находящийся на МО с большим d-вкладом атома металла. Интересно, что, несмотря на такой тип ионизации и уменьшение значения $N_{e}^{{{\text{tot}}}}$, вклад атома металла в EDDBH(r) для сэндвичевых катионов, как правило, выше, чем для нейтральных молекул (табл. 1, 2). Это обусловлено переносом электронной плотности с лиганда на металл при релаксации сэндвичевой молекулы после отрыва электрона [24]. Соответственно, вклад лигандов в электронную плотность делокализованных связей в сэндвичевых катионах уменьшается, а вклад металла возрастает (табл. 1, 2). Разная природа ионизируемых МО в свободных лигандах и сэндвичевых комплексах приводит к тому, что в первом случае при отрыве электрона циклическая π-ароматическая система распадается на фрагменты, а во втором таких драматических изменений не происходит (рис. 4).
При спиновых переходах с увеличением мультиплетности, как и при ионизации, число делокализованных электронов в большинстве исследованных сэндвичевых комплексов несколько снижается, причем происходит это в основном из-за уменьшения вклада металла $N_{e}^{{\text{M}}}$ (табл. 1, 2). Наибольшее изменение значения $N_{e}^{{\text{M}}}$ отмечается при дублет-секстетном переходе в молекуле (Cp)2Mn (табл. 1), для которой высокоспиновое состояние характеризуется повышенной степенью ионности связи металл-лиганд [29]. Типичная картина изменения пространственного распределения функции EDDBH(r) при синглет-триплетном переходе в сэндвичевой молекуле показана на примере ферроцена (рис. 5). Делокализованная электронная плотность существенно уменьшается в окрестностях атома железа и в области между металлом и лигандом. При этом с внешней стороны карбоцикла EDDBH(r) несколько возрастает. В целом, функция EDDBH(r) в сэндвичевом комплексе изменяется не столь существенно, как при синглет-триплетном переходе в свободном анионе ${{{\text{С}}}_{{\text{5}}}}{\text{H}}_{5}^{ - }$, где в триплетном состоянии очень сильно искажается молекулярная геометрия, а ароматическая π-система распадается (рис. 5). Бензол, который при переходе в триплетное состояние становится антиароматическим [30], также характеризуется более резким уменьшением числа делокализованных электронов по сравнению с бис-бензольными комплексами (табл. 2).
Рис. 5.
Контурные карты функции EDDBH(r) для (Cp)2Fe в синглетном (a) и триплетном (б) электронном состоянии. Сечение проходит в плоскости CFeC, перпендикулярной карбоциклам. Для сравнения приведены аналогичные карты для иона (C5H5)– в синглетном (в) и триплетном (г) электронном состоянии. Интервал значений EDDBH(r) 0.01–0.05 ат. ед., шаг 0.005 ат. ед. Обозначены максимальные значения в области экстремумов. Уровень расчета B3PW91/TZVP.
Значения $N_{e}^{{\text{L}}}$, описывающие вклад лиганда в делокализацию электронной плотности, для металлоценов всегда ниже, чем величина $N_{e}^{{{\text{tot}}}}$ для свободного ароматического аниона ${{{\text{С}}}_{{\text{5}}}}{\text{H}}_{5}^{ - }$ (табл. 1). В отличие от этого, для комплексов (BZ)2M вклады лиганда в делокализованную электронную плотность близки к значению $N_{e}^{{{\text{tot}}}}$ для молекулы бензола, а в случае производных ванадия и титана превосходят эту величину (табл. 2). На первый взгляд, такие значения $N_{e}^{{\text{L}}}$ могут говорить о большей степени ароматичности лигандов в молекулах (BZ)2Ti и (BZ)2V по сравнению с бензолом. Однако более подробный анализ показал, что делокализация электронов в карбоциклах сэндвичевых комплексов во многом обусловлена взаимодействиями не внутри карбоциклического фрагмента, а между металлом и лигандом (рис. 3, 5).
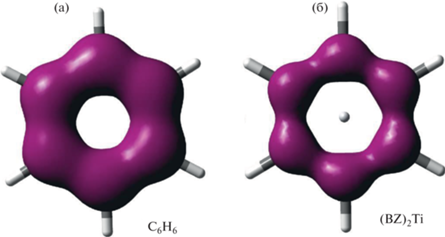
Расчет функции EDDBP(r) [16, 17], описывающей делокализацию электронной плотности вдоль циклического пути, в случае свободного бензола не приводит к заметному изменению числа делокализованных электронов. В то же время, для (BZ)2Ti количество электронов, участвующих в делокализации по карбоциклическому контуру в соответствии с EDDBP(r) (3.04), существенно меньше вклада лиганда, рассчитанного на уровне B3PW91/TZVP из EDDBH(r) (6.13). Таким образом, делокализованная электронная плотность, обусловленная взаимодействиями внутри карбоциклического фрагмента, заметно уменьшается при переходе от свободного ароматического лиганда к сэндвичевому комплексу, что отражается на граничных поверхностях EDDBP(r) (рис. 6).
ЗАКЛЮЧЕНИЕ
Использование метода электронной плотности делокализованных связей (EDDB) в исследовании строения сэндвичевых соединений открывает новые аспекты влияния природы металла и лиганда, а также ионизации и спиновых переходов на характер делокализации электронной плотности в металлоорганических молекулах. Выполненные в настоящей работе расчеты функций EDDBH(r) и EDDBP(r) выявили принципиальные различия природы взаимодействий, влияющие на число делокализованных электронов в сэндвичевых системах и свободных лигандах. В металлокомплексах ключевое значение для участия кар-боциклов в делоклизации имеют взаимодействия металл-лиганд. Последовательное заполнение d-электронной оболочки при увеличении атомного номера металла в ряду однотипных комплексов, как правило, ведет к монотонному уменьшению степени делокализации электронной плотности. Ионизация нейтральных молекул также снижает число делокализованных электронов в сэндвичевых системах, но при этом вклад атома металла в делокализацию в большинстве случаев возрастает. Синглет-триплетные переходы в металлоценах и бис-бензольных комплексах понижают электронную плотность делокализованных связей, но в меньшей степени, чем в свободных лигандах ${{{\text{С}}}_{{\text{5}}}}{\text{H}}_{5}^{ - }$ и C6H6. В отличие от ионизации, такие переходы обычно приводят к уменьшению вклада атома металла в делокализованную электронную плотность. Различные механизмы делокализации электронов в сэндвичевых молекулах и свободных лигандах необходимо учитывать при анализе свойств металлоценов и родственных соединений в рамках современных теорий ароматичности.
Список литературы
Wilkinson G., Rosenblum M., Whiting M.C., Woodward R.B. // J. Am. Chem. Soc. 1952. V. 74. P. 2125.
Fischer E.O., Pfab W. // Z. Naturforsch. 1952. V. 7. P. 377.
Comprehensive Organometallic Chemistry II: a Review of the Literature 1982–1994 / Eds. Abel E.W., Stone F.G.A., Wilkinson G. Oxford, N.Y.: Pergamon, 1995. Vols. 5–9.
Laszlo P., Hoffmann R. // Angew. Chem. Int. Ed. 2000. V. 39. P. 123.
Bochman M. // Organometallics and Catalysis: An Introduction. Oxford: Oxford University Press. 2015, 432 p.
Plachida P., Evans D.R., Solanki R. / In: Nanoelectronic Device Applications Handbook. Eds. Morris J. E., Iniewski K. Boca Raton: CRC Press, 2013, pp. 409–420.
Ihara T. / In: Advances in Bioorganometallic Chemistry. Eds. Hirao T., Moriuchi T. Elsevier, Amsterdam, 2019. Chapter 14. pp. 277–303.
Scottwell S.O., Barnsley J.E., McAdam C.J., Gordon K.C., Crowley J.D. // Chem. Commun. 2017. V. 53. P. 7628.
Woodward R.B., Rosenblum M., Whiting M.C. // J. Am. Chem. Soc. 1952. V. 74. P. 3458.
Clack D.W., Warren K.D. // Struct. Bond. 1980. V. 39. P. 1.
Elschenbroich C. Organometallics. 3rd edn. Wiley-VCH, Weinheim, 2006. Chapter 15, pp. 528–549.
Aromaticity: Modern Computational Methods and Applications / Ed. Fernandez I. Elsevier Science, 2021. 499 p.
Bean D.E., Fowler P.W., Morris M.J. // J. Organomet. Chem. 2011. V. 696. P. 2093.
Grocka I., Latos-Grazynski L., Stepien M. // Angew. Chem. Int. Ed. 2013. V. 52. P. 1044.
Valiev R.R., Kurten T., Valiulina L.I., Ketkov S.Y., Cherepanov V.N., Dimitrova M., Sundholm D. // Phys. Chem. Chem. Phys. 2022. V. 24. P. 1666.
Szczepanik D.W., Zak E.J., Dyduch K., Mrozek J. // Chem. Phys. Lett. 2014. V. 593. P. 154.
Szczepanik D.W., Andrzejak M., Dominikowska J., Pawelek B., Krygowski T.M., Szatylowicz H., Sola M. // Phys. Chem. Chem. Phys. 2017. V. 19. P. 28970.
Green J.C. // Struct. Bond. 2019. V. 181. P. 81.
Reed A.E., Curtiss L.A., Weinhold F. // Chem. Rev. 1988. V. 88. P. 899.
Frisch M.J. et al. GAUSSIAN 09 (Revision D.01), Gaussian Inc., Wallingford CT. 2010.
Ketkov S.Y., Selzle H.L. // Angew. Chem. Int. Ed. 2012. V. 51. P. 11527.
Ketkov S. // Dalton Trans. 2020. V. 49. P. 569.
Ketkov S.Y., Rychagova E.A., Zhigulin G.Y., Tzeng S.Y., Tzeng W.B. // High Energ. Chem. 2020. V. 54. P. 414.
Ketkov S.Y., Tzeng S.Y., Rychagova E.A., Markin G.V., Makarov S.G., Tzeng W.B. // Dalton Trans. 2021. V. 50. P. 10729.
Zhao Y., Truhlar D.G. // Theor. Chem. Acc. 2008. V. 120. P. 215.
Sun Y., Chen H. // J. Chem. Theory Comput. 2013. V. 9. P. 4735.
Szczepanik D.W. RunEDDB. Available at: http://www.eddb.pl
Rasmussen S.C. // ChemTexts. 2015. V. 1. № 1. Article 10.
Layfield R.A. // Chem. Soc. Rev. 2008. V. 37. P. 1098.
Baird N.C. // J. Am. Chem. Soc. 1972. V. 94. P. 4941.
Дополнительные материалы отсутствуют.
Инструменты
Химия высоких энергий